

Abstract
Rationale
Disruption of endothelial barrier function and neutrophil-mediated injury are two major mechanisms underlying the pathophysiology of sepsis-induced acute lung injury (ALI). Recently we reported that endotoxin induced activation of RhoA in mice lungs that led to the disruption of endothelial barrier and lung edema formation; however the molecular mechanism of this phenomenon remained unknown.
Objective
We reasoned that LIMK1, which participates in the regulation of endothelial cell contractility and is activated by RhoA/Rho kinase pathway, could mediate RhoA-dependent disruption of endothelial barrier function in mouse lungs during ALI. And if that is the case, then attenuation of endothelial cell contractility by down-regulating LIMK1 may lead to the enhancement of endothelial barrier function, which could protect mice from endotoxin-induced ALI.
Methods and Results
Here we report that LIMK1 deficiency in mice significantly reduced mortality induced by endotoxin. Data showed that lung edema formation, lung microvascular permeability, and neutrophil infiltration into the lungs were suppressed in limk1−/− mice.
Conclusions
We identified that improvement of endothelial barrier function along with impaired neutrophil chemotaxis were the underlying mechanisms that reduced severity of ALI in limk1−/− mice, pointing to a new therapeutic target for diseases associated with acute inflammation of the lungs.
Keywords: LIMK1, RhoA, endothelial barrier function, inflammation, acute lung injury, neutrophil infiltration, actin cytoskeleton
Introduction
Sepsis is the most common cause of acute lung injury (ALI), a syndrome that results from acute pulmonary edema and inflammation1. The pathogenesis of ALI includes disruption of endothelial barrier that leads to lung edema2; and activation of neutrophils by cytokines, which results in neutrophil infiltration and tissue damage3, 4.
RhoA regulates both endothelial barrier function and neutrophil function. In endothelial cells, RhoA promotes actin polymerization and actomyosin-based cell contractility, which leads to the disruption of endothelial barrier function5. In neutrophils, RhoA regulates cell migration through signaling that leads to myosin light chain phosphorylation and actin polymerization6.
We reported recently that endotoxin (LPS) induced activation of RhoA in mouse lungs in vivo, which resulted in the disruption of the endothelial barrier and lung edema formation7. RhoA/Rho kinase (ROCK) signaling cascade was involved in the disruption of the endothelial barrier function in mouse lungs since LPS-8 and peptide agonist corresponding to the tethered ligand sequence of human PAR-1 (SFLLRN) (PAR-1)-induced7 increases in vascular permeability were reversible by pharmacological inhibition of ROCK.
We hypothesized that LIMK1, which downstream of ROCK9 mediates endothelial cells contractility10, regulates RhoA-dependent disruption of endothelial barrier function in mouse lungs during ALI. We also hypothesized that attenuation of endothelial cell contractility by downregulation of LIMK1 may lead to the enhancement of endothelial barrier function, which ultimately could protect mice during endotoxin-induced acute lung injury. In addition, as actin polymerization is required for the efficient neutrophils chemotaxis, we reasoned that lack of LIMK1 would impair neutrophil migration, leading to decreased diapedesis.
Here we demonstrated that LPS induced activation of RhoA and LIMK1 in the mouse lungs in vivo. To study the role of LIMK1 in ALI, we used limk1−/− mice. Importantly, the severity of ALI was decreased in limk1−/− mice, as was observed by increased survival rate, decreased edema formation and polymorphonuclear leukocytes (PMN) infiltration in the lungs. We found that LIMK1 regulated RhoA-mediated LPS-induced increase in microvascular permeability. Our data showed that downregulation of LIMK1 resulted in the improvement of the endothelial barrier function both in-vivo and in cell culture models. We demonstrated that, mechanistically, LIMK1 mediated endothelial barrier disruption through phosphorylation of its downstream target, cofilin. Finally, we determined that limk1−/− PMN exhibited decreased chemotactic ability due to reduced actin polymerization.
Taking into account that genetic deletion of LIMK1 in mice resulted only in minor systemic effects11, we propose that organ-specific (targeted) downregulation or inhibition of kinase activity of LIMK1 could be an effective strategy to (i) improve endothelial barrier function, leading to less permeable blood vessels; and to (ii) decrease neutrophil infiltration in lungs that might be beneficial during acute lung injury.
Materials and Methods
Detailed description of materials; animal procedures; lung preparation and Kfc measurements; tissue collection and preparation for biochemical assays; electron microscopy studies; immunohistochemistry; ECIS studies; PMN isolation and superoxide production, degranulation and chemotaxis assays are provided in the Online Data Supplement.
Animals
All animal procedures were carried out following the Public Health Service Policy on Humane Care of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of University of Illinois at Chicago.
Statistical analysis
For statistical analysis, paired two-tailed Student’s t-test was used to compare data between two groups. ANOVA test was used to compare several groups. Log-rank test was used to assess differences in mortalities. Values are expressed as mean ± SEM. p < 0.05 was considered statistically significant.
Results
Genetic deletion of LIMK1 decreases severity of ALI in mice
LIMK1 and LIMK2 proteins are expressed in lungs and in neutrophils both in mice and humans (Figure 1A, B, C Ref.12, 13). Genetic deletion of LIMK1 did not result in the up-regulation of LIMK2 expression (Figure 1B, C).
Figure 1. Genetic deletion of LIMK1 decreases endotoxin-induced mortality and lung edema.
