

Abstract
The RV144 vaccine trial in Thailand demonstrated that an HIV vaccine could prevent infection in humans and highlights the importance of understanding protective immunity against HIV. We used a nonhuman primate model to define immune and genetic mechanisms of protection against mucosal infection by the simian immunodeficiency virus (SIV). A plasmid DNA prime/recombinant adenovirus serotype 5 (rAd5) boost vaccine regimen was evaluated for its ability to protect monkeys from infection by SIVmac251 or SIVsmE660 isolates after repeat intrarectal challenges. Although this prime-boost vaccine regimen failed to protect against SIVmac251 infection, 50% of vaccinated monkeys were protected from infection with SIVsmE660. Among SIVsmE660-infected animals, there was an about one-log reduction in peak plasma virus RNA in monkeys expressing the major histocompatibility complex class I allele Mamu-A*01, implicating cytotoxic T lymphocytes in the control of SIV replication once infection is established. Among Mamu-A*01–negative monkeys challenged with SIVsmE660, no CD8+ T cell response or innate immune response was associated with protection against virus acquisition. However, low levels of neutralizing antibodies and an envelope-specific CD4+ T cell response were associated with vaccine protection in these monkeys. Moreover, monkeys that expressed two TRIM5 alleles that restrict SIV replication were more likely to be protected from infection than monkeys that expressed at least one permissive TRIM5 allele. This study begins to elucidate the mechanism of vaccine protection against immunodeficiency viruses and highlights the need to analyze these immune and genetic correlates of protection in future trials of HIV vaccine strategies.
INTRODUCTION
The goal for an effective AIDS vaccine is to generate immunity that prevents the acquisition of HIV-1. However, achieving this goal has proven elusive. Two clinical trials of an HIV-1 envelope (Env) protein immunogen showed no efficacy of this vaccine, and the administration of a replication-defective adenovirus vector expressing HIV-1 genes was associated with increased acquisition of HIV-1 infections in a subpopulation of vaccinees (1–4). However, the recent modest success of a two-modality vaccine regimen (an HIV-1 Env protein and a recombinant canary pox construct expressing HIV-1 Env) in a population of Thai volunteers (the RV144 trial) has renewed the focus of investigators on developing a vaccine that can prevent infection with HIV-1 (5). The results of this trial showed vaccine efficacy up to 3 years after initiating vaccination, with a 31.2% reduction in infection rate.
An effective or even a partially effective AIDS vaccine in humans or monkeys could prove enormously instructive because it would provide an opportunity to explore immune and genetic correlates of protection against virus acquisition. Elucidating the vaccine-elicited immune responses that are associated with protection in vaccinees would create a rationale for focusing attention on developing immunogens that generate such immune responses. However, attempts to pursue such studies have been frustrated by the absence of even partial protection against acquisition of HIV in humans or simian immunodeficiency virus (SIV) in monkeys.
The present study was initiated to examine the protective efficacy of a plasmid DNA prime/recombinant adenovirus serotype 5 (rAd5) boost vaccine regimen against acquisition of two divergent SIV isolates by monkeys after low-dose repeat mucosal exposures to the virus. Although this vaccine regimen differs from that used in the RV144 Thai trial, it has been shown to induce robust immune responses to HIV-1 in humans and to SIV in monkeys. Because the plasmid DNA prime/rAd5 boost vaccine regimen provided partial protection against one of the SIV isolates in the study, we examined both immune and genetic correlates of vaccine protection against SIV acquisition in this nonhuman primate model.
RESULTS
To evaluate vaccine protection in rhesus monkeys against repeated, mucosal challenge with a pathogenic SIV isolate, we immunized Indian-origin rhesus monkeys with plasmid DNA (4 mg per construct, intra-muscularly, at weeks 0, 4, and 8) and rAd5 vectors (1 × 1011 particles per construct, intramuscularly, at week 26) using vectors expressing SIVmac239 env and gag-pol. To assess vaccine protection against different SIV isolates, we challenged monkeys about 4 months after the rAd5 boost with a quasi-species of either the closely related SIV-mac251 or the genetically distant SIVsmE660.
The mucosal virus challenges were carried out weekly for 12 consecutive weeks by intrarectal inoculations with one AID50 (median animal infectious dose) of virus challenge stock. Plasma SIV RNA levels were assessed each week, and monkeys that had detectable plasma virus were excluded from subsequent virus challenges. Three parallel studies were performed: (i) a two-arm experiment using Mamu-A*01–, Mamu-B*08–, and Mamu-B*17–negative Indian-origin rhesus monkeys with an SIVmac251 challenge (n = 40) [these monkeys did not express major histocompatibility complex (MHC) class I alleles that are associated with the development of potent cytotoxic T lymphocyte responses that confer protection against rapid SIV replication]; (ii) a two-arm experiment using Mamu-A*01–, Mamu-B*08–, and Mamu-B*17–negative Indian-origin rhesus monkeys with an SIVsmE660 challenge (n = 50); and (iii) a two-arm experiment using Mamu-A*01–positive Indian-origin rhesus monkeys, with an SIVsmE660 challenge (n = 39). The control arm of each of these experiments was composed of sham-vaccinated animals. The conditions used in these virus challenge studies provided an 85% power to detect a 50% reduction in the per-challenge risk of virus acquisition for the 50-monkey experiment and about a 75% power to detect this effect size for the 40-and 39-monkey experiments (6).
The endpoints in each of these experiments were acquisition of infection and peak plasma virus RNA concentrations. In the first experiment, vaccination had no impact on acquisition of infection with SIVmac251, a virus that is very close in genetic sequence to the virus used in the construction of the vaccine immunogens (Fig. 1A). Nevertheless, the experimentally vaccinated group of monkeys had about a one-log10 lower peak plasma virus RNA level than the group of sham-vaccinated monkeys (Fig. 1A).
Fig. 1.

Protection against SIV acquisition and peak plasma virus RNA concentrations after infection. Low-dose virus challenges involved weekly intrarectal inoculations with one AID50 of virus challenge stock for 12 consecutive weeks. Plasma SIV RNA concentrations were assessed weekly, and monkeys that had detectable plasma virus were excluded from subsequent virus challenges. Shown are Kaplan-Meier curves for SIV acquisition and peak plasma SIV RNA concentrations during primary infection. (A to C) Three parallel studies were performed: (A) a two-arm experiment with Mamu-A*01–negative rhesus monkeys and an SIVmac251 challenge (n = 40); (B) a two-arm experiment with Mamu-A*01–negative rhesus monkeys and an SIVsmE660 challenge (n = 50); and (C) a two-arm experiment with Mamu-A*01–positive rhesus monkeys and an SIVsmE660 challenge (n = 39). The control arm of each of these experiments comprised sham-vaccinated animals. (D) The results of the two SIVsmE660 challenge studies were combined and displayed as a single study. The differences between the two groups of monkeys in acquisition of infection and in peak plasma virus RNA concentrations were analyzed by the log-rank test and by the Wilcoxon rank-sum test, respectively.
