

Abstract
The 2 465 177 bp genome of Sulfolobus islandicus LAL14/1, host of the model rudivirus SIRV2, was sequenced. Exhaustive comparative genomic analysis of S. islandicus LAL14/1 and the nine other completely sequenced S. islandicus strains isolated from Iceland, Russia and USA revealed a highly syntenic common core genome of approximately 2 Mb and a long hyperplastic region containing most of the strain-specific genes. In LAL14/1, the latter region is enriched in insertion sequences, CRISPR (clustered regularly interspaced short palindromic repeats), glycosyl transferase genes, toxin–antitoxin genes and MITE (miniature inverted-repeat transposable elements). The tRNA genes of LAL14/1 are preferential targets for the integration of mobile elements but clusters of atypical genes (CAG) are also integrated elsewhere in the genome. LAL14/1 carries five CRISPR loci with 10 per cent of spacers matching perfectly or imperfectly the genomes of archaeal viruses and plasmids found in the Icelandic hot springs. Strikingly, the CRISPR_2 region of LAL14/1 carries an unusually long 1.9 kb spacer interspersed between two repeat regions and displays a high similarity to pING1-like conjugative plasmids. Finally, we have developed a genetic system for S. islandicus LAL14/1 and created ΔpyrEF and ΔCRISPR_1 mutants using double cross-over and pop-in/pop-out approaches, respectively. Thus, LAL14/1 is a promising model to study virus–host interactions and the CRISPR/Cas defence mechanism in Archaea.
Keywords: Archaea, Sulfolobus islandicus LAL14/1, genome analysis, genetics, CRISPR
2. Introduction
The genus Sulfolobus was first described by Brock et al. in 1972 [1] and includes thermoacidophilic Archaea that grow at 70–85°C and pH 2–3 under aerobic conditions either chemolithotrophically by oxidizing elementary sulfur/hydrogen sulfide or heterotrophically [2]. Sulfolobus strains have been isolated from various acidic thermal habitats (in the USA, Italy, Iceland, Russia and elsewhere). They are easily maintained under laboratory conditions, making them convenient models to study the molecular organization of the archaeal cell [3].
The sequences of 12 Sulfolobus genomes are currently available. They include Sulfolobus solfataricus P2 [4], Sulfolobus tokodaii [5], Sulfolobus acidocaldarius [6] and nine strains of Sulfolobus islandicus: HV10/4 and REY15A [7] (isolated from hot springs in Iceland) [8]; M.14.25, M.16.27 and M.16.4 (from hot springs at the Mutnovsky Volcano, Kamchatka, Russia); Y.N.15.51 and Y.G.57.14 (from hot springs in Yellowstone National Park, USA); and L.D.8.5 and L.S.2.15 (from Lassen National Park, USA) [9].
The strain S. islandicus LAL14/1 was isolated in 1995 from a solfataric field in Iceland by the group of Zillig [8]. Its geographical origin, growth requirements and physiology indicate that LAL14/1 is a close relative of two S. islandicus strains also isolated from Iceland, HVE10/4 and REY15A. However, LAL14/1 has a particular pattern of sensitivity to various archaeal viruses. LAL14/1 is resistant to the rudivirus SIRV1 but can be efficiently infected by its close relative SIRV2 [10], which has a complex cycle of development in the host cells. At the end of the infection cycle, that lasts about 14 h, specific pyramid-like structures are formed on the cell surface facilitating release of virus particles [11–13]. These unique characteristics make S. islandicus LAL14/1 an interesting model to study virus–host interactions in Archaea.
Effective genetic tools have been developed for a limited number of Sulfolobus species [14], including S. solfataricus P1 and 98/2 [15,16], S. acidocaldarius [17,18], S. islandicus REY15A [19–21] and S. islandicus M.16.4 [22]. However, genetic approaches have not previously been available for LAL14/1.
In this study, we report the results of the in silico analysis of the genome sequence of S. islandicus LAL14/1 and detailed comparisons with other available S. islandicus strains, and in particular the closely related strains, REY15A and HVE10/4. We also have established genetic tools for this strain by creating both ΔpyrEF and ΔCRISPR_1 mutants. This work has made substantial progress towards the possibility of applying powerful global approaches (for example, transcriptome, RNAseq and proteome analyses) to elucidate the interplay between host and viral genes and proteins during the viral infection cycle.
3. Material and methods
3.1. Strains growth
Sulfolobus islandicus strains were grown aerobically at 80°C and under constant agitation in rich medium containing 0.2 g l−1 of Tryptone Peptone, 2 g l−1 of sucrose and 1 g l−1 of yeast extract. The minimal medium used to select Ura+ variant isolates was as described previously [2]. Ura− mutants were selected on rich solid medium in the presence of 50 mg l−1 of 5′-fluoroorotic acid.
3.2. Genetic experiment
3.2.1. PCR amplification
pyrEF mutant. The following primers were used to amplify the locus, including the pyrEF operon and the upstream (1 kb) and downstream (1 kb) situated regions of the S. islandicus E233S chromosome: oligoUP, CAGTAGCTAAAACAATTGAAAGAGTAGGTG; oligoDOWN, CTAATGATGCTTGATAGAAGTATTTAGCGT. The PCR amplification was performed in 50 µl of reaction mixture containing 10 µM of each primer, 1 µl template, 10 µl 5× HF Phusion Buffer (Finzyme), 10 nM dNTPs and 0.5 µl pfu DNA polymerase (Finzyme) with the following conditions: 30 s at 98°C, 60 s at 55°C and 90 s at 72°C for 35 cycles.
CRISPR mutant. The following primers were used to amplify the DNA fragments IN (oligoup1, AAAAAACCATGGTACGATTCCGCTTAAGCC; oligodown2, AAAAAAGGATCCGTAATGAGAGCTTGGTTT); OUT (oligoup3, AAAAAGTCGACTACTACCGTGTACTTCCCC; oligodown4, AAAAACCATGGTGCGTTAATGAGGCAAGGT) and TARGET (oligoup5, AAAAAAGCATGCTTCTGCTCAAAAGGAGGA; oligodown6, AAAAAACTGCAGTAGAAGAAGATAGCCCAC). The positions of these fragments is indicated in figure 8.
Figure 8.

Genetic map of the CRISPR_1 region deleted by the pop-in/pop-out approach. (a) Genetic map of the CRISPR_1 region. The deleted region is situated between the regions OUT and TARGET. (b) A scheme representing two stages of recombination events generating the mutant ΔCRISPR_1Δcsx1**Δcas4Δ pyrEF. Two paralogues of csx1 are present in this region. The gene csx1* corresponds to the gene Sil_0392 and csx1** to Sil_0393.
Transformation. Electroporation of S. islandicus LAL14/1 was performed as described by Deng et al. [19].
3.3. Genome sequencing
Total DNA was extracted from the cells using phenol–chloroform and ethanol precipitation. The sequencing was done by Fidelity System Inc. using Illumina technology and assembled using the software Velvet v. 1.2 [23]. The genome was automatically annotated and refined manually. Open reading frames (ORFs) were predicted using phred, phrap and consed software [24–26] and tRNA with tRNAscan-SE [27]. Putative insertion sequence (IS) elements were identified by BLASTn search against the IS Finder Database (http://www.is-biotoul.fr/). Annotations were manually curated using Ugene software [28].