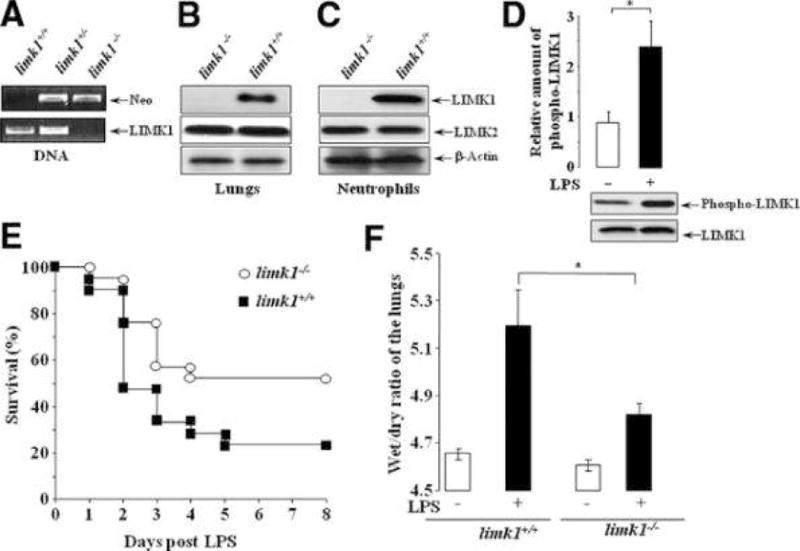
A) Genomic DNA was extracted from mice tails and analyzed by PCR for the presence of LIMK1 (330-bp product) and neomycin (370-bp product) DNAs in genomic DNA extracts. B) Lungs were extracted, homogenized and lysed, or C) neutrophils were isolated from the bone marrow and lysed, and total lysates were analyzed by Western blotting with anti-LIMK1, anti-LIMK2 and anti-actin antibodies. D) LPS 23 mg/kg was given i.p. for 3 hours. Lungs were extracted, homogenized lysed, and total LIMK1 was immunoprecipitated using anti-LIMK1 antibody and protein A/G agarose. Immunoprecipitates were analyzed by Western blotting with anti-LIMK1 and anti-phospho LIMK1/LIMK2 antibody. Three independent experiments yielded similar results. n = 3 lungs per experiment. E) Survival after LPS 23 mg/kg given i.p. to limk1+/+ and limk1−/− mice (n = 21, data were pooled from two experiments; differences in mortalities were assessed by log-rank test p = 0.0468). F) Lung edema formation at 12 hours after 23 mg/kg LPS. Lungs were removed, weighed (wet weight) and placed in an oven at 70ºC. Their weight was determined every morning until two readings were identical (dry weight). The wet/dry ratio was determined by dividing the wet weight to the dry weight. Data were pooled from two independent experiments. The average was calculated from n = 8 for limk1+/+ mice, and n = 5 for limk1−/− mice. P = 0.0393.
As a model of ALI, where lung is injured indirectly, and which targets primarily the capillary endothelium14, we chose intra-peritoneal administration of LPS. Initially we determined if LIMK1 was activated in this model of ALI. LPS, at 23 mg/kg (LD50 for limk1−/− mice (data not shown)), induced activation and phosphorylation of LIMK1 in limk1+/+ mice (Figure 1D). To address the question whether genetic deletion of LIMK1 may attenuate the severity of ALI, LPS was given to the limk1+/+ and limk1−/− mice and the survival rate was assessed over period of 7 days. Limk1−/− mice displayed an increased survival rate as compared to limk1+/+ littermates (from 23.8% limk1+/+ to 52.38% limk1−/−; p = 0.0468) (Figure 1E). Pulmonary edema, which serves as a hallmark of this disease15, is one of the factors contributing to the mortality of patients with ALI1. LPS challenge resulted in the formation of pulmonary edema16 that was significantly reduced limk1−/− mice (from 5.192 limk1+/+ to 4.82 limk1−/−)(Figure 1F).
Downregulation of LIMK1 enhances endothelial barrier function in vivo and in cell culture models
Disruption of the endothelial barrier function is a major contributor to the lung edema development2. To compare the endothelial barrier function in limk1+/+ and limk1−/− mice, we determined the pulmonary microvessel filtration coefficient (Kfc), a quantitative measure of vascular permeability, using isolated-perfused lung model17. Genetic deletion of LIMK1 resulted in a significant decrease of Kfc in a basal state (Figure 2A). LPS-induced Kfc was also significantly smaller in limk1−/− mice (Figure 2A). Observed decrease in vascular permeability may serve as an explanation for the decreased lung edema formation in limk1−/− mice.
Figure 2. Downregulation of LIMK1 enhances endothelial barrier functionin vivoand in cell culture models.