In the second experiment, after 12 successive challenges, experimentally vaccinated monkeys had about a 50% lower rate of infection with SIVsmE660 than the sham-vaccinated monkeys (48% versus 88%, P = 0.001; Fig. 1B). This challenge virus is as distant genetically from the sequence of the virus used in the construction of the vaccine immunogens as two of the most genetically disparate clade-identical HIV-1 isolates (7). Vaccination of these Mamu-A*01–negative rhesus monkeys had no effect on peak plasma virus RNA concentrations after infection (Fig. 1B). In the third experiment in Mamu-A*01–positive rhesus monkeys, experimentally vaccinated monkeys also had about a 50% lower rate of infection with SIVsmE660 than the sham-vaccinated monkeys (Fig. 1C). In addition, these experimentally vaccinated monkeys had about a one-log10 lower peak plasma virus RNA concentration than the group of sham-vaccinated monkeys (37% versus 75%, P = 0.009; Fig. 1C). When the results of the second and third vaccine experiment were combined for analysis, the statistical significance of the difference in SIVsmE660 acquisition between experimentally vaccinated and sham-vaccinated monkeys was significant (Fig. 1D, P = 4 × 10−5 with a log-rank test). Therefore, these experiments demonstrated protection against acquisition of the SIVsmE660 isolate but not the SIVmac251 isolate. Moreover, they showed that the effects of vaccination on virus acquisition and virus load after infection are distinct, but that both effects can be manifest in the same group of vaccinated challenged monkeys. No effect of the vaccine was apparent on set-point plasma virus RNA concentration or loss of CD4+ T lymphocytes after SIV challenge in these cohorts of monkeys (figs. S1 and S2).
The availability of these experimentally vaccinated monkeys in which about 50% were protected from SIVsmE660 infection provided an opportunity to explore the immune correlates associated with protection against acquisition of the virus. To examine possible correlates without imposing a bias on the findings, we evaluated vaccine-elicited cellular immune responses, innate responses, antibody responses, and host genetic factors. These studies were done on both the Mamu-A*01–negative and the Mamu-A*01–positive animals challenged with SIVsmE660. Given that these studies were done to define immune and genetic determinants of vaccine protection, only the experimentally vaccinated monkeys were evaluated. These vaccinated monkeys were divided into two groups: those that were protected and those that became infected.
SIV-specific cellular immune responses in the experimentally vaccinated monkeys were evaluated by interferon-γ (IFN-γ) enzyme-linked immunospot (ELISpot) and intracellular cytokine staining; total CD4+ and CD8+ T cell populations were characterized by phenotypic profiling. Unfractionated peripheral blood mononuclear cells (PBMCs) evaluated in a pooled peptide ELISpot assay for SIVmac239 Env-, Gag-, and Pol-specific cellular immune responses on the day of challenge demonstrated no differences between the infected and the uninfected monkeys (Fig. 2). PBMCs sampled at week 46, 16 weeks after the rAd boost, were also evaluated by intracellular cytokine staining after pooled peptide stimulation for SIV Env- and Gag-specific T cell responses. These studies were performed with both SIVmac239 and SIVsmE543 peptide pools. No statistically significant differences were noted between the infected and the uninfected monkeys in the magnitude of either the CD4+ or the CD8+ T cell responses (Fig. 3). An evaluation of the polyfunctionality of these cellular responses also did not differentiate between the monkeys that became infected and the monkeys that did not become infected (Fig. 3). Finally, a phenotypic characterization of the total CD4+ for expression of cell surface molecules associated with memory did not differentiate the monkeys that became infected from the monkeys that did not become infected (fig. S3).
Fig. 2.

Vaccine-induced pooled peptide ELISpot responses to SIV proteins. Monkey PBMCs were assessed in a pooled peptide ELISpot assay for SIVmac239 Env-, Gag-, and Pol-specific cellular immune responses on the day of challenge. Each plotted data point represents the sum of spot-forming cell (SFC) responses for PBMCs of an individual monkey specific for SIVmac239 Env, Gag, and Pol. Data points are for two studies: the Mamu-A*01–negative monkeys challenged with SIVsmE660 and the Mamu-A*01–positive monkeys challenged with SIVsmE660. In each study, the monkeys were divided into two groups: vaccinated monkeys that became infected and vaccinated monkeys that did not become infected. No statistically significant differences between the magnitudes of the responses in these groups in each study were noted as determined by the Mann-Whitney test.
Fig. 3.
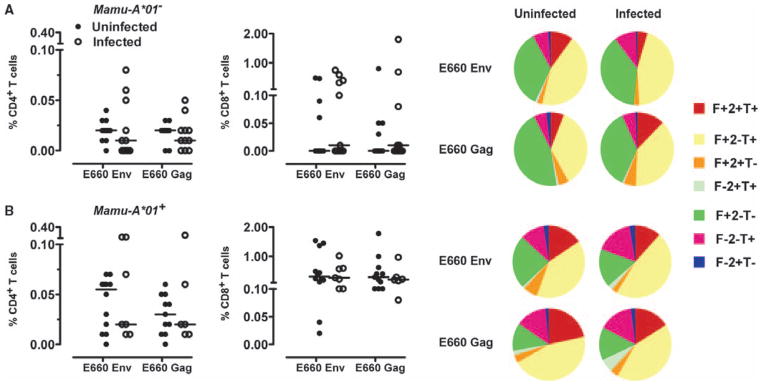
Vaccine-induced virus-specific cellular immune responses. (A and B) PBMCs isolated from Mamu-A*01–negative (A) and Mamu-A*01–positive (B) monkeys after the boost immunization were exposed to pools of overlapping peptides spanning the Gag or Env proteins of SIVsmE660, and the fractions of CD4+ or CD8+ T cells producing IFN-γ (F), TNF-α (T), or IL-2 (2) were determined by intracellular cytokine staining. Data are presented as the frequency of cytokine-producing CD4+ or CD8+ T cells from the groups of monkeys. The cytokine profiles of these cells were determined by expressing each cytokine response as a proportion of the total antigen-specific cytokine-producing CD4+ and CD8+ T cell response. Data were analyzed with the SPICE software and are presented in pie charts as the mean values from the experimental groups of monkeys. No statistically significant differences between the magnitudes of the responses in these groups in each study were noted as determined by the Mann-Whitney test.