3.4. Phylogenetic analysis
The genome sequences of nine S. islandicus strains were downloaded as Genbank files from the NCBI database (NC_012588, NC_012623, NC_017275, NC_013769, NC_012589, NC_012632, NC_012726, NC_017276 and NC_012622).
The DNA sequences of all genomes were aligned using the progressiveMauve algorithm with default parameters and analysed with stripSubsetLCBs. The S. islandicus core genome sequence was used for phylogenetic dating based on the standard rate of accumulation of random mutations in hyperthermophilic Archaea (4.66 × 10−9 substitution per site per year; [9]). Clonal genealogy was inferred using the ClonalFrame algorithm three times.
3.5. Dot-Plot
The Ugene Dot-Plot algorithm with the option ‘search for inverted repeats’ enabled was used to generate dot-plots from pairs of sequences. Sequences from the 10 S. islandicus strains were aligned with the progressiveMauve algorithm with scoring parameters divided by four to ensure the recognition of large conserved regions.
3.6. Replication origins
Zplotter applet v. 2.0 was used to calculate Zcurves (http://tubic.tju.edu.cn/zcurve/).
OriC1 and OriC2 structures were determined by alignment of OriC1 and OriC2 sequences in S. solfataricus P2 with S. islandicus LAL14/1 sequences using the BLASTn algorithm with default parameters. OriC3 structures were determined by mapping UCM (UnCharacterized Motif) sequences in the genome of LAL14/1 using the Ugene search tool with default parameters.
3.7. Exceptional motifs
R'MES software [29] was used to evaluate the significance of motif frequency in S. islandicus genomes. This statistical method compares the observed count of each motif to the count predicted by a reference probabilistic Markovian model. Exceptional motifs of lengths two to eight were analysed choosing models according to the authors’ guidelines (http://migale.jouy.inra.fr/?q=method) and using the highest possible order each time (usually I-2, where I is the length of the studied motif).
3.8. Spacer data
All available CRISPR spacers from S. islandicus LAL14/1 were determined using CRISPRfinder (http://crispr.u-psud.fr/Server/) [30].
3.9. PAM motifs
To find PAM motifs, protospacers corresponding to the selected spacers listed in the electronic supplementary material, table S15 were aligned and visualized with WebLogo (http://weblogo.berkeley.edu/logo.cgi). For each protospacer, the regions analysed were 10 nucleotide-long sequences immediately upstream and downstream from the region identical or similar to the spacer.
3.10. Sulfolobus islandicus pan-genome characterization
The pan-genome of the 10 available S. islandicus strains was obtained with an in-house program [31]. Briefly, the proteins encoded by the 10 genomes were compared using two-way BLASTp analysis and ranked in families of orthologous proteins according to the criteria defined by Lerat et al. [32].
4. Results
4.1. General features of the Sulfolobus islandicus LAL14/1 genome
The genome of S. islandicus strain LAL14/1 (NCBI accession no. CP003928) was sequenced at 104-fold coverage by Fidelity System (http://fidelitysystems.com/) using Illumina technology. The protein-coding genes were annotated using Ugene software [28]. The S. islandicus LAL14/1 genome consists of a single circular chromosome of 2 465 177 bp; 85.6 per cent of the genome is coding. The chromosome carries 2601 protein-coding genes and has a GC content of 35 per cent. The general properties of the S. islandicus LAL14/1 genome composition are summarized in table 1.
Table 1.
General properties and composition of the genome of S. islandicus LAL14/1.
| genome size | 2 465 177 bp |
| protein-coding genes | 2601 (85.6%) |
| CDS average size | 815 bp |
| average size of intergenic regions | 190 bp |
| tRNA-coding genes | 45 functional tRNA |
4.2. Phylogenetic position of Sulfolobus islandicus LAL14/1
The phylogenetic tree of available S. islandicus strains was established by comparison of chromosomal DNA sequences shared by all 10 strains (figure 1) as described in §3. The strain LAL14/1 is phylogenetically very close to the strains HVE10/4 and REY15A, also isolated from Iceland. On the basis of the standard rate of random mutation accumulation in S. islandicus [9], we estimate that strain REY15A separated from the clade LAL + HVE about 460 000 years ago and strain LAL14/1 diverged from the clade LAL + HVE 60 000 years later. The tree is divided into three main clades corresponding to the geographical origins of the strains: clade I from Iceland; clade M from Kamchatka (Russia) and clade L/Y from Lassen and Yellowstone (USA) [9,33].
Figure 1.
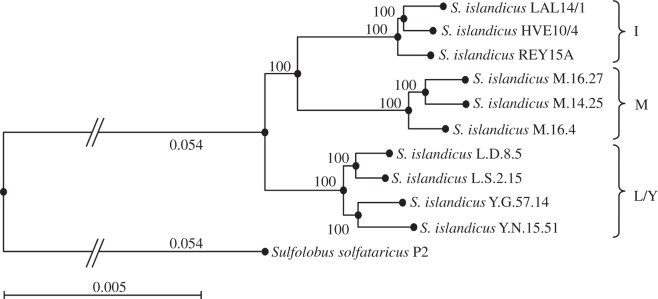
Phylogenetic position of S. islandicus LAL14/1 among other sequenced S. islandicus strains. Sulfolobus solfataricus P2 is used as an external group. The length of branches is proportional to the phylogenetic distance between the strains. Bootstrap values are indicated.
The general features of the 10 S. islandicus genomes are reported in table 2. LAL14/1 has the smallest genome, apparently due to the small number of horizontally transferred CAG regions (see below). It carries a remarkably complex CRISPR system, with more spacers than the other S. islandicus genomes, some matching the virus SIRV1 perfectly.
Table 2.
Comparison of genomic patterns of 10 S. islandicus strains (compilation of our own data and those from [7,9]. CAG are discussed elsewhere in the text. SIRV1 and SIRV2 are rudiviruses described in [10,34,35]. R, resistant; S, sensitive; nd, no data. CRISPR and Cmr families are indicated following established classification [36,37].
| LAL14/1 | REY15A | HVE10/4 | L.D.8.5 | L.S.2.15 | M.16.4 | M.16.27 | M.14.25 | Y.G.57.14 | Y.N.15.51 | |
|---|---|---|---|---|---|---|---|---|---|---|
| genome size (Mb) | 2.4 | 2.7 | 2.5 | 2.7 | 2.7 | 2.6 | 2.7 | 2.6 | 2.7 | 2.8 |
| gene no. | 2601 | 2666 | 2745 | 2939 | 2760 | 2759 | 2680 | 2632 | 2928 | 2868 |
| CAG no. | 3 | 4 | 7 | 16 | 13 | 5 | 5 | 11 | 16 | 22 |
| CRISPR/Cas families (no. of spacers) | I (199) III (84) |
I (208) | I (215) III (47) |
I (223) III (42) |
I (225) II (7) |
I (139) | I (164) II (72) |
I (143) II (39) |
I (57) | I (99) |
| Cmr families | B,F | B | 2 × B | B | B | 2 × B | B | B,D | 3 × B | E |
| perfect spacers against SIRV1 and phenotype | 2 (R) | 0 (R) | 0 (R) | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd |
| perfect spacers against SIRV2 and phenotype | 0 (S) | 2 (R) | 3 (R) | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd | 0 nd |
4.3. The Sulfolobus islandicus pan-genome and specific genomic pattern of the strain LAL14/1
The first version of the S. islandicus pan-genome was published in 2009, based on the analysis of seven strains; it includes 20 610 proteins [9]. Three additional S. islandicus genome sequences have since become available (REY15A, HVE10/4 [7] and LAL14/1 (present work)). The new updated version of the S. islandicus pan-genome has 27 578 proteins (in silico prediction) that can be divided into 3492 families of orthologous proteins (see §3). The statistics of the family distribution is presented in the electronic supplementary material, figure S1. There are 1892 ubiquitous families, present in at least one copy in all of the S. islandicus genomes. This group, indicated in the annotation by arCOG + number, constitutes the S. islandicus core-genome.