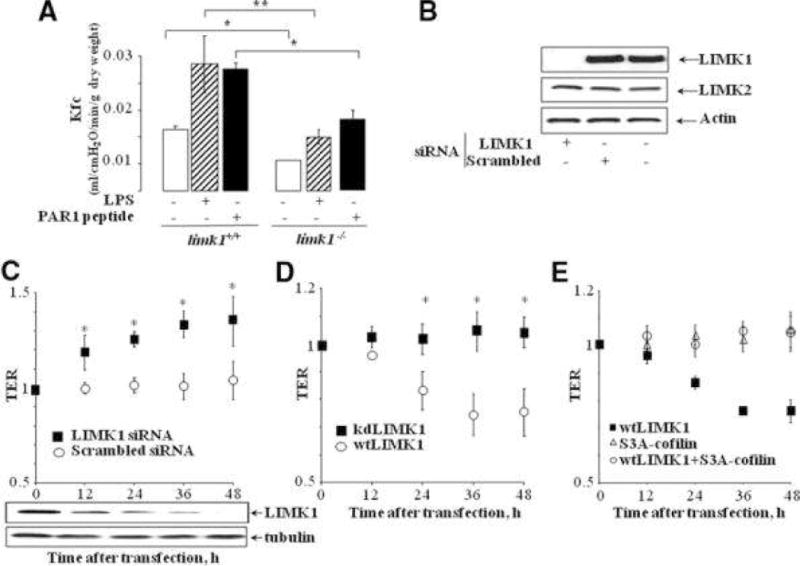
A) Kfc values in lungs of limk1+/+ and limk1−/− mice under basal conditions; 6 hour after 23 mg/kg i.p. LPS; 5 minutes after PAR-1 agonist peptide infusion into the lung preparation (final concentration, 6 μM). Individual Kfc values are the mean of 4 consecutive determinations per lung preparation. Error bars represent SEM. (Basal conditions: n = 4 lung preparations per bar, p = 0.00903; LPS treatment: n = 7 for limk1+/+, n = 5 for limk1−/−, p = 0.017717; PAR-1 peptide infusion: n = 3 lung preparations per bar, p = 0.025113). B) Downregulation of endogenous LIMK1 protein in HUVECs using siRNA at 48h. C) Electrical resistance measurements of endothelial permeability. HUVECs grown on gold electrodes were transfected with (C) siRNA against LIMK1 or scrambled control siRNA (total amount of LIMK1 protein upon siRNA-knockdown is indicated below the graph); (D) kinase dead LIMK1 (kdLIMK1) or wild type LIMK1 (wtLIMK1); or (E) wild type LIMK1 (wtLIMK1), or S3A-cofilin, or wtLIMK1+S3A-cofilin. The data shown was collected from three independent experiments performed in duplicates. Error bars represent SEM. (* p < 0.05)
RhoA family GTPases regulate endothelial barrier function via modulation of the actin cytoskeleton dynamics5. Activation of RhoA destabilizes, whereas activation of Rac1 and Cdc42 signaling pathways enhance the barrier function5.
We addressed the possibility of compensatory upregulation/activation of RhoA proteins in limk1−/− mice, by evaluating the total and GTP-bound amounts of RhoA, Rac1 and Cdc42 in the lungs in the basal and LPS-stimulated conditions. No differences were observed between limk1+/+ and limk1−/− genotypes in either condition (Online Figure I).
Importantly, although LPS still induced activation of RhoA in the lungs of limk1−/− mice, the disruption of endothelial barrier function was significantly decreased in this genotype, therefore placing LIMK1 as a crucial regulator of endothelial permeability downstream of RhoA.
The mechanism of how LPS induces activation of RhoA is not understood and probably includes autocrine signaling from several receptors. For that reason, as a model to study the mechanism of LIMK1-mediated attenuation of RhoA-dependent disruption of endothelial barrier function in mouse lungs, we used a PAR-1 peptide, which has been shown to disrupt the endothelial barrier function both in vivo and in vitro18 almost exclusively through activation of RhoA signaling cascade19. Although PAR-1 peptide induced the increase in Kfc in either genotype, in PAR-1-challenged limk1−/− mice, Kfc was significantly smaller, and comparable to the Kfc of limk1+/+ mice in the basal conditions (Figure 2A).
To rule out other possible compensatory effects that might be associated with gene deletion in mice, we addressed the question if downregulation of LIMK1 would decrease permeability in cultured endothelial cells. Endogenous LIMK1 was downregulated in human umbilical vein endothelial cells (HUVECs) using siRNA (Figure 2B). Endothelial barrier function was evaluated using electric cell-substrate impedance sensor (ECIS)20. Downregulation of LIMK1 in HUVECs resulted in the enhancement of endothelial barrier function (Figure 2C).
We previously showed that overexpression of wild type LIMK1 in HUVECs that possesses kinase activity resulted in the production of actin stress fibers and therefore in increased contractility of endothelial cells10. To determine if kinase activity of LIMK1 plays role in the disruption endothelial barrier, we overexpressed the wild type (wt) or kinase-dead (kd)10 mutants of LIMK1 in HUVECs. kdLIMK1 had no effect, whereas overexpression of wtLIMK1 significantly decreased endothelial permeability (Figure 2D). The known target of LIMK1 is cofilin, an actin-depolymerizing factor21. To elucidate the mechanism of how LIMK1 regulates endothelial barrier disruption, we co-expressed non-phosphorylatable mutant of cofilin S3A-cofilin21 with wtLIMK1. Importantly, co-expression of S3A-cofilin resulted in the complete recovery of phenotype induced by wtLIMK1 (Figure 2E), suggesting that LIMK1 exerts its effect on endothelial barrier disruption through phosphorylation of cofilin.
LIMK1 destabilizes inter-endothelial junctions
Barrier breakdown that is observed during inflammation, is regulated, in part, by the integrity of intercellular junctions5. To analyze ultra-structural changes of the endothelial barrier in limk1−/− mice, we used quantitative transmission electron microscopy. Tight junctions, formed between adjacent endothelial cells in the capillary segment of the lungs, were detected as characteristic central dark element (fused outer layers) separated symmetrically by two light zones (central, lipid layers of unit) from two dark lines which are the cytoplasmic layers of unit membrane of each endothelial cell22.