Mediators of innate immune responses were also evaluated in these monkeys 4 weeks after the rAd boost. Natural killer (NK) cell activity, assessed by determining CD3−CD20−CD8+NKG2A+ PBMC expression of molecules associated with NK cell activation and function, was comparable in the monkeys that did and did not become infected (fig. S4). Further, Luminex technology was used to assess plasma concentrations of a diversity of cytokines 4 weeks after the rAd boost. No differences were documented in the patterns of cytokine expression in the vaccinated monkeys that became infected and in the vaccinated monkeys that did not become infected (fig. S5). The anti-Env antibodies generated by these vaccinated monkeys were assessed with binding, antibody-dependent cellular cytotoxicity (ADCC), and neutralization assays. To evaluate serum binding to the challenge virus, we determined ED50 (median effective dose) values for serum binding to SIVsmE660 gp140 using serum sampled from both Mamu-A*01–positive and Mamu-A*01–negative monkeys that were challenged with SIVsmE660 at both week 34 and day of challenge time points. No statistically significant differences in these values were observed between the vaccinated monkeys that became infected and those that did not become infected (Fig. 4). The avidity of the anti-Env antibodies in these two groups of monkeys, measured by sodium thiocyanate (NaSCN) sensitivity of antibody binding to Env protein, did not differ (Fig. 4). Further, ADCC activity, as measured with recombinant Env protein–pulsed target cells, was not different in these two groups of animals (Fig. 5).
Fig. 4.
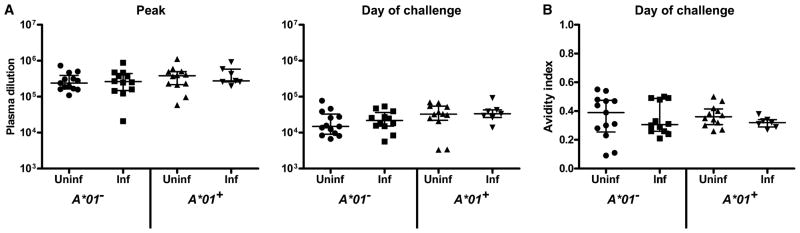
Vaccine-induced antibody binding to SIVsmE660. (A) Sera sampled 2 weeks after the rAd5 boost (week 34) and from the day of first SIVsmE660 viral challenge (week 53) were assayed by ELISA for the level of antibody binding to SIVsmE660 gp140. Values are the reciprocal plasma dilution yielding 50% half-maximal binding (ED50). (B) Avidity index values from sera on the day of challenge. Avidity indices were calculated by dividing ED50 values with NaSCN treatment by ED50 values without treatment. P values were calculated with a Mann-Whitney rank-sum test.
Fig. 5.

Vaccine-induced antibody-dependent cell-mediated cytotoxicity responses to SIVmac251gp120. CD3−CD20−CD56+ human PBMCs (NK cells) were incubated with SIVmac251gp120-coated CEM-NKr-CCR5 target cells and plasma sampled from the monkeys on the day of challenge. The expression of CD107a, IFNγ, TNF-α, and MIP-1β by the NK cells was evaluated by intracellular cytokine staining. Data are expressed as percentage of NK cells expressing these molecules. No statistically significant differences between the magnitudes of the responses in these groups in each study were noted as determined by the Mann-Whitney test.
Before initiating studies of vaccine-induced neutralizing antibody responses in these vaccinated monkeys, we assessed the neutralization sensitivity of the challenge viruses. Although the neutralization resistance of the SIVmac251 challenge stock has been well documented, the neutralization phenotype of the SIVsmE660 stock had not been well characterized. To evaluate the neutralization sensitivity of this SIVsmE660 virus stock, we assayed pseudovirions generated from Env proteins of selected members of the virus quasi-species in TZM-bl cells, and we also assayed the challenge stock itself in TZM-bl and rhesus monkey PBMCs. Although some of the SIVsmE660 Env-based pseudovirions were easy to neutralize (fig. S6A), the CR54-PK-2A5 pseudovirion displayed a more difficult to neutralize (tier 2–like) phenotype (fig. S6B). The challenge stock virus quasi-species was easy to neutralize in TZM-bl cells (fig. S7A), but was tier 2–like in rhesus monkey PBMCs (fig. S7B). Therefore, this SIVsmE660 challenge virus exhibited intermediate sensitivity to neutralization compared to the resistance to neutralization of most SIVmac239/251 stocks.
To evaluate serum neutralization in pseudovirion-based assays, we generated pseudovirions using two different transmitted/founder env genes selected from a separate cohort of monkeys that were infected intrarectally; one pseudovirion had a tier 1– and the other a tier 2–like phenotype (Fig. 6). Assays were performed to assess neutralization by serum sampled from these monkeys at peak immunity and day of challenge time points. Comparable neutralization of the tier 1–like pseudovirion was demonstrated by serum from the vaccinated monkeys that became infected and those that did not become infected (Fig. 6). The sera sampled on the day of challenge from these two groups of monkeys also mediated comparable neutralization of the tier 2–like pseudovirion when assessed by determining ID50 (50% inhibitory dilution) values. However, a significantly lower titer-neutralizing antibody response was measured in the day-of-challenge serum of the Mamu-A*01–negative monkeys that became infected when assessed with the tier 2–like pseudovirion and expressed as percent neutralization with a 1:10 serum dilution (P = 0.03) (Fig. 6).
Fig. 6.

Neutralization of tiers 1 and 2 SIVsmE660 Env pseudoviruses. (A to D) Serum samples were obtained from Mamu-A*01–negative (A and B) or Mamu-A*01–positive (C and D) monkeys 2 weeks after rAd boost immunization or on the day of challenge (DOC) and were tested for neutralizing activity against tier 1 (CP3C-P-A8) or tier 2 (CR54-P-2A5) SIVsmE660 Env pseudoviruses with the TZM-bl assay. Data are presented as serum ID50 titer (A and C) or percent neutralization (B and D) at a 1:10 serum dilution. Immunized monkeys are grouped retrospectively according to their infection status after SIVsmE660 challenge. Serum-neutralizing antibody activity from sham-vaccinated monkeys (control) is shown in (B) and (D). Data points represent responses from individual monkeys, with the median response indicated by the bar. P values were calculated with a Mann-Whitney rank-sum test.