There are 1030 families present in two or more (but not all) strains. The best-represented families of S. islandicus include various transposases, ABC transporters and CoA pathway genes (see the electronic supplementary material, table S1). The majority of these families are present in the LAL14/1 genome, and some are overrepresented (more frequent than predicted from the average pan-genome statistics), for example, the transposases belonging to the IS1 family. Others, for example transposases of families ISH3 and IS110, are clearly underrepresented.
There are 570 families classified as singletons. They are strictly strain-specific and present in only one copy per genome (see the electronic supplementary material, figure S2 and table S2). The S. islandicus LAL14/1 genome contains 65 singletons, and specific functions could be predicted for 14 of them. They include a putative transcription regulator of the MarR family (SiL_0405), a putative acyl-coenzyme A synthetase/AMP fatty acid ligase (SiL_0481), a small subunit of the methyltransferase (SiL_0587), a putative glycosyltransferase (SiL_0818), a membrane protein involved in the export of O-antigen and teichoic acid (SiL_0839), a putative secreted endonuclease distantly related to the archaeal Holliday junction resolvase (SiL_1319), an Fe–S oxidoreductase (SiL_1473), and seven families of Cmr proteins related to CRISPRs: Cmr3 (SiL_0600), Cas10 (SiL_0601), Cmr6 (SiL_0602), Cmr5 (SiL_0603), Cmr1 (SiL_0604), Cmr4 (SiL_0605) and Csm6-like protein (SiL_0630).
The taxonomical distribution of the individual genes and singletons of S. islandicus LAL14/1 is summarized in table 3. Of the 2601 annotated protein-coding genes, only 4.7 per cent are exclusive to this strain; 10 per cent are only found in S. islandicus species and 37.6 per cent are specific to Sulfolobales. The 51 S. islandicus LAL14/1-specific singletons, and the seven S. islandicus-specific and seven Crenarchaeota-specific singletons are listed in the electronic supplementary material, tables S3A–C.
Table 3.
Taxonomic specificity of the protein-coding genes of S. islandicus LAL14/1.
| gene specificity |
S. islandicus LAL14/1 |
|
|---|---|---|
| total gene distribution | singleton distribution | |
| S. islandicus LAL14/1-exclusive | 123 | 51 |
| S. islandicus-exclusive (10 strains) | 134 | 0 |
| Sulfolobus-specific | 719 | 7 |
| Crenarchaeota-specific | 238 | 7 |
| Archaea-specific | 409 | 0 |
| Archaea + Bacteria-specific | 535 | 0 |
| Archaea + Eukarya specific | 72 | 0 |
| universal | 371 | 0 |
| total | 2601 | 65 |
The specific genomic pattern of LAL14/1 is presented in table 4. Like all the other studied S. islandicus genomes, LAL14/1 codes for a large number of transposases representing families composed of multiple paralogues. In this strain, the most represented family is the protein OrfB encoded by IS200/IS605. The transposon ISC1048 of the family IS607 is also overrepresented. However, other transposase families, such as families IS110 and ISH3, are clearly underrepresented in LAL14/1.
Table 4.
Major groups of paralogues in S. islandicus LAL14/1 and their representation in 10 S. islandicus genomes.
|
S. islandicus LAL14/1 |
protein families | all S. islandicus |
||
|---|---|---|---|---|
| range | protein no. | protein no. | range | |
| 1 | 16 | IS200/605 families protein OrfBa | 129 | 3 |
| 2 | 13 | oligopeptide/dipeptide ABC transporter, ATPase subunita | 113 | 4 |
| 3 | 10 | IS630 family transposasea | 67 | 8 |
| 4 | 9 | IS1 family transposase | 53 | 7 |
| 5 | 6 | IS110 family transposasea | 151 | 1 |
| 6 | 6 | 3-hydroxyacyl-CoA dehydrogenase NAD-binding proteina | 78 | 5 |
| 7 | 5 | inosine/uridine nucleoside hydrolase | 32 | 30 |
| 8 | 4 | high-affinity nickel-transporter | 21 | 68 |
| 9 | 4 | dTDP-4-dehydrorhamnose 3,5-epimerase | 18 | 145 |
aIncludes several nodes.
The families of orthologous proteins were compared between the S. islandicus strains LAL14/1, REY15A and HVE10/4, all isolated from the same geographical location. This revealed substantial similarity between the genomes of these strains: of the 2770 families analysed, 2130 (77%) are shared by these three S. islandicus genomes. A surprisingly large number of families are unique for each of these strains (figure 2), constituting a specific genomic signature for each of the strains.
Figure 2.
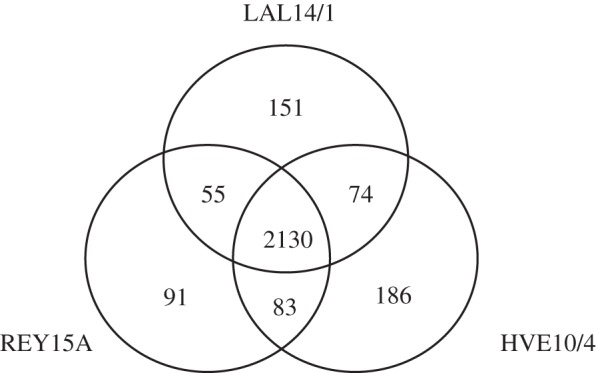
Conservation of protein nodes in three closely related S. islandicus strains. A total of 2130 families are shared by three strains. Each of the strain is characterized by the presence of a specific set of families: 151 for LAL14/1; 91 for REY15A and 186 for HVE10/4.
4.4. Exceptional motifs in Sulfolobus islandicus LAL14/1 genome
Many non-coding motifs have specific biological functions in genomes and statistical analyses of oligomer frequencies in genome sequences can identify possibly significant motifs (e.g. reviewed in [38] for bacteria). Similar analyses have also been very useful for studying the evolution of genome functions and regulation [39].
Systematic analyses of short oligonucleotides of fixed composition (usually called words) were conducted with the LAL14/1 and other S. islandicus genomes to identify non-coding functional motif candidates. The word frequency and preference patterns are very similar in the three closely related S. islandicus strains, suggesting that their functional motifs and associated mechanisms are generally similar. Some more pronounced differences were observed for longer words, and this was mainly associated with the different genomic content of the large variable regions.
As commonly observed in archaeal and bacterial genomes, palindromic motifs are generally avoided (e.g. electronic supplementary material, table S4 for LAL14/1 and electronic supplementary material, table S5), possibly as a consequence of the presence of restriction–modification systems encoded in the genome [40] or of other biological phenomena such as the control of chromosome replication [38]. Among the analysed words (see the electronic supplementary material, tables S4 and S5), some are clearly overrepresented because of their presence in the repeats of the CRISPR sequences. For example, the overrepresented word ACTATAGA, included in the CRISPR repeats, is repeated 196 times (see the electronic supplementary material, figure S3).