We did not observe differences in the structure of endothelium in the lungs of non-stimulated wild type and limk1−/− mice (Figure 3A, B). PAR1 peptide induced opening of inter-endothelial junctions in both genotypes, however, quantitative morphometric analysis of the inter-endothelial junctions revealed that the number of open junctions was significantly smaller in limk1−/− mice (Fig. 3C).
Figure 3. LIMK1 destabilizes interendotheial junctions.
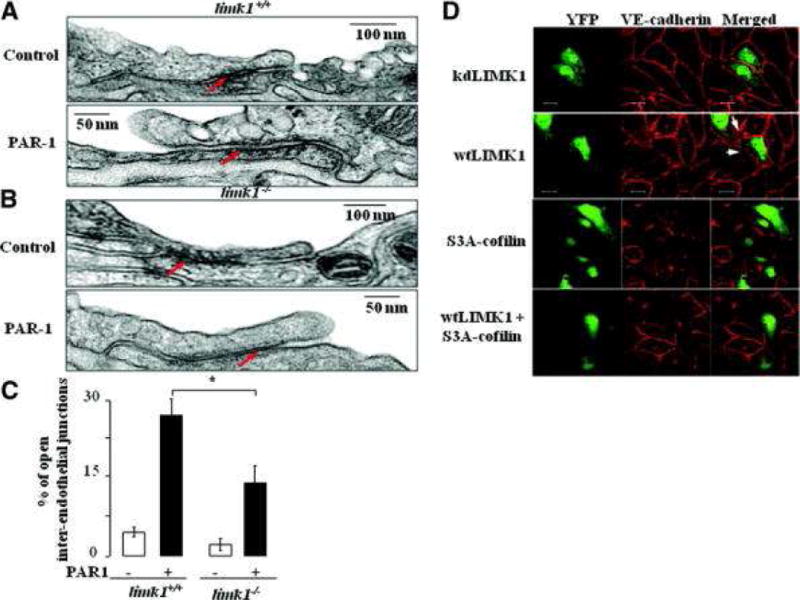
A) Electron microscopy of interendothelial junction of the limk1+/+, and limk1−/− mice (B) treated with PAR-1 peptide, where indicated. Arrows point to the sealed or opened junctions. Magnification X 60 000. C) Morphometric analysis of lung inter-endothelial junctions. Tight junctions were detected as a characteristic central dark element (fused outer layers) separated symmetrically by two light zones (central, lipid layers of unit) from two dark lines which are the cytoplasmic layers of unit membrane of each endothelial cell. Individual values are the mean of 50 junctions counted from each of 3 lung preparations per bar (p = 0.010197). Counting was performed in a blind manner to the mice genotype and experimental conditions. Error bars represent SEM. D) Kinase dead LIMK1 mutant (kdLIMK1) (upper panel), or wild type LIMK1 (wtLIMK1) (second panel from the top), or S3A-cofilin (third panel), or wtLIMK1+S3A-cofilin (lower panel) were overexpressed in HUVECs for 24 hours. YFP was co-transfected in each experiment to make available the visualization of transfected cells. Cells were then fixed and stained with the anti-VE-cadherin antibody and appropriate secondary antibody. Images were taken using dual-wavelength laser scanning confocal microscope. Intercellular gap formation was observed between any adjacent cells transfected with wild type LIMK1. At least 20 pairs of transfected cells were analyzed per sample. Experiments were performed three times yielding similar results.
To address the question about the role of LIMK1 kinase activity in morphology of endothelial junctions, we overexpressed kdLIMK1 or wtLIMK1 in HUVECs. Continuous junctional immunostaining was displayed by staining with anti-VE-cadherin antibody in HUVECs transfected with the kdLIMK1 (Figure 3D). On the contrary, overexpression of wtLIMK1 resulted in appearance of intercellular gaps (Figure 3D). Importantly, destabilization of endothelial junctions, induced by wtLIMK1, was eliminated by co-expression of S3A-cofilin together with wtLIMK1 (Figure 3D). Similar results were obtained by staining with anti-Zonula Occludence-1 (ZO-1) antibody (data not shown).
To summarize, these data indicated that down-regulation of LIMK1 enhanced endothelial barrier function both in-vivo and in cell culture models. Lesser number of the open inter-endothelial junctions, as observed after PAR1 stimulation, could be the underlying cause of enhanced endothelial barrier function in limk1−/− mice. The decrease in vascular permeability, due to the improvement of endothelial barrier function, could have contributed to the attenuation of edema formation in limk1−/− mice. Mechanistically, LIMK1 exerted its effect on the disruption of endothelial barrier through phosphorylation of actin-depolymerizing factor, cofilin.
Reduced diapedesis of limk1−/−PMN in the lungs during ALI
It is well established that infiltration of neutrophils in lungs, followed by tissue damage and lung edema3, 4, plays a crucial role in the pathogenesis of ALI23–26. Increased production of reactive oxygen species and proteolytic enzymes by neutrophils are known mechanisms underlying the development of tissue damage in ALI3.
We tested whether production of reactive oxygen species or degranulation were decreased in limk1−/− mice. Data showed that fMLP- or PMA-induced O2− production in neutrophils as well as the total amount of superoxide produced did not differ between limk1+/+ and limk1−/− mice (Online Figure II A). Neutrophil degranulation, a measure of proteolytic enzymes release, induced by fMLP or C5a (an anaphylatoxin, which is produced upon compliment activation in sepsis27, was similar in limk1+/+ and limk1−/− PMN (Online Figure II B).