A 1:50 dilution of plasma sampled from these monkeys at the time of the peak vaccine-elicited immuneresponse was assessed in a neutralization assay with human PBMCs as target cells and the challenge stock of SIVsmE660 as the replicating virus. A statistically significant decrease in virus replication was observed with plasma from Mamu-A*01–negative vaccinated monkeys that became infected compared with plasma from Mamu-A*01–negative vaccinated monkeys that did not become infected (P = 0.008)(Fig. 7).Although a similar analysis of the plasmas sampled on the day of challenge did not show a statistically significant difference in neutralization between these groups of monkeys in this PBMC-based assay, the relative levels of neutralizing antibodies in the plasma of each monkey in this assay were moderately correlated with those from week 38 (estimated Spearman correlation = 0.40; P = 0.05). No correlate of protection against acquisition of SIVsmE660 was observed when the vaccine-elicited immune responses in the Mamu-A*01–positive rhesus monkeys were assessed in either the pseudovirion- or the PBMC target cell–based neutralization assay.
Fig. 7.

Neutralization of SIVsmE660 in human PBMCs. CD8+ T cell–depleted, concanavalin A–stimulated human PBMCs were infected at a low MOI with SIVsmE660. Infected PBMCs were subsequently cultured in the presence of a 1:50 dilution of plasma collected from the Mamu-A*01–negative and Mamu-A*01–positive study animals at 6 weeks after rAd vaccination (peak). Virus from the infected PBMC-plasma coculture supernatant was quantified with the TZM-bl assay. Luciferase assay results were normalized to a control plasma and plotted as a relative viral replication. P values were calculated with a Mann-Whitney rank-sum test.
We also evaluated genetic determinants that might have contributed to the protection from infection observed in the monkeys. We have recently shown that the expression of particular TRIM5 alleles (encoding the host restriction factor TRIM5) by rhesus monkeys is associated with control of SIVmac251 replication both in vitro and in vivo (8, 9). Two alleles are codominantly expressed at the TRIM5 locus of the rhesus monkey. If one or both of those alleles are from the group of alleles 6 to 11 (permissive), SIV replication occurs at a higher level than if both are from the group of alleles 1 to 5 (restrictive). To evaluate the contribution of the expression of particular TRIM5 alleles to protection against infection, we retrospectively determined the TRIM5 genotypes for all of the monkeys in these studies (tables S2 and S3). We separated the common variants of TRIM5 into two groups: (i) restrictive (1-5/1-5 or 1-5/TRIMCypA) and (ii) permissive (1-5/6-11, 6-11/6-11, or 6-11/TRIMCypA). We excluded monkeys that had two TRIMcypA alleles (TRIMCypA/TRIMCypA) from this analysis because TRIM5 alleles were deleted in these monkeys (3 of 89 monkeys).
When the sham-immunized monkeys were evaluated together as a single group regardless of their Mamu-A*01 status, monkeys with at least one permissive TRIM5 allele were more likely to acquire SIVsmE660 infection than those monkeys expressing restrictive alleles (Fig. 8A) (P = 0.0023). Similarly, when vaccinated monkeys (Mamu-A*01− and Mamu-A*01+) that were challenged with SIVsmE660 were assessed as one cohort, we observed a statistically significant association between the expression of two restrictive alleles and protection against virus acquisition (Fig. 8B) (P = 0.0013). Therefore, the expression of TRIM5 alleles 1 to 5 was associated with protection from infection after low-dose SIVsmE660 intrarectal exposures in rhesus monkeys regardless of vaccination status. Because this was a retrospective analysis, there were not enough monkeys that were homozygous for TRIM5 alleles 6 to 11 to differentiate between the effect of having one versus two permissive alleles on mucosal acquisition of infection. A protective effect of the vaccine was observed in vaccinated monkeys regardless of their TRIM5 genotypes (Fig. 9).
Fig. 8.
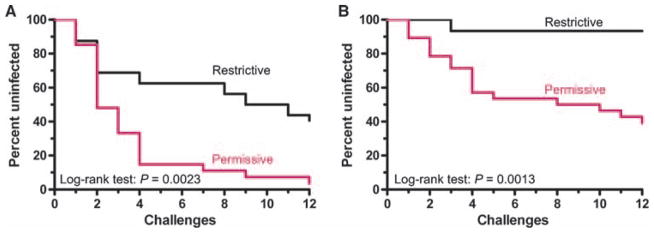
Effect of TRIM5 genotype on SIV mucosal infection in naïve and vaccinated monkeys. The effect of restrictive or permissive TRIM5 alleles on the percentage of monkeys that remained un-infected after each weekly intrarectal SIVsmE660 exposure is shown in Kaplan-Meier curves. (A and B) Naïve (A) and vaccinated (B) monkeys were divided into two groups: one group expressing only restrictive TRIM5 alleles (black) and the other expressing at least one permissive allele (red). In the control group, 16 monkeys expressed restrictive TRIM5 alleles, whereas 27 monkeys expressed permissive alleles. In the vaccine group, 15 monkeys expressed restrictive alleles, whereas 28 monkeys expressed permissive alleles. The statistical comparisons between monkeys with restrictive and permissive TRIM5 genotypes in control and vaccinated arms were determined by the log-rank test.
Fig. 9.
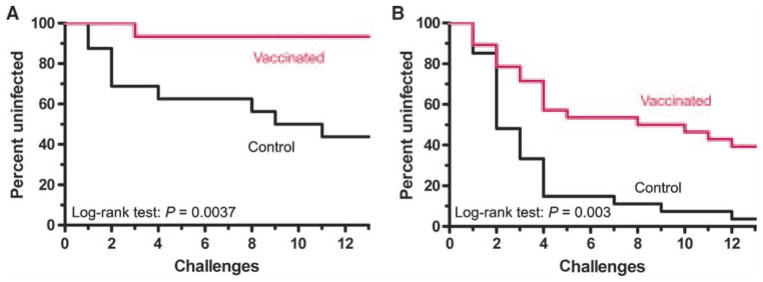
Effect of vaccination on SIV mucosal infection. The effect of vaccination on the percentage of monkeys with either restrictive or permissive TRIM5 genotypes that remain uninfected after each weekly intrarectal SIVsmE660 exposure is shown in Kaplan-Meier curves. (A and B) TRIM5 restrictive (A) or permissive (B) monkeys were divided into control (black) and vaccinated (red) groups. The statistical comparisons between monkeys in control and vaccinated arms for TRIM5 restrictive and permissive genotypes were determined by the log-rank test.