For most of the other candidates, no obvious biological function could directly be inferred. There is evidence for eukaryotes that the non-random pattern of short words (two to four letters) may be due to evolutionary changes in informational processes such as DNA replication and repair, and the pattern of long words (eight letters) may reflect evolutionary changes in gene regulatory machinery. The presence of such long words may reflect a non-random frequency of the DNA-binding sites specific for transcription factors [39]. This observation might indicate the presence of eukaryotic-like transcriptional regulation in Archaea.
4.5. Structure and dynamics of the Sulfolobus islandicus genomes
The structural comparison of two closely related genomes, HVE10/4 and REY15A, was published in 2011 [7]. It revealed a high level of synteny of gene content for these genomes and the presence of two variable regions, one of about 0.5–0.7 Mb and a second corresponding to a 200 kb inversion. The structure of the LAL14/1 genome was compared with those of the HVE10/4 and REY15A genomes by dot-plot analyses (see the electronic supplementary material, figure S4). This revealed a well-conserved 2 Mb core region common to all three genomes, with substantial synteny conservation. Many rearrangements were detected in the variable regions of each genome. The localization, length and genetic context of all major differences detected by the dot-plot approach are listed in the electronic supplementary material, table S6. Previous analysis revealed a large inversion of 0.5 Mb as one of the major differences between HVE10/4 and REY15A genomes [7]. Genome sequencing of the LAL14/1 in combination with the phylogenetic analysis (figure 1) allowed us to infer that the inversion occurred in the ancestor of the HVE10/4, while LAL14/1 retained the ancestral genome organization.
The program progressiveMauve [41] was used for alignments of the 10 S. islandicus genomes and visualization of genome rearrangements (figure 3). All the genomes have a common general organization, with a well-preserved part covering about 75 per cent of the genome and a long variable region. Small strain-specific variable regions are scattered throughout the conserved regions of all of the 10 genomes analysed; many correspond to the insertion of heterologous genes transferred horizontally (see below).
Figure 3.

Alignment of 10 S. islandicus genomes by progressiveMauve approach. The blocs of the same colours indicate the regions of synteny. The unique regions are indicated by the absence of blocs.
Each genome contains several relatively small specific regions and all of them have a unique long variable region situated in the same segment of the genome (see the electronic supplementary material, table S7). These variable regions range in size from 587 to 802 kb and have very heterogeneous genetic contexts. The largest variable region in S. islandicus LAL14/1 is 608 kb (between positions 282 and 890 kb) and represents 24.7 per cent of the genome. It does not contain any known essential genes, such as those for tRNA, rRNA or ribosomal proteins, or the replication origin sites (ori). Variable regions are usually preferential sites for integration and accumulation of non-essential genes in the genome [7] and the LAL14/1 genome is not an exception. The variable region of LAL14/1 carries most of its integrative elements, including both functional and inactivated IS, MITEs (miniature inverted-repeat transposable elements), the two largest CAG regions, all of the identified CRISPR/cas and cmr modules, half of the toxin/antitoxin genes, and many of the putative glycosylase genes (figure 4).
Figure 4.
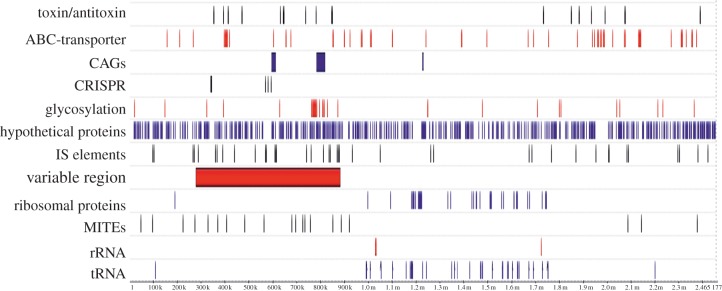
Distribution of some families of genes in the S. islandicus LAL14/1 genome compared with the position of the large variable region.
4.6. Analysis of tRNA genes, integration events and horizontal gene transfer
The pattern of tRNA genes in S. islandicus LAL14/1 is the same as those in S. islandicus REY15A and HVE10/4 [7] and very similar to those in other sequenced S. islandicus strains. The LAL14/1 chromosome carries 45 functional tRNA genes (see the electronic supplementary material, table S8) all located in the conserved regions. Sixteen of the tRNA genes include intron sequences (12–65 nt long) immediately downstream from the anticodon triplet. The genes for the tRNAglu[CTC] and tRNAglu[TTC] each have an insertion in their D-loop.
In Sulfolobales, the tRNA genes are preferential sites of integration of conjugative plasmids and fuselloviruses [4,42,43]. The mechanism of integration usually involves site-specific recombination between the tRNA gene target and the integrase gene (int) carried by an extrachromosomal element [44]. The presence of remnants of the corresponding int gene, overlapping the sequence of the tRNA gene target, often serves as a strong indication for an ancestral integrative event. The sequences of the remnants of the integrative elements are often incomplete or extensively degenerated, making their in silico identification challenging.
To identify the potential integrated extrachromosomal elements in the LAL14/1 genome, we set out to locate gene clusters enriched in homologues of proteins encoded by archaeal plasmids and viruses. For this purpose, all LAL14/1 proteins were compared (BLASTp) against the local protein database containing sequences of publicly available archaeal viruses (fifty-five) and crenarchaeal plasmids (twenty-five). Genomic loci containing at least five plasmid/viral homologues per 20 kb region were retained and manually inspected. This approach led to identification of four putative integrated elements. Notably, none of them appears to be functional, as judged from their incomplete gene complements when compared with ‘autonomous’ elements and lack of identifiable attachment sites. Three elements (SiL-E1 [SiL_0398..SiL_0402], SiL-E2 [SiL_1310..SiL_1321], SiL-E3 [SiL_1467..SiL_1481]) are likely to be remnants of conjugative plasmids, while the fourth one (SiL_2367..SiL_2371) is related to SSV-like fuselloviruses. SiL-E2, SiL-E3 and the SSV-like element are located in the proximity of different tRNA genes, which probably served as their respective integration targets, while SiL-E1 was found within the CRISPR_2 locus (see below).
In order to uncover the potentially more ancient integration events at the tRNA genes, which could have eluded identification using the criteria detailed above, we have inspected the genomic regions proximal to other LAL14/1 tRNA genes for the presence of plasmid/viral homologues and compared the obtained patterns with those of HVE10/4 and REY15A.
In general, the pattern of insertions linked to the tRNA genes in the LAL14/1 strain is similar, but not identical, to those in REY15A and HVE10/4 (table 5).
Table 5.