Next, we evaluated the extent of PMN extravasation into the lung tissues using hemotaxilin/eosin and leukocyte-specific Leder staining. In both limk1+/+ and limk1−/− mice, lung sections showed infiltration of neutrophils at 6 h after LPS challenge. Notably, the number of neutrophils in the lungs of limk1−/− mice was significantly smaller (Figure 4A, B, C).
Figure 4. Migratory PMN function is impaired inlimk1−/− mice.
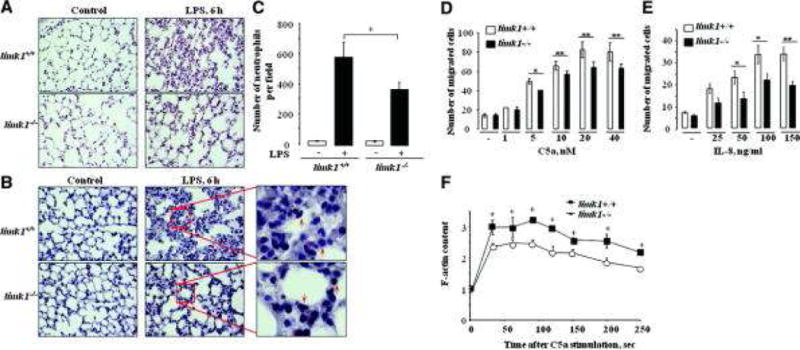
A) Representative photomicrographs of lung sections stained with hemotaxilin/eosin (magnification 40X). B) Representative photomicrographs of lung sections stained with Leder staining (naphtol AS-D chloroacetate esterase, to detect neutrophils). Arrows point to neutrophils. C) Quantitative analysis of photomicrographs, each bar represents mean count of five different sections from each of 3 mice lung preparations per bar (p = 0.011492). Counting was performed in a blinded manner to the mice genotype and experimental conditions. Error bars represent SEM. D, E) Chemotactic response of limk1+/+ and limk1−/− PMN. Neutrophil chemotaxis was determined in a Transwell assay. Culture medium with or without C5a (D) or IL-8 (E) was added to the lower well and cells were placed in the upper well for 40 min. Spontaneous cell motility was determined with culture medium both in the upper and lower wells. Data represents relative amount of cells that reached the lower well. Experiments were performed three times, in triplicates; each time neutrophils were isolated and pooled from 3 mice per group. Counting was performed in a blinded manner to the mice genotype and experimental conditions. Error bars represent SEM. F) C5a-induced actin polymerization in neutrophils isolated from limk1+/+ and limk1−/− mice. Neutrophils were stimulated with 20 nM C5a for the indicated period of time at room temperature, cells were fixed and permeabilized. Actin polymerization was determined by FITC-phalloidin staining and FACS. Experiments were performed three times, in duplicates; each time neutrophils were isolated and pooled from 3 mice per group. Error bars represent SEM.
Decreased number of neutrophils in lungs of limk1−/− mice was not due to a decrease of the total amount of circulating neutrophils, since hemograms obtained before and after LPS injection showed no difference between limk1+/+ and limk1−/− genotypes (Online Table I).
ALI causes increased secretion of cytokines that promote neutrophil infiltration in the lungs3. We determined whether decreased cytokine production accounted for decreased neutrophil infiltration in limk1−/− mice. Data showed that IL-6, MIP-1α, TNFα and RANTES production in response to LPS did not differ between limk1+/+ and limk1−/− mice (Online Figure III).
Next, we investigated whether altered chemotactic activity in response to C5a and IL-8 (a chemokine that mediates the influx of neutrophils in the lungs during ALI28, 29) contributed to decreased neutrophil infiltration in the lungs of limk1−/− mice. Interestingly, directed chemotaxis of limk1−/− PMNs was decreased by ~20% in response to C5a (Figure 4D); and by ~40% in response to IL-8 (Figure 4E). That led us to conclusion that reduced number of infiltrated PMN in the lungs of limk1−/− mice could be attributed to defects in limk1−/− PMN chemotaxis.
Polymerization of actin is one of the early events during neutrophils activation and is required for the efficient chemotaxis30, 31. Considering a significant role of LIMK1 in actin polymerization, we analyzed C5a-induced changes in F-actin content in neutrophils from limk1+/+ and limk1−/− mice. C5a-induced actin polymerization in limk1−/− PMNs was significantly decreased and less sustained than in limk1+/+ PMNs (Figure 4F), thus providing an explanation for the defects in limk1−/− PMN chemotaxis.
To summarize, genetic deletion of LIMK1 reduced the severity of ALI as was indicated by increased survival rate, decreased lung edema formation, and decreased PMN infiltration in the lungs. We propose that enhancement of the endothelial barrier function in lungs of limk1−/− mice, through attenuation of RhoA-dependent signaling, along with a decrease of limk1−/− PMN chemotaxis, due to defects in actin polymerization, were the underlying mechanisms that reduced the severity of ALI.