Finally, using a logistic regression analysis, we assessed the relative contributions of vaccine-elicited immune responses and genetic determinants of protection from infection in vaccinated monkeys (details of analysis in the Supplemental Material). SIVsmE660 infection in vaccinated monkeys was the outcome variable used in the logistic regression models. In the Mamu-A*01–positive animal study, the permissive TRIM5 phenotype was the most significant covariate (Table 1; P = 0.008, estimated odds ratio (OR) of infection = 18). Once the permissive TRIM5 phenotype was in the model, no other covariate added significantly to the model. In the Mamu-A*01–negative animal study, the first covariate added was lack of SIVsmE660 neutralization as measured in PBMCs at week 38, the time of peak vaccine-induced immune responses (P = 0.028, OR = 18.6). The second covariate added was low SIVsmE660 Env-specific CD4+ T cell responses (P = 0.032, OR = 9.8). No other covariates were subsequently significant. Therefore, low levels of virus neutralization and low Env-specific CD4+ T cell responses were strong predictors of infection in the Mamu-A*01–negative animals. When both the Mamu-A*01–positive and the Mamu-A*01–negative monkeys were grouped together for analysis, the first covariate added to the model (P = 0.005, OR = 31.7) was lack of restrictive TRIM5 alleles. Other statistically significant covariates included Env-specific CD4+ T cell responses and neutralizing antibody responses as measured with the pseudovirion/TZM-bl cell assay (Table 1).
Table 1.
Best models for protection and infection after vaccination. OR, odds ratio.
| Variable | Category | P | Final model OR estimate | Correct prediction of protection (%) | Correct prediction of infection (%) |
|---|---|---|---|---|---|
| Mamu-A*01+–vaccinated animals | |||||
| TRIM5 phenotype | Permissive | 0.008 | 18.0 | 75 | 86 |
| Mamu-A*01−–vaccinated animals | |||||
| PBMC neut WK38* | >0.50§ | 0.028 | 18.6 | 92 | 75 |
| EnvCD4+ T cell† | ≤0.01|| | 0.032 | 9.8 | ||
| EnvCD4+ T cell | Unknown | 1.3 | |||
| Mamu-A*01±–vaccinated animals | |||||
| TRIM5 genotype | Lack of restrictive allele | 0.005 | 31.7 | 76 | 95 |
| EnvCD4+ T cell | ≤0.01¶ | 0.012 | 12.7 | ||
| EnvCD4+ T cell | >0.06# | 0.044 | 23.2 | ||
| EnvCD4+ T cell | Unknown | 0.2 | |||
| Neutralizing antibody‡ | ≤17** | 0.020 | 7.9 | ||
Plasma neutralization of SIVsmE660 using PBMCs. Values represent relative viral replication after addition of plasma.
SIVsmE660 Env-specific CD4+ T cell responses as measured by intracellular cytokine staining.
Plasma neutralization of SIVsmE660 using pseudovirion/TZM-bl assay. Values represent titer.
>25th percentile.
≤25th percentile.
≤25th percentile.
>75th percentile.
≤Median (50th percentile).
DISCUSSION
Here, vaccination was associated with protection against infection with SIVsmE660 but not SIVmac251. The sequences of SIVmac239, the virus used for generating the vaccine immunogens, and the SIVmac251 isolate used for challenging the monkeys are very similar (10). In contrast, the SIVsmE660 virus sequence is quite divergent from that of SIVmac239 (7). The sequence distance between SIVmac239 and SIVsmE660 is comparable to that of two sequence-divergent isolates of clade B viruses. One might have expected better protection against SIVmac251 challenge because vaccine-induced immune responses to SIVmac239 should have been more cross-reactive with SIVmac251 than SIVsmE660. The protection from mucosal acquisition of SIVsmE660 but not SIVmac251 in this vaccine study may have been a consequence of differences in susceptibility to immune pressure, tropism, or the replication capacity of these two viruses.
Although SIVmac239 and SIVmac251 are relatively resistant to antibody-mediated neutralization, the neutralization profile of SIVsmE660 has not been well studied (11). We demonstrated that pseudovirions generated with env genes isolated from a number of rhesus monkeys infected with this virus quasi-species can be either highly sensitive (tier 1–like) or relatively resistant (tier 2–like) to neutralization in a pseudovirion/TZM-bl neutralization assay. We have also shown that the challenge stock of SIVsmE660 used in the present study has tier 2–like properties when assessed for neutralization using PBMC target cells.
Although mucosal exposure of rhesus monkeys to SIV isolates is a powerful animal model for studying sexual transmission of HIV-1 in humans, this model does have limitations. For example, HIV-1 transmission is often facilitated in humans by cofactors such as local mucosal lesions and coinfections. Because the transmission model used in the current study does not include the induction of mucosal lesions in the experimental monkeys, it may prove easier to block mucosal infections in this animal model than in humans.
Although the recently completed RV144 Thai trial demonstrated vaccine-associated protection in 31.2% of vaccinees at 3 years after the initiation of vaccination, no diminution of virus load was seen in vaccinees who became infected (5). This observation has led to the suggestion that it may be possible to elicit an immune response that can abort an HIV-1/SIV infection at the portal of virus entry without an effect on subsequent virus replication if an infection is initiated in a vaccinee. Here, in vaccinated monkeys that became infected after repeated, low-dose intrarectal exposures to virus, the monkeys challenged with SIVmac251 had a lower peak plasma virus RNA level compared with control monkeys. This same effect was seen in the Mamu-A*01–positive but not the Mamu-A*01–negative vaccinated monkeys challenged with SIVsmE660. These data suggest that the relationship between vaccine protection against acquisition of infection and virus replication after infection are independent, one possibly mediated by the humoral immune response and the other by cellular immunity.
We observed an association between a vaccine-induced neutralizing antibody response and protection against SIVsmE660 acquisition, but this was found in Mamu-A*01–negative but not Mamu-A*01–positive monkeys. The more robust antibody association was the neutralizing antibody response measured in a PBMC neutralization assay, evaluated at the time of peak antibody response after vaccination. An association between vaccine-induced neutralizing antibodies and protection against virus acquisition was also observed with Env pseudo-viruses that had a tier 2–like neutralization profile but not pseudoviruses with a tier 1–like profile. Moreover, an association was seen between protection and vaccine-induced Env-specific CD4+ T cell responses. This cellular immune response might have provided help in the generation of an anti-Env antibody response. Thus, three complementary findings suggest that neutralizing antibodies may indeed be contributing to vaccine protection in this model.
Although we have demonstrated in the present study that neutralizing antibodies may be contributing to the protection of vaccinated monkeys, this finding does not rule out the possibility that other vaccine modalities may provide protection against an AIDS virus infection through different immune mechanisms. For example, it has been reported that rhesus monkeys can be protected against SIVmac239 infection by vaccination with a recombinant cytomegalovirus (CMV) vaccine vector (12). Because this vector induces memory CD8+ T cell responses in mucosal tissues, it is possible that the protection induced by recombinant CMV immunization is mediated by this cellular mechanism. It is also possible that other vaccine modalities may confer more robust protection than the DNA/rAd5 vaccine used here.