Overview of integration events targeting the tRNA genes in three S. islandicus strains, LAL14/1 (present work) and REY15A and HVE10/4 [7]. For the corresponding lanes of the table, the sequences found in three strains are given in footnotes a–c. In the CAG column the main digit gives the number of genes and the upper small digit indicates the CAG score rated from 1 (most atypical) to 10 (not atypical). CAGs written in bold are detailed in table 6.
| tRNA | intron | integration events |
|||||
|---|---|---|---|---|---|---|---|
| REY15A |
HVE10/4 |
LAL14/1 |
|||||
| BLASTp | CAG | BLASTp | CAG | BLASTp | CAG | ||
| Val-TAC | no | SiRe1242–1247 conj. plasmid | 52–6 | SiH1814 | 11 | — | — |
| Phe-GAAa | no | SiRe1321–SiRe1323 conj. plasmid | 91–7 | SiH1399–1402 conj. plasmid | 45–7 | SiL1310–1321 Sil-E2 conj. plasmid | 61–5 CAG3 |
| Met-CAT | yes | SiRe1465–SiRe1479 | 151–7 CAG4 | SiH1557–1560 | 42–7 | SiL1467–1481 Sil-E3 conj. plasmid | 131–5 |
| Glu-TTCb | no | intN fragmentc SiRe1484–1490 conj. plasmid | 73–7 | intN fragmentc SiH1561–1574 conj. plasmid | 121–7 | intN fragment SiL1484–1485 conj. plasmid | 51–3 |
| Ala-GGCa | no | intN fragment | 16 | intN fragment | 17 | intN fragment | 15 |
| Thr-GGT | no | SiRe2413–2417 SSV-like | 31 | SiH2464–2472 SSV-like | 51 to 2 CAG 7 | SiL2367–2371 SSV-like | 31 |
| Pro-GGGa | yes | intN fragment | 13 | intN fragment | 13 | intN fragment | 13 |
| His-GTGb | no | SiRe1787–1792 | 121–7 | SiH1866–SiH1871 | 51–6 | — | — |
| Leu-TAAa | yes | SiRe1255; SiRe1257 | 25 and 4 | SiH1333; SiH1335 | 25 and 4 | SiL1246–SiL1248 | 32–7 |
| Arg-GCG | no | — | — | SiH1544 transposase | 15 | — | — |
| Ser-GGAa | no | SiRe1778–1779 | 23 and 5 | SiH1858–1859 | 23 and 5 | SiL1773–SiL1774 | 23 and 5 |
| Lys-CTTb | yes | — | — | SiH1917 transposase | 11 | SiL1826; SiL1828 | 22 and 4 |
| Ala-CGC | no | SiRe1038 hyp. protein | 11 | — | — | — | — |
| Val-GAC | no | SiRe1734 hyp. protein | 11 | — | — | — | — |
| Pro-TGG | no | SiRe1936–1939 transposase and hyp proteins | 41–5 | — | — | — | — |
| Leu-CAG | no | — | — | — | — | — | 11 |
| Gly-GCC | no | — | — | — | — | — | 11 |
| Arg-CCT | yes | — | — | — | — | — | 11 |
Some of the insertions are clearly strain-specific (insertions into the tRNAAla[CGC], tRNAVal[GAC], tRNAPro[TGG], tRNALeu[CAG], tRNAGly[GCC], tRNAArg[CCT] and tRNAArg[GCG] genes). Other are present in two (insertions in tRNALys[CTT] and tRNAHis[GTG]) or all three strains (tRNALeu[TAA] and tRNASer[GGA] genes). In the case of 7 tRNA genes (tRNAPhe[GAA], tRNAGlu[TTC], tRNAAla[GGC], tRNAThr[GGT], tRNAPro[GGG], tRNALeu[TAA] and tRNASer[GGA]), the three strains carry nearly identical remnants of the same integrated elements. This suggests that the respective integration events occurred in the common ancestor of REY15A, HVE10/4 and LAL14/1.
Proviruses are common companions of archaeal genomes [45–47]. Thus, it was somewhat surprising not to find potentially functional proviruses in the LAL14/1 genome. The only virus-derived element of LAL14/1 integrated in the tRNAThr[GGT] gene is a highly degenerated remnant of an SSV-like fusellovirus. Notably, the element does not appear to be closely related to any particular fusellovirus, since different genes display affinities to distinct fuselloviruses. To gain an insight into the timeframe of this viral integration event, we analysed the equivalent loci in all available S. islandicus genomes. The traces of SSV integration were found in all S. islandicus strains, except for the M.16.27, which contained a gene for the pNOB8-type integrase at the equivalent position [48]. Interestingly, in all cases the elements were severely degenerated; a selection of genomic alignments can be found in the electronic supplementary material, figure S5. The most parsimonious scenario for the observed distribution of SSV-like remnants in S. islandicus genomes involves a single event of SSV-like virus genome integration into the tRNAThr[GGT] gene, followed by gradual deterioration of the provirus along the evolutionary history of S. islandicus species. The integration has probably occurred following the divergence of S. islandicus and S. solfataricus from their common ancestor, since S. solfataricus lacks a detectable SSV-like element at the equivalent genomic locus.
Identification of insertions by BLASTp analysis may be hampered by the insufficient conservation of the inserted genes or by limited coverage of the diversity of archaeal mobile genetic elements. Furthermore, some of the insertions could occur in loci other than the tRNA genes. To overcome these caveats, we have applied a BLASTp-independent approach based on the search of CAGs [17,49]. Following this approach, the putative integrated elements could be identified following their atypical codon usage compared with that of the conserved part of the host chromosome. For LAL14/1, HVE10/4 and REY15A the results obtained by this approach are summarized in table 6, and a brief general comparison of CAG distribution in 10 S. islandicus genomes is present in the electronic supplementary material, table S9. Notably, nearly all insertions detected in LAL14/1, HVE10/4 and REY15A by the BLASTp analyses were confirmed by the CAG approach; in addition, some of the integrative events were only predicted by the CAG search. LAL14/1 has three CAG regions (CAG1–3) of 14.6, 32.5 and 2.5 kb that carry 43 genes with atypical codon usage. CAG3 was found to correspond to SiL-E2 element integrated into tRNAPhe[GAA] identified by BLASTp analysis, while CAG1 and CAG2 could not be predicted by other approaches.
Table 6.
Major CAG regions and corresponding predicted functions in S. islandicus LAL14/1, HVE10/4 and REY15A.
| strain/CAG | position | no. of atypical genesa | description |
|---|---|---|---|
| S. islandicus HVE10/4 | |||
| CAG 1 | 497317–519235 | 15 (5) | transposase IS200/IS605 |
| CAG2 | 554497–563952 | 8 (4) | gene orfB |
| CAG3 | 725905–750622 | 17 (4) | genes vapBC, CRISPR_3 of family III and six genes cas |
| CAG4-1 | 895122–912372 | 20 (6) | genes of hydrogenases and ABC transporter; HVE10/4 specific |
| CAG4-2 | 921672–938729 | 16 (9) | genes of hydrogenases; HVE10/4 specific |
| CAG5 | 967655–977019 | 11 (9) | HVE10/4 specific |
| CAG6 | 1399248–1420774 | 14 (7) | restriction–modification system of type I found HVE10/4 specific |
| CAG7 | 2380543–2386271 | 6 (6) | insertion in tRNA[Thr]GGT |
| S. islandicus REY15A | |||
| CAG 1 | 555326–562352 | 7 (4) | partially similar to CAG1 of LAL14/1 and to CAG3 of HVE10/4; vapBC |
| CAG2 | 722610–725192 | 5 (4) | REY15A specific |
| CAG3-1 | 790437–804374 | 17 (13) | transposase IS5 |
| CAG3-2 | 829169–837249 | 6 (4) | glycosyl transferase gene |
| CAG3-3 | 845351–852630 | 7 (1) | IS200/605; vapBC; genes cas |
| CAG4 | 1372809–1383519 | 12 (6) | insertion in tRNA[Met]CAT; vapBC |
| S. islandicus LAL14/1 | |||
| CAG1 | 600528–615132 | 10 (4) | vapBC; gene csm6-like |
| CAG2 | 789437–821915 | 26 (16) | genes of methyltransferases and glycosyltransferases |
| CAG3 | 1232632–1235126 | 7 (5) | inserts in tRNA[Phe]GAA |
aThe first digit indicates the total number of genes in the CAG region and the digit shown in the parenthesis indicates those for which the function could not be predicted.