Discussion
In this study we demonstrated that genetic deletion of LIMK1 resulted in increased survival rate and decreased lung edema formation during ALI. For the first time, we provided the evidence that LIMK1 mediated RhoA-dependent endothelial barrier disruption during inflammation, induced by LPS or PAR-1 peptide. We suggested that improvement of the endothelial barrier function through attenuation of LIMK1-mediated contractility; and decrease in PMN chemotaxis due to defects in actin polymerization were the underlying mechanisms that led to the reduction of the inflammatory responses in limk1−/− mice during ALI. Downregulation of LIMK1 in vivo and in cell culture models resulted in the improvement of endothelial barrier function. Mechanistically, LIMK1 mediated disruption of endothelial barrier through phosphorylation of actin-depolymerizing factor, cofilin.
The pathogenesis of ALI includes disruption of endothelial barrier that leads to protein-rich edema2, which ultimately contributes to significant mortality of the patients affected with ALI1, 32, 33. Two actin cytoskeleton-dependent events are linked in a positive feed-forward cycle and are primarily responsible for the development of inflammatory response and lung edema during ALI: impairment of the endothelial barrier function and neutrophil-dependent tissue damage23–26.
Endothelial barrier function: actin cytoskeleton and LIMK1
Endothelial barrier function is controlled by the actomyosin cytoskeleton via regulation of the stability of intercellular junctions and tensile force within the monolayer34–36. Here, for the first time, we showed that downregulation of LIMK1 resulted in decreased microvascular permeability in mouse lungs and improved endothelial barrier function both in vivo and in cell culture models. The lower basal permeability induced by downregulation of LIMK1 could be explained by reduced homeostatic tensile force in endothelial cells due to reduced actin polymerization. Our current data also showed that LIMK1 regulated disruption of endothelial barrier function primarily through phosphorylation of actin depolymerizing factor, cofilin (Figure 2E).
RhoA-ROCK-LIMK1 signaling axis regulates endothelial barrier disruption during inflammation
It has been speculated that RhoA–mediated increase in actomyosin contractility plays a significant role in the disruption of endothelial barrier during inflammation36. Our current data together with the previous data from our lab7 and others8 provided the evidence that RhoA, through RhoA-ROCK-LIMK1 pathway, induced disruption of endothelial barrier during inflammation in mouse lungs in vivo. We showed here that LPS induced activation of both RhoA and LIMK1 in mouse lungs (Online Figure I and Figure 1D). Notably, genetic deletion of LIMK1 significantly decreased LPS- and PAR-1-induced RhoA-mediated effects on the disruption of endothelial barrier function, without affecting the activation cycle of small GTPases per se (Online Figure I). The mechanism of LPS-induced RhoA activation in endothelial cells in vivo still remains to be determined. It may include autocrine signaling through G protein-coupled receptors (GPCRs) facilitated by anaphylatoxins, which are products of complement activation37, 38, or by chemokines, since it has been shown that chemokines exert their effects through at least nineteen GPCRs39, 40.
The role of other organs and cell types in the pathogenesis of ALI in limk1−/− mice
Endothelial cells and neutrophils are two cell types primarily involved in the development of inflammatory response in lungs during ALI, therefore it is plausible to conclude that decreased microvascular permeability and PMN infiltration in LIMK1-/- mice played the major role in increased survival rate during ALI. However, there is still a possibility that defects in other cell types also contributed to the protection against mortality, since LIMK1 is ubiquitously expressed in all tissues13. On the other hand, although we observed that LPS-induced lung edema formation was significantly suppressed (Figure 1G), about half of limk1−/− mice were mortal by LPS treatment. This phenomenon can be also explained by the fact that the development of LPS-induced sepsis is manifested by multiple organ failure that includes kidney, lung, liver, etc.41. Therefore, defects in multiple organs contribute to overall mortality, and suppression of lung edema formation may not be enough to completely abolish LPS-induced mortality.
PMN diapedesis during ALI
Neutrophil migration into the lung tissue plays a crucial role in the pathogenesis of ALI23–26. Activation of PMN results in the responses such as migration, degranulation and phagocytosis, which are mediated by actin cytoskeleton42. We studied the role of LIMK1 during LPS-induced acute lung injury in two classes of cellular responses of PMN: 1) migratory (chemotaxis and transmigration) and 2) secretory (generation of oxygen radicals and secretion of lysosomal enzymes).
The transendothelial migration (diapedesis) is governed by coordinated action of both endothelial cells and neutrophils. In endothelial cells Rho proteins-dependent43 remodeling of the F-actin cytoskeleton and actin-associated interendothelial adherens junctions has been observed as the leukocyte completes diapedesis step44, 45. In neutrophils sequential activation of RhoA family small GTPases by microenvironmental signals leads to the actin cytoskeleton, which results in cell orientation and extension of extrapolary filopodia in the gap between endothelial cells30.
Here we show that genetic deletion of LIMK1 decreased PMN infiltration in mouse lungs in vivo and produced defects in PMN chemotaxis in vitro (Figure 4A, B, C, D, E). Observed reduction in limk1−/− PMN chemotaxis can be explained by perturbances in the dynamics of actin polymerization (Figure 4F).
It is reasonable to attribute decreased limk1−/− PMN infiltration in lungs to defects in PMN chemotaxis. However, it is also possible that impaired actin cytoskeleton dynamics in endothelial cells contributed to reduced diapedesis, since it was shown that disruption of actin cytoskeleton in endothelial cells resulted in reduced transendothelial migration of monocytes46.
We suggest that impaired actin cytoskeleton dynamics, which resulted in the enhancement of endothelial barrier, together with decreased PMN chemotaxis contributed to the decreased PMN diapedesis. The relative degree to which improvement of the endothelial barrier or decreased PMN chemotaxis contributed to the reduced PMN infiltration and lung injury remains to be determined.