We have previously shown that the TRIM5 alleles expressed by individual monkeys have a significant effect on the control of SIV replication in those animals, both in their lymphocytes in vitro and in vivo after experimental infection (8, 9). Here, we show a strong trend toward an effect of the expression of restricting TRIM5 alleles on mucosal acquisition of SIVsmE660 infection even in the absence of vaccination. The complementary contributions of TRIM5 haplotype and vaccine-induced immune responses for protection have potentially important mechanistic implications. The antibody induced by vaccination did not by itself confer sterile protection against infection. Given that a large multiplicity of infection (MOI) of HIV-1 or SIV can override TRIM5α restriction, these findings suggest that antibody binding to the SIV virions decreased the effective inoculum of infectious virus, making the residual infectious virions susceptible to the TRIM5α-mediated restriction.
The vaccination approach used in this study, a plasmid DNA prime/rAd5 boost, was developed to induce both CD8+ T lymphocyte responses and Env-specific antibody responses. The exploration of immune correlates in the present study suggests that a neutralizing antibody may be the response that conferred protection against mucosal acquisition of SIVsmE660. Our ability to also detect the genetic contribution to vaccine protection may have been a consequence of the marginal protection afforded by these vaccine modalities. It is possible that this vaccine may have protected monkeys from a smaller inoculum of SIVmac251. It is also possible that a vaccine that elicits a potent neutralizing antibody response may confer robust protection in vaccinees regardless of their TRIM5 alleles.
MATERIALS AND METHODS
Animals
Monkeys used in this study were housed in accordance with the guidelines of the Institutional Animal Care and Use Committee for Harvard Medical School and the Guide for the Care and Use of Laboratory Animals [Institute of Laboratory Animal Resources (U.S.) Committee on Care and Use of Laboratory Animals, 1996]. This study was approved by the Harvard Medical Area Standing Committee on Animals (protocol number 03724; assurance number A3431-01). Procedures were conducted under ketamine anesthesia, and all efforts were made to minimize suffering.
Plasma SIV RNA concentrations
Plasma SIV RNA concentrations were determined with a modified two-step quantitative reverse transcription–polymerase chain reaction (qRT-PCR) process based on that described previously (13, 14). Experimental samples were run in parallel with an SIV-gag RNA standard on an Applied Biosystems StepOne Real Time PCR System.
PBMC ELISpot assay
Multiscreen 96-well plates were coated overnight with 100 μl per well of anti-human IFN-γ (5 μg/ml) (B27; BD Pharmingen) in endotoxin-free Dulbecco’s phosphate-buffered saline (D-PBS). The plates were then washed three times with D-PBS containing 0.1% Tween 20, blocked for 2 hours with D-PBS containing 5% fetal bovine serum (FBS) to remove the Tween 20, and incubated with peptide pools and 2 × 105 PBMCs in triplicate in 100-μl reaction volumes. The peptide pools spanning the entire SIVmac239 Env, Gag, or Pol proteins comprised 15–amino acid peptides overlapping by 11 amino acids. Each peptide in a pool was present at a 1 μg/ml concentration. After an 18-hour incubation at 37°C, the plates were washed nine times with D-PBS containing 0.1% Tween 20 and once with distilled water. The plates were then incubated with biotinylated rabbit anti-human IFN-γ (2 μg/ml) (Biosource) for 2 hours at room temperature, washed five times with D-PBS containing 0.1% Tween 20, and incubated for 2.5 hours with a 1:500 dilution of streptavidin–alkaline phosphatase (Southern Biotechnology). After three washes with D-PBS containing 0.1% Tween 20 and three washes with D-PBS alone, the plates were developed with bromochloroindolyl phosphate–nitro blue tetra-zolium (BCIP-NBT) chromogen (Pierce), stopped by washing with tap water, air-dried, and read with an ELISPOT reader (Cellular Technology Ltd.).
PBMC stimulation and intracellular cytokine staining
PBMCs were isolated from EDTA-anticoagulated blood and frozen in the vapor phase of liquid nitrogen. Cells were later thawed and allowed to rest for 6 hours at 37°C in a 5% CO2 environment. The viability of these cells was >90%. PBMCs were then incubated at 37°C in a 5% CO2 environment for 6 hours in the presence of RPMI/10% fetal calf serum alone (unstimulated), a pool of 15-nucleotide oligomer Gag or Env peptides (2 μg/ml of each peptide), or staphylococcal enterotoxin B (SEB) (5 μg/ml, Sigma-Aldrich) as a positive control. All cultures contained monensin (GolgiStop; BD Biosciences) as well as anti-CD28 (1 μg/ml) and anti-CD49d (1 μg/ml) (BD Biosciences). The cultured cells were stained with monoclonal antibodies (mAbs) specific for cell surface molecules including CD3, CD4, and CD8. After fixing with Cytofix/Cytoperm solution (BD Biosciences), cells were permeabilized and stained with antibodies specific for IFN-γ, tumor necrosis factor–α (TNF-α), and interleukin-2 (IL-2). Labeled cells were fixed in 1% formaldehyde–PBS.
Antibodies
The antibodies used in this study were directly coupled to fluorescein isothiocyanate (FITC), phycoerythrin (PE), PE–Texas red (ECD), peridinium chlorophyll protein (PerCP)–Cy5.5, PE-Cy7, AmCyan, Pacific Blue, allophycocyanin (APC), Alexa Fluor 700, and APC-Cy7. All reagents were validated and titrated with rhesus monkey PBMCs. The following mAbs were used: anti–TNF-α (MAb11), anti–IFN-γ (B27), anti–IL-2 (MQ1-17H12), anti-CD28 (L293), anti-CD95 (DX2), anti-CD3 (SP34-2), anti-CD8α (RPA-T8), anti-CD4 (L200), anti-CD107a (H4A3), anti–MIP-1β (macrophage inflammatory protein–1β) (D21-1351), anti-Ki67 (B56), anti-CCR5 (3A9), and anti-β7 (FIB504) (all from BD Biosciences). A violet fluorescent reactive dye (ViViD; Invitrogen) was also used as a viability marker to exclude dead cells in the analysis.
Intracellular cytokine staining assay for ADCC activity
An intracellular cytokine staining assay was used to measure SIV antibody–mediated NK cell cytokine expression and degranulation. Target cells were prepared by incubation with monomeric gp120 derived from SIVmac251 for 1 hour at room temperature, followed by washing twice with ice-cold R10. Purified human NK cells were used as effector cells. Target cells (250,000) were dispensed into each well of a 96-well plate. Diluted plasma (100 μl) was added to each well, and the target and antibody mixture was incubated for 15 min at room temperature. Effector cells (250,000) were added to each well at an effector/target ratio of 1:1. Cells were incubated for 4 hours in the presence of monensin and anti-CD107a. At the end of the incubation, cells were washed and stained with antibodies specific for CD3, CD20, CD56, and CD16 and the intracellular molecules IFN-γ, TNF-α, and MIP-1β. Cells were washed and fixed in 1% paraformaldehyde. Human CD3−CD20−CD56+ NK cells were evaluated for the expression of CD107a, IFN-γ, TNF-α, and MIP-1β.