Some of the functions identified as being associated with the CAG loci are: the restriction–modification system I characteristic of HVE10/4; some elements of CRISPR-based immunity in HVE10/4 and LAL14/1; and various enzyme families (methyl- and glycosyltransferases, hydrogenases). The genes transferred horizontally and integrated into the chromosomes of these S. islandicus strains include many transposons and toxin/antitoxin gene pairs of the vapBC family.
To summarize, LAL14/1 carries the remnants of 13 insertion events into the tRNA genes, and three additional elements (SiL-E1, CAG1 and CAG2) are integrated into other loci; the same or a similar number of insertions is found in HVE10/4 and REY15A. These results further illustrate the fact that tRNA genes are frequently attacked by various mobile genetic elements. The observation that all (or at least the majority) of the integrated elements appear to be non-functional suggests that LAL14/1 possesses an efficient mechanism of purging its genome of unwelcomed insertions.
4.7. Replication origins, oriC
The positions of the replication origins in the chromosome of LAL14/1 were predicted by two independent approaches, Z-curve [50,51] and ACCA-plot [52], that produced very similar results (see the electronic supplementary material, figure S6). Consistent with all other Sulfolobales genomes analysed in silico [7,53] or in vivo [54–56], three oriC origins of replication were detected. Their positions and genomic contexts are well conserved with respect to other Sulfolobus genomes (see the electronic supplementary material, table S10 and [7]). The oriC1 site (mapped at position 1.59 Mb) is linked to the cdc6-1 gene (SiL_0002), oriC2 (position 800 bp) is linked to the cdc6-3 gene (SiL_1733), and oriC3 (position 1.15 Mb) to the cdc6-2/whiP genes (SiL_1228/SiL_1206).
The structures of the oriC1 and oriC2 sites in S. solfataricus P2 are well characterized [56,57], and these two replication origins are organized similarly in S. islandicus LAL14/1 (figure 5). Their central AT-rich UCM sequence (uncharacterized motif) is surrounded by the characteristic ORB sequences (origin recognition box). The oriC1 site also contains additional specific palindromic sequences, called C2 and C3, that are recognized by the replication initiation proteins Cdc6-1, Cdc6-2 and Cdc6-3 [56].
Figure 5.

Conserved sequences and structural organization of replication origins oriC in S. islandicus. (a) Consensus DNA sequences present in oriC sites. The size of the letters is directly proportional to the residue conservation. (b) Structural organization of three oriC in S. islandicus.
In Sulfolobales, the oriC3 site is usually linked to the whiP gene. A comparative analysis of oriC3 in the 10 S. islandicus strains and in S. solfataricus P2 provided new insights into the organization of this region: we identified three conserved ORB-like sites (nORB) upstream and downstream from the typical UCM site (figure 5). The oriC2 and oriC3 sites are organized similarly and, unlike oriC1, do not contain the C sequences.
4.8. Toxin–antitoxin systems
A family II (VapBC) toxin–antitoxin (TA) system is present in many Archaea and is very abundant in Sulfolobales [7,58,59]. All S. islandicus strains carry many TA gene pairs of the VapBC family as well as genes of another family considered to play a TA role [60]: HEPN-NT (Higher Eukaryotes and Prokaryotes Nucleotide-binding-Nucleotidyl Transferase; electronic supplementary material, table S11).
Eight of the 15 vapBC gene pairs in S. islandicus LAL14/1 map in the variable region of the genome. The other seven vapBC gene pairs map in the conserved part of the genome and all share a similar genetic context in the three strains analysed. For four of these loci (SiL_2040/2041, SiL_2042/2043, SiL_2080/2081, SiL_2253/2254), the genomic context is particularly well preserved. All vapBC loci, except SiL_2575/2576, are flanked by degenerated copies of IS elements, probably involved in the transposition of vapBC.
As observed in HVE10/4 and REY15A [7], the vapB (toxin) and vapC (antitoxin) genes of different subtypes were found in S. islandicus LAL14/1 in various combinations giving different variants of the vapBC operon (data not shown). This combinatorial diversity of vapBC gene pairs may indicate the existence of several types of the toxin/antitoxin mechanisms. All vapB and vapC gene combinations found in S. islandicus LAL14/1 are also found in HVE10/4 and REY15A, except for SiL_0413/0414 and SiL_0631/0632 combinations present in LAL14/1 and HVE10/4 but not in REY15A.
Members of the toxin/antitoxin family HEPN-NT were detected in all S. islandicus strains, with multiple copies of the corresponding genes (see the electronic supplementary material, table S12). Unlike the vapBC system, HEPN-NT gene pairs are stable and each HEPN gene type is strictly associated with its specific NT gene type. HEPN-NT operons are classified into two subfamilies, I and II. Subfamily I is ubiquitous and all of its representatives in the 10 S. islandicus genomes analysed both occupy the same genetic regions and are always localized in conserved parts of the genomes. Subfamily II is much more diverse. Its representatives in S. islandicus map in both conserved and variable regions of the chromosome. Note that many copies of HEPN-HT family II pairs include only truncated forms of the HEPN gene and are not functional.
The production of sulfolobicins, a type of toxin that inhibits the growth of sensitive Sulfolobus strains, is characteristic of two other well-studied Sulfolobales, S. acidocaldarius and S. tokodaii [61–63]. No sulfolobicin-encoding genes, such as sulA, sulB and sulC, were found in any of the 10 S. islandicus genomes, indicating the absence of this toxin system from these species. Nevertheless, a truncated copy of the sulA gene, which obviously cannot code for a functional toxin, is present in S. islandicus REY15A [61].
4.9. UV-inducible type IV pili
Many Sulfolobales (ex. S. solfataricus, S. tokodaii and S. acidocaldarius) code for a UV-inducible type IV pilus system that promotes cellular aggregation and efficient exchange of chromosomal markers [64,65]. The formation of pili is controlled by the UV-inducible ups operon which comprises five genes: upsX, upsE, upsF, upsA and upsB [4–6]. This operon is present in all 10 S. islandicus strains (and all other sequenced species of Sulfolobales, electronic supplementary material, table S13) and is in all cases in the conserved part of the genome. The Ups proteins encoded by all S. islandicus are very similar and form a specific phylogenetic group within the Ups family in Sulfolobales (see the electronic supplementary material, figure S7).
The in silico data strongly suggest that the ups locus of S. islandicus is functional in vivo, and it very probably plays the same biological role as in S. solfataricus, S. tokodaii and S. acidocaldarius.
4.10. Insertion sequence elements and miniature inverted-repeat transposable elements
Sulfolobus islandicus LAL14/1, as HVE10/4 and REY15A, contains several families of IS elements with members present in multiple copies [7] (see the electronic supplementary material, table S14).