PMN superoxide production and degranulation
PMN cytoskeletal dynamics plays a crucial role in superoxide production and degranulation30. It was shown that inhibition of actin polymerization in activated neutrophils resulted in augmentation of superoxide anion production and degranulation induced by different agents47–49. Interestingly, we found that stimulus-induced superoxide production and degranulation in limk1−/− PMN did not differ from limk1+/+ cells, although actin polymerization was impaired. Possible explanation underlying this phenomenon might be in the difference of the extent to which actin cytoskeleton dynamics was perturbed, since there are multiple ways to regulate actin cytoskeleton dynamics and LIMK1 affects only one of those.
To summarize, we demonstrated that the severity of acute lung injury was decreased in limk1−/− mice, as was observed by increased survival rate, decreased edema formation and PMN infiltration in the lungs. Based on our findings, we propose that organ-specific downregulation or inhibition of LIMK1 activity could be a potential strategy to reduce vascular leakage during many life-threatening diseases including ALI.
Supplementary Material
Acknowledgments
Sources of funding
This work was supported by National Institutes of Health Grants GM56159 and P01 HL60678.
Non-standard Abbreviations and Acronyms
- ALI
Acute lung injury
- C5a
Complement component 5a
- fMLP
N-Formylmethionyl-leucyl-phenylalanine
- GPCR
G protein-coupled receptor
- HUVEC
Human umbilical vein endothelial cells
- LIMK1
LIM domain kinase 1
- LPS
Lipopolysaccharide
- PAR-1 peptide
peptide agonist corresponding to the tethered ligand sequence of human PAR-1 (SFLLRN)
- PMA
Phorbol myristate acetate
- PMN
Polymorphonuclear leucocytes
- ROCK
Rho kinase
- VE-cadherin
Vascular endothelial cadherin
- ZO-1
Zonula occludence-1
Footnotes
Disclosures
None
References
- 1.Ware LB, Matthay MA. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med. 2005;353(26):2788–2796. doi: 10.1056/NEJMcp052699. [DOI] [PubMed] [Google Scholar]
- 3.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7(1):1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007;9(11):1991–2002. doi: 10.1089/ars.2007.1785. [DOI] [PubMed] [Google Scholar]
- 5.Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;2007(412):re8. doi: 10.1126/stke.4122007re8. [DOI] [PubMed] [Google Scholar]
- 6.Niggli V. Rho-kinase in human neutrophils: a role in signalling for myosin light chain phosphorylation and cell migration. FEBS Lett. 1999;445(1):69–72. doi: 10.1016/s0014-5793(99)00098-8. [DOI] [PubMed] [Google Scholar]
- 7.Gorovoy M, Neamu R, Niu J, Vogel S, Predescu D, Miyoshi J, Takai Y, Kini V, Mehta D, Malik AB, Voyno-Yasenetskaya T. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007;101(1):50–58. doi: 10.1161/CIRCRESAHA.106.145847. [DOI] [PubMed] [Google Scholar]
- 8.Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, Ishizaka A. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol. 2005;32(6):504–510. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- 9.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275(5):3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 10.Gorovoy M, Niu J, Bernard O, Profirovic J, Minshall R, Neamu R, Voyno-Yasenetskaya T. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J Biol Chem. 2005;280(28):26533–26542. doi: 10.1074/jbc.M502921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35(1):121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 12.Lian JP, Marks PG, Wang JY, Falls DL, Badwey JA. A protein kinase from neutrophils that specifically recognizes Ser-3 in cofilin. J Biol Chem. 2000;275(4):2869–2876. doi: 10.1074/jbc.275.4.2869. [DOI] [PubMed] [Google Scholar]
- 13.Foletta VC, Moussi N, Sarmiere PD, Bamburg JR, Bernard O. LIM kinase 1, a key regulator of actin dynamics, is widely expressed in embryonic and adult tissues. Exp Cell Res. 2004;294(2):392–405. doi: 10.1016/j.yexcr.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Matute-Bello G, Frevert CW, Martin TR. Animal Models of Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol. 2008 doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rojas M, Woods CR, Mora AL, Xu J, Brigham KL. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol. 2005;288(2):L333–341. doi: 10.1152/ajplung.00334.2004. [DOI] [PubMed] [Google Scholar]
- 16.Parker JC, Townsley MI. Evaluation of lung injury in rats and mice. Am J Physiol Lung Cell Mol Physiol. 2004;286(2):L231–246. doi: 10.1152/ajplung.00049.2003. [DOI] [PubMed] [Google Scholar]
- 17.Vogel SM, Gao X, Mehta D, Ye RD, John TA, Andrade-Gordon P, Tiruppathi C, Malik AB. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics. 2000;4(2):137–145. doi: 10.1152/physiolgenomics.2000.4.2.137. [DOI] [PubMed] [Google Scholar]
- 18.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 19.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273(34):21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 20.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA. 1992;89(17):7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393(6687):805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 22.Luft JH. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed Proc. 1966;25(6):1773–1783. [PubMed] [Google Scholar]