Protein expression and enzyme-linked immunosorbent assay
SIVsmE660 gp140 protein was expressed with the expression plasmid SIVsmE660CR54-PK.2A5 via transient transfection of FreeStyle 293-F cells (Invitrogen). The His-tagged protein was purified over a diethyl-aminoethyl (DEAE) Sepharose resin followed by immobilized metal affinity chromatography (IMAC) with a nickel chelation resin (Sigma). Reacti-Bind ELISA (enzyme-linked immunosorbent assay) plates (Pierce) were coated with 200 ng of protein, and the plates were blocked with 200 μl per well of 150 mM NaCl, 50 mM tris-HCl, 1 mM EDTA, 3.3% FBS, 2% bovine albumin, and 0.07% Tween 20 (B3T buffer) for 1 hour. Serial dilutions of plasma samples were diluted in B3T buffer and incubated on assay plates for 1 hour. Biotin-conjugated goat anti-monkey IgG (immunoglobulin G) antibody (Rockland Immunochemicals) was used as the secondary antibody, followed by peroxidase-labeled streptavidin (Kirkegaard and Perry Laboratories). The color reaction was carried out with 3,3′,5,5′-tetramethylbenzidine (Kirkegaard and Perry Laboratories) for 30 min at ambient temperature, and the reaction was stopped with 1 N H2SO4. All other ELISA incubations were at performed 37°C. The reciprocal serum dilution that demonstrated half-maximal binding (ED50) was calculated with a four-parameter nonlinear regression equation with GraphPad Prism. Avidity ELISAs were performed in a similar manner but included the chaotropic agent NaSCN. After addition of plasma samples, plates were incubated with 1.5 M NaSCN for 15 min. Control plates were incubated with PBS. Avidity indices were calculated by dividing ED50 values with NaSCN treatment by ED50 values without treatment.
Cytokine secretion
Twenty-three cytokines were measured in plasma with the nonhuman primate cytokine Milliplex kit (Millipore) according to the manufacturer’s instructions. The cytokines included IL-1α, IL-1RA (IL-1 receptor antagonist), IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12/23 (p40), IL-13, IL-15, IL-17, IL-18, IFN-γ, G-CSF (granulocyte colony-stimulating factor), GM-CSF (granulocyte-macrophage colony-stimulating factor), MCP-1 (monocyte chemotactic protein-1), MIP-1α (macrophage inflammatory protein 1α), MIP-1β, TNF-α, TGF-β (transforming growth factor–β), sCD40L (soluble CD40 ligand), and VEGF (vascular endothelial growth factor). Plasma was incubated with antibody-coupled beads overnight followed by incubation with biotinylated detection antibody and, finally, incubation with streptavidin-PE. Each sample was assayed in duplicate, and cytokine standards supplied by the manufacturer were run on each plate. Multianalyte profiling was performed with a Luminex-100 system, and data were analyzed with BioPlex manager software (Bio-Rad). One additional analyte was measured with a human IFN-α ELISA kit (PBL Interferon Source).
Pseudovirion-based neutralization assay
Neutralizing antibody responses against SIVsmE660 Env pseudo-viruses were measured with a luciferase-based assay in TZM-bl cells [obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), NIH from J. C. Kappes, X. Wu, and Tranzyme Inc.] as previously described (15, 16). Briefly, threefold serial dilutions of pre- and post-immune sera were performed in duplicate (96-well flat bottom plate) in 10% DMEM (Dulbecco’s modified Eagle’s medium) growth medium (100 μl per well). Two hundred TCID50 (median tissue culture infectious dose) of virus was added to each well in a volume of 50 μl, and the plates were incubated for 1 hour at 37°C. TZM-bl cells were then added (1 × 104 per well in 100-μl volume) in 10% DMEM growth medium containing DEAE-dextran (Sigma) at a final concentration of 11 μg/ml. Assay controls included replicate wells of TZM-bl cells alone (cell control) and TZM-bl cells with virus (virus control). After a 48-hour incubation at 37°C, 150 μl of assay medium was removed from each well and 100 μl of Bright-Glo luciferase reagent (Promega) was added and luminescence was measured. The ID50 titer was calculated as the serum dilution that caused a 50% reduction in relative luminescence units (RLUs) compared to the virus control wells after subtraction of cell control RLUs. Assay stocks of molecularly cloned SIVsmE660 CP3C-P-A8 (tier 1) and CR54-PK-2A5 (tier 2) Env-pseudotyped viruses were prepared by cotransfecting 293T/17 cells with an Env-expressing plasmid and an Env-deficient backbone plasmid (SIVmac239deltaEnv), as previously described (15, 17).
PBMC-based neutralization assays
Whole blood (50 ml) was obtained from healthy human donors (Research Blood Components), and PBMCs were purified by Ficoll-Hypaque centrifugation. PBMCs were incubated in concanavalin A (6.25 μg/ml) and IL-2 (20 U/ml) for 2 days and then depleted of CD8+ T cells with the EasySep Human CD8 Positive Selection Kit (Stem Cell Technologies). CD8+ T cell–depleted PBMCs were subsequently infected with 0.01 MOI of SIVsmE660 and incubated for 2 days. Plasma from vaccinated uninfected animals was heat-inactivated for 60 min at 50°C. SIVsmE660-infected PBMCs (1 × 105) were incubated with 1:50, 1:250, or 1:1250 dilutions of heat-inactivated plasma for 5 days. Culture supernatants (5 μl) were harvested and applied to 2 × 104 TZM-bl cells and incubated for an additional 2 days. TZM-bl cells were analyzed for luciferase activity with the Steady-Glo Luciferase Assay System (Promega). Relative viral replication was reported as the luciferase activity generated by culture supernatants of infected PBMCs incubated with plasma from vaccinated animals divided by the activity generated by infected PBMCs cultured with plasma from a control unvaccinated animal. Neutralization assays in rhesus PBMCs were performed as described (18), with the exception that PBMCs were stimulated overnight with a higher concentration of PHA-P (phytohemagglutinin P) (10 μg/ml). Neutralization potency was determined by reductions in p27 Gag antigen synthesis (Advanced BioScience Laboratories Inc., catalog no. 5450).
TRIM5 genotyping
Genomic DNA was isolated from lymphocytes from rhesus monkeys with QIAamp DNA kit (Qiagen) and sequenced for TRIM5 exons.