The orfB-containing IS (families IS605 and IS200/605) considered to be ancestral for the archaeal domain [66,67] are overrepresented in all three S. islandicus strains analysed: 95% of the copies of the orfB gene not linked to orfA in HVE10/4, REY15 and LAL14/1 were predicted to be functional.
Only seven of the 53 structurally valid IS (IS with intact inverted terminal repeats (ITRs)) in the LAL14/1 genome are predicted to code for functional transposases; transposase genes in the remaining 46 IS are truncated. The IS patterns of the two other strains, HVE10/4 and REY15A, are very different. Most of the IS in these genomes (45/76 in HVE10/4 and 52/96 in REY15A) code for a full-length transposase and are therefore predicted to be functional. However, many of the mutated IS may be mobilized by transposases of the same family encoded in trans [68]. Thus, 62 of the 76 IS (81.6%) detected in HVE10/4 and 87 of the 96 IS (90.6%) in REY15A could, in theory, be mobile (see the electronic supplementary material, table S14); the proportion is lower for LAL14/1, for which 31 of the 53 IS (58.4%) are potentially active.
This may indicate greater genetic stability of S. islandicus LAL14/1 than of either HVE10/4 or REY15A. Were this the case, strain LAL14/1 would be the most attractive model for genetic manipulations.
Another group of mobile elements in S. islandicus is MITEs, believed to correspond to truncated derivatives of autonomous DNA transposons [69–73]. MITEs exhibit the structural features of DNA transposons, containing terminal inverted repeats flanked by small direct repeats. The internal sequences of MITEs are short and devoid of ORFs. As non-autonomous elements, the transposition of MITEs is totally dependent on trans-acting transposases [68,74,75]. Only two classes of MITEs, SMN1 (320 bp) and SM3A (164 bp), were detected in the 10 S. islandicus genomes (see the electronic supplementary material, table S15). The SM3A family is more numerous in LAL14/1 than any of the other S. islandicus strains. The three S. islandicus strains from Iceland share two identical SM3A, but LAL14/1 also carries nine extra copies of SM3A that share only 95% similarity with other two SM3A copies. SMN1 transposition is dependent on the presence of a functional ISC1733 transposase and SM3A transposition on ISC1058 [68,76,77]. The MITEs of the SMN1 type in LAL14/1, HVE10/4 and REY15A could be mobilized by the ISC1733 type transposase [76] predicted to be functional in these strains. The observation of the mobilization of SMN1 in S. islandicus REN1H1 is consistent with this prediction [76]. None of the three S. islandicus strains analysed codes for a functional ISC1058 transposase, suggesting that SM3A, although present, cannot transpose in HVE10/4, REY15A and LAL14/1.
4.11. CRISPRs: structure, targets and phenotype
All sequences and genes related to the CRISPR system present in S. islandicus LAL14/1 (CRISPR arrays, cas and cmr gene cassettes) map within the large variable genomic region. It carries five CRISPR loci, three cas gene cassettes associated with subtype I-A and two cmr gene cassettes associated with subtype III-B [78,79]. Following the leader and repeat sequence compositions, the five CRISPRs of S. islandicus LAL14/1 could be divided into two families, I and III [36], while the two cmr modules belong to families B and F ([37]; figure 6 and table 7).
Figure 6.

CRISPR organization in S. islandicus LAL14/1. (a) Comparison of the CRISPR structures and composition in 10 S. islandicus strains. (b) Structure and composition of CRISPRs in S. islandicus LAL14/1. Large-scale presentation: the orientation, position and size of CRISPR arrays (black arrows) as well as cas (red arrows) and cmr (blue arrows) gene modules are indicated. Detailed presentation: all perfectly matching spacers (large arrows) as well as selected imperfect spacers are indicated by small coloured arrows specified in the legend included in the figure body. (c) The pING1-like insertion in the CRISPR_2 locus is interspersed precisely between two complete 24 bp repeats, resembling by its position typical CRISPR spacers.
Table 7.
Composition of CRISPRs in S. islandicus LAL14/1 and their putative targets. SIRV1 VIII and SIRV1 XX are different subtypes of the virus SIRV1 [34].
| CRISPRs and their families | repetition | position and direction | no. of spacers | spacers with 100% identity to the indicated putative targets |
|---|---|---|---|---|
| CRISPR_1 I | GCTAATCTACTATAGAATTGAAAG | 344177..349768 ←L |
86 | 11/86 SIRV1 VIII |
| CRISPR_2 I | GCTAATCTACTATAGAATTGAAAG | 353820..363037 L→ |
113 | 93/113 pARN3 |
| CRISPR_3 III | GTAACAACACAAAGAAACTAAAAC | 573686..575526 ←L |
31 | — |
| CRISPR_4 III | GTAACAACACAAAGAAACTAAAAC | 584662..586213 ←L |
25 | — |
| CRISPR_5 III | GTAACAACACAAAGAAACTAAAAC | 597345..599148 L→ |
30 | 1/29 SIRV1 VIII and SIRV1 XX |
The family I CRISPR locus comprises two oppositely oriented blocks of repeat-spacer arrays separated by the first module of Cas genes. The second module is situated at the end of one of the repeat-spacer arrays (figure 6a). The additional Cmr module is not linked to this part and maps several hundred kilobases downstream in the genome. The family III CRISPR (figure 6a) comprises three clusters of spacers/repeats associated with two gene modules, one including the cas gene and another cmr. The cmr gene order in this module is the same as that in the Cmr module associated with the family I CRISPR.
The analysis of 285 spacers forming the CRISPR array of LAL14/1 revealed the presence of a surprisingly high number of spacers that perfectly (in three cases) or imperfectly (30 cases; electronic supplementary material, table S16) match the genomes of rudiviruses, fuselloviruses and conjugative plasmids previously described in the Icelandic hot spring environments [34,35]. The two perfectly matching spacers carried by CRISPR_1 and CRISPR_5 target the genome of the rudivirus SIRV1 [80]. Interestingly, the spacer in CRISPR_1 matches a SIRV1 gene encoding a protein, P98, responsible for formation of pyramidal structures involved in virion egress [11–13]. No spacers with 100 per cent identity to a closely related virus, SIRV2, were detected. The third perfectly matching spacer is identical to a sequence in the conjugative plasmid pARN3 [81]. Unexpectedly, we have identified an about 2 kb insertion, SiL-E1, in the CRISPR_2 locus. SiL-E1 resembles the typical CRISPR spacers in that it is interspersed between two identical repeats and is followed by additional spacer-repeat units (figure 6). This pseudo-spacer encompasses five ORFs and displays high sequence similarity to and collinearity with pING1-like conjugative plasmids of S. islandicus [81–83]. More specifically, SiL-E1 shares overall 82 per cent identity with plasmids pING1 [83] and pHVE14 [81]. SiL-E1 is not present in other S. islandicus strains. Notably, sequence similarity between SiL-E1 and pING1-like plasmids extends throughout the length of SiL-E1, leaving no unaccounted positions between the inserted sequence and the repeat regions (figure 6c). This suggests that SiL-E1 is unlikely to be a result of illegitimate recombination.
Some of the spacers imperfectly match DNA regions present in the genomes of other S. islandicus strains (M.14.25, M.16.4, Y.N.15.51, Y.G.57.14, L.D.8.5, L.S.2.15) and in S. solfataricus 98/2 and P2. The functions of the corresponding genes are unknown, and even the biological significance of this observation is unclear. Possibly, the genomic loci matched by these spacers represent the remnants of unknown viruses or plasmids integrated into the corresponding genomes.