- 23.Martin TR. Neutrophils and lung injury: getting it right. J Clin Invest. 2002;110(11):1603–1605. doi: 10.1172/JCI17302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogg JC, Doerschuk CM. Leukocyte traffic in the lung. Annu Rev Physiol. 1995;57:97–114. doi: 10.1146/annurev.ph.57.030195.000525. [DOI] [PubMed] [Google Scholar]
- 25.Doerschuk CM, Mizgerd JP, Kubo H, Qin L, Kumasaka T. Adhesion molecules and cellular biomechanical changes in acute lung injury: Giles F. Filley Lecture. Chest. 1999;116(1 Suppl):37S–43S. doi: 10.1378/chest.116.suppl_1.37s-a. [DOI] [PubMed] [Google Scholar]
- 26.Martin TR, Pistorese BP, Hudson LD, Maunder RJ. The function of lung and blood neutrophils in patients with the adult respiratory distress syndrome. Implications for the pathogenesis of lung infections. Am Rev Respir Dis. 1991;144(2):254–262. doi: 10.1164/ajrccm/144.2.254. [DOI] [PubMed] [Google Scholar]
- 27.Shin HS, Snyderman R, Friedman E, Mellors A, Mayer MM. Chemotactic and anaphylatoxic fragment cleaved from the fifth component of guinea pig complement. Science. 1968;162(851):361–363. doi: 10.1126/science.162.3851.361. [DOI] [PubMed] [Google Scholar]
- 28.Miller EJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. 1992;146(2):427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 29.Martin TR. Lung cytokines and ARDS: Roger S. Mitchell Lecture. Chest. 1999;116(1 Suppl):2S–8S. doi: 10.1378/chest.116.suppl_1.2s. [DOI] [PubMed] [Google Scholar]
- 30.Vicente-Manzanares M, Sanchez-Madrid F. Role of the cytoskeleton during leukocyte responses. Nat Rev Immunol. 2004;4(2):110–122. doi: 10.1038/nri1268. [DOI] [PubMed] [Google Scholar]
- 31.Jones GE. Cellular signaling in macrophage migration and chemotaxis. J Leukoc Biol. 2000;68(5):593–602. [PubMed] [Google Scholar]
- 32.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151(2 Pt 1):293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 33.Vincent JL, Sakr Y, Ranieri VM. Epidemiology and outcome of acute respiratory failure in intensive care unit patients. Crit Care Med. 2003;31(4 Suppl):S296–299. doi: 10.1097/01.CCM.0000057906.89552.8F. [DOI] [PubMed] [Google Scholar]
- 34.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11(12):471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 35.Braga VM. Cell-cell adhesion and signalling. Curr Opin Cell Biol. 2002;14(5):546–556. doi: 10.1016/s0955-0674(02)00373-3. [DOI] [PubMed] [Google Scholar]
- 36.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39(4-5):187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 37.Vallota EH, Muller-Eberhard HJ. Formation of C3a and C5a anaphylatoxins in whole human serum after inhibition of the anaphylatoxin inactivator. J Exp Med. 1973;137(5):1109–1123. doi: 10.1084/jem.137.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol. 2002;169(4):2102–2110. doi: 10.4049/jimmunol.169.4.2102. [DOI] [PubMed] [Google Scholar]
- 39.New DC, Wong YH. CC chemokine receptor-coupled signalling pathways. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2003;35(9):779–788. [PubMed] [Google Scholar]
- 40.Johnson Z, Power CA, Weiss C, Rintelen F, Ji H, Ruckle T, Camps M, Wells TN, Schwarz MK, Proudfoot AE, Rommel C. Chemokine inhibition--why, when, where, which and how? Biochem Soc Trans. 2004;32(Pt 2):366–377. doi: 10.1042/bst0320366. [DOI] [PubMed] [Google Scholar]
- 41.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 42.Omann GM, Allen RA, Bokoch GM, Painter RG, Traynor AE, Sklar LA. Signal transduction and cytoskeletal activation in the neutrophil. Physiol Rev. 1987;67(1):285–322. doi: 10.1152/physrev.1987.67.1.285. [DOI] [PubMed] [Google Scholar]
- 43.Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol. 1999;162(5):2964–2973. [PubMed] [Google Scholar]
- 44.Sandig M, Korvemaker ML, Ionescu CV, Negrou E, Rogers KA. Transendothelial migration of monocytes in rat aorta: distribution of F-actin, alpha-catnin, LFA-1, and PECAM-1. Biotech Histochem. 1999;74(6):276–293. doi: 10.3109/10520299909034666. [DOI] [PubMed] [Google Scholar]
- 45.Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167(4):2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 46.Kielbassa K, Schmitz C, Gerke V. Disruption of endothelial microfilaments selectively reduces the transendothelial migration of monocytes. Exp Cell Res. 1998;243(1):129–141. doi: 10.1006/excr.1998.4133. [DOI] [PubMed] [Google Scholar]
- 47.Al-Mohanna FA, Hallett MB. Actin polymerization modifies stimulus-oxidase coupling in rat neutrophils. Biochim Biophys Acta. 1987;927(3):366–371. doi: 10.1016/0167-4889(87)90101-7. [DOI] [PubMed] [Google Scholar]
- 48.Al-Mohanna FA, Ohishi I, Hallett MB. Botulinum C2 toxin potentiates activation of the neutrophil oxidase. Further evidence of a role for actin polymerization. FEBS Lett. 1987;219(1):40–44. doi: 10.1016/0014-5793(87)81187-0. [DOI] [PubMed] [Google Scholar]
- 49.Norgauer J, Kownatzki E, Seifert R, Aktories K. Botulinum C2 toxin ADP-ribosylates actin and enhances O2- production and secretion but inhibits migration of activated human neutrophils. J Clin Invest. 1988;82(4):1376–1382. doi: 10.1172/JCI113741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.