Supplementary Material
Fig. S1. Set-point plasma virus RNA levels following infection after repeated intrarectal challenge.
Fig. S2. The loss of total CD4+ T cells and CM CD4+ T cells after infection.
Fig. S3. Expression of maturation-associated molecules on CD4+ T cells.
Fig. S4. NK cell phenotyping.
Fig. S5. Vaccine-induced cytokine responses.
Fig. S6. Neutralization phenotype of SIVsmE660 Env pseudoviruses.
Fig. S7. Neutralization phenotype of SIVsmE660 challenge virus.
Table S1. Plasma virus RNA levels in the cohorts of rhesus monkeys in the study.
Table S2. TRIM5 genotyping of the 129 rhesus monkeys in the study.
Table S3. The distribution of TRIM5 alleles in the cohorts of rhesus monkeys in the study.
Acknowledgments
We thank V. Hirsch for providing the SIVsmE660 used for preparation of the challenge stock of virus; S. Bao and J. Noor for the viral inoculation preparations; A. Zajac and A. Ault for Animal Care and Use Committee approvals and logistics; BioQual including M. Lewis and staff for animal experiments conducted at the BioQual Inc. contract facility; and L. Granpre for her contributions to the neutralizing antibody studies.
Funding: This work was supported by funds from the intramural research program of the Vaccine Research Center, NIAID, NIH; the Harvard Medical School Center for AIDS Research grant AI060354; NIAID K08 AI069995 (W.W.Y.); NIH grant N01-AI30033; and the NIAID Center for HIV/AIDS Vaccine Immunology grant AI067854.
Footnotes
Author contributions: N.L.L., J.R.M., G.J.N., D.C.M., P.J.N., M.S.S., and S.S.R. conceived and designed the experiments. Y.S., W.W.Y., M.A., S.-Y.L., L.S., M.L., S.K., A.P.B., L.V.M., J.Z., H.B., S.D.S., J.-P.T., and A.D. performed the experiments. J.R.M., R.S.G., J.B.W., C.S., M.L., M.G.H., and P.B.G. analyzed the data. G.M.S. contributed reagents, materials, and analysis tools. N.L.L. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF. rgp120 HIV Vaccine Study Group, Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J Infect Dis. 2005;191:654–665. doi: 10.1086/428404. [DOI] [PubMed] [Google Scholar]
- 2.Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, Hu D, Tappero JW, Choopanya K. Bangkok Vaccine Evaluation Group, Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194:1661–1671. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- 3.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN. Step Study Protocol Team, Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. Step Study Protocol Team, HIV-1 vaccine-induced immunity in the test-of-concept Step Study: A case-cohort analysis. Lancet. 2008;372:1894–1905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. MOPH-TAVEG Investigators, Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 6.Hudgens MG, Gilbert PB, Mascola JR, Wu CD, Barouch DH, Self SG. Power to detect the effects of HIV vaccination in repeated low-dose challenge experiments. J Infect Dis. 2009;200:609–613. doi: 10.1086/600891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh WW, Jaru-Ampornpan P, Nevidomskyte D, Asmal M, Rao SS, Buzby AP, Montefiori DC, Korber BT, Letvin NL. Partial protection of Simian immunodeficiency virus (SIV)-infected rhesus monkeys against superinfection with a heterologous SIV isolate. J Virol. 2009;83:2686–2696. doi: 10.1128/JVI.02237-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL. TRIM5α modulates immunodeficiency virus control in rhesus monkeys. PLoS Pathog. 2010;6:e1000738. doi: 10.1371/journal.ppat.1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim SY, Chan T, Gelman RS, Whitney JB, O’Brien KL, Barouch DH, Goldstein DB, Haynes BF, Letvin NL. Contributions of Mamu-A*01 status and TRIM5 allele expression, but not CCL3L copy number variation, to the control of SIVmac251 replication in Indian-origin rhesus monkeys. PLoS Genet. 2010;6:e1000997. doi: 10.1371/journal.pgen.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.HIV Sequence Compendium 2010.
- 11.Johnson WE, Morgan J, Reitter J, Puffer BA, Czajak S, Doms RW, Desrosiers RC. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J Virol. 2002;76:2075–2086. doi: 10.1128/jvi.76.5.2075-2086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M, Jr, Lifson JD, Nelson JA, Jarvis MA, Picker LJ. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15:293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cline AN, Bess JW, Piatak M, Jr, Lifson JD. Highly sensitive SIV plasma viral load assay: Practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 14.Whitney JB, Luedemann C, Bao S, Miura A, Rao SS, Mascola JR, Letvin NL. Monitoring HIV vaccine trial participants for primary infection: Studies in the SIV/macaque model. AIDS. 2009;23:1453–1460. doi: 10.1097/QAD.0b013e32832b43d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, Coico R, editors. Current Protocols in Immunology. John Wiley and Sons; New York: 2004. pp. 12.11.11–12.11.15. [DOI] [PubMed] [Google Scholar]
- 17.Basavapathruni A, Yeh WW, Coffey RT, Whitney JB, Hraber PT, Giri A, Korber BT, Rao SS, Nabel GJ, Mascola JR, Seaman MS, Letvin NL. Envelope vaccination shapes viral envelope evolution following simian immunodeficiency virus infection in rhesus monkeys. J Virol. 2010;84:953–963. doi: 10.1128/JVI.01679-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bures R, Gaitan A, Zhu T, Graziosi C, McGrath MK, Tartaglia J, Caudrelier P, ElHabib R, Klein M, Lazzarin A, Stablein DM, Deers M, Corey L, Greenberg ML, Schwartz DH, Montefiori DC. Immunization with recombinant canarypox vectors expressing membrane-anchored glycoprotein 120 followed by glycoprotein 160 boosting fails to generate antibodies that neutralize R5 primary isolates of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2000;16:2019–2035. doi: 10.1089/088922200750054756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Set-point plasma virus RNA levels following infection after repeated intrarectal challenge.
Fig. S2. The loss of total CD4+ T cells and CM CD4+ T cells after infection.
Fig. S3. Expression of maturation-associated molecules on CD4+ T cells.
Fig. S4. NK cell phenotyping.
Fig. S5. Vaccine-induced cytokine responses.
Fig. S6. Neutralization phenotype of SIVsmE660 Env pseudoviruses.
Fig. S7. Neutralization phenotype of SIVsmE660 challenge virus.
Table S1. Plasma virus RNA levels in the cohorts of rhesus monkeys in the study.
Table S2. TRIM5 genotyping of the 129 rhesus monkeys in the study.
Table S3. The distribution of TRIM5 alleles in the cohorts of rhesus monkeys in the study.