The analysis of the protospacer corresponding to the spacers listed in the electronic supplementary material, table S15 allows the identification of the PAM sequence (protospacer adjacent motif). These sequences situated at the proximity of protospacers are crucial for two essential steps of CRISPR-based immunity: adaptation [84] and interference [37,85,86]. They also play an important role in the mechanism of target discrimination that prevents the recognition of chromosomal spacers as valid targets [87].
For the LAL14/1 CRISPRs of the family I, we found the same PAM motif, CC, in the position (−3, −2) at the 5′ end as was already described by Gudbergsdottir et al. [88] (figure 7a). No specific PAM motif was detected at the 3’ end of these protospacers (figure 7b). Little is known about the protospacers corresponding to the CRISPRs of the family III. Our data indicate the existence of a conserved motif [T/A]GT occupying the position (−4, −3, −2) at the 5′ end of the protospacer (figure 7c). The 3′ end of these protospacers is very rich in A/T nucleotides (figure 7d).
Figure 7.
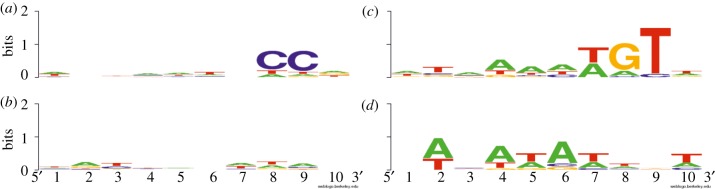
Conserved motifs in the protospacer sequences for CRISPRs I ((a) 5′ and (b) 3′) and CRISPRs III ((c) 5′ and (d) 3′).
4.12. Development of a genetic model
Sulfolobus islandicus LAL14/1 is a promising model for studying virus–host interaction in Archaea. LAL14/1 cells can be infected by SIRV2, a model rod-shaped virus [89] that codes for a unique mechanism of virion release: pyramidal structures form on the host cell surface, breaking the S-layer and allowing the virions to escape from the cells [11–13,90]. Investigations on SIRV2 cycle regulation and SIRV2–host interaction will require genetic tools for strain LAL14/1 as no available genetic models of Sulfolobus can be infected by SIRV2.
One of the most commonly used genetic markers in Archaea is the pyrEF operon. Pyrimidine prototrophs (Pyr+) can be easily selected, on minimal medium without uracil, after transformation of a pyrEF–host strain by a plasmid or viral vector carrying the wild-type pyrEF operon [19,91–94]. A non-reversing spontaneous pyrEF-deletion mutant of S. islandicus REY15A is widely used as a host for genetic manipulations [20]. No such mutant of S. islandicus LAL14/1 was available, so we constructed a ΔpyrEF mutant via allelic replacement approach.
To generate a pyrEF disruption mutant, a knockout cassette containing the ΔpyrEF allele from S. islandicus REY15A, strain E233S [19] and 1 kb regions situated downstream and upstream from pyrEF was obtained by PCR amplification (see §3). The S. islandicus LAL14/1 cells were transformed by this linear DNA fragment of 2233 bp and the ΔpyrEF mutants resulting from the replacement of the wild-type copy of the pyrEF operon on the host chromosome by a double cross-over were selected on 5′FOA (5′-fluoroorotic acid). Twenty transformants were selected and the pyrEF operon was analysed by PCR and sequencing; 15 of the analysed colonies carried the expected ΔpyrEF deletion. This mutant strain, called S. islandicus LAL14/1-CD, showed the same virus resistance/sensitivity phenotype as the parental strain (data not shown). We confirmed that this mutant is indeed derived from strain LAL14/1 by sequencing the gene coding for the A subunit of the cytochrome b558/566 (Sil_2350; the sequence of this gene is not identical in REY15A, HV10/4 and LAL14/1).
Sulfolobus islandicus LAL14/1-CD could be efficiently transformed with the pHZ2 (pRN2 replicon) [19] autonomously replicating in S. islandicus and carrying a wild-type copy of the pyrEF operon. The transformation efficiency was 102–103 colonies/μg of DNA.
A powerful genetic pop-in/pop-out approach was previously developed for another genetic model, S. islandicus REY15A [19]. It allows rapid and efficient creation of knockout mutants. To show that this approach is efficient in LAL14/1, we have chosen to delete one of the CRISPR loci (CRISPR_1), because CRISPR-coded functions are usually not essential for the cells in the absence of viruses and their deletion mutants are expected to be viable. Also, one of the spacers of the CRISPR_1 matches perfectly the SIRV1 virus for which LAL14/1 is resistant. If the resistance is linked to the CRISPR activity, its inactivation could decrease the level of resistance giving a detectable phenotype to this mutant.
The recombinant plasmid used to inactivate the CRISPR_1 and the positions of the regions IN (867 bp), OUT (919 bp) and TARGET (776 bp) in the vector pSEF described by Deng et al. [19] are indicated in figure 8.
Two successive rounds of recombination (figure 8) deleted the chromosomal fragment situated between the IN and OUT regions producing a pyrEF+ derivative from which the CRISPR_1/cas region has been deleted (ΔCRISPR_1/Δcsx1Δcas4; csx1 is annotated as Sil_0393). The deletion was confirmed by PCR analysis (data not shown). Interestingly, ΔCRISPR_1/Δcsx1Δcas4 was as resistant to infection by SIRV1 as S. islandicus LAL14/1-CD. The presence of a second spacer and functional CRISPR_3 may explain this result.
The observed efficient transformation of LAL14/1 as well the ease of creation and selection of its deletion mutants indicate that LAL14/1 represents an excellent genetic model.
5. Discussion
Sulfolobus islandicus LAL14/1 is a promising model for studies on virus–host interaction and CRISPR/cas-based acquired immunity in hyperthermophilic Archaea. We report an extensive comparative in silico analysis of its genome and established this strain as genetic model.
The genome of LAL14/1 is the 10th of the species S. islandicus to be sequenced [9], and the third of an S. islandicus strain isolated in Iceland [8]. Strain LAL14/1 has the smallest known S. islandicus genome: it has only 2601 genes carried by a 2.47 Mb chromosome. With other sequenced S. islandicus strains LAL14/1 shares the same major groups of paralogous genes, most of which are transposases of various families (table 4). Its genome also encodes a large number of diverse ABC transporters, including the oligopeptide transporter. This is consistent with the high frequency of isolation of S. islandicus strains from enrichment cultures using rich organic media [7]. Indeed, S. islandicus LAL14/1 grows heterotrophically on standard laboratory liquid media with yeast extract as main carbon source.
The S. islandicus pan-genome contains 570 singletons representing a strain-specific set of proteins; 65 are only found in S. islandicus LAL14/1. As has been widely documented for other prokaryotic virus/host models (see review [95]), it is possible that some of the particular features of LAL14/1, for example, virus–host range, could be linked to the presence of specific genes or gene repertoires absent from other S. islandicus strains. A large proportion of the strain-specific genes map in a large variable region. In each of the S. islandicus genomes analysed this region covers more than 25 per cent of the chromosome length, and contains most of the transposons promoting the horizontal gene transfer and all CRISPR sequences (figure 4).
Our in silico analysis of the relics of mobile elements in the genomes of the three closely related S. islandicus strains confirms that mobile genetic elements preferentially integrate into tRNA genes. Nevertheless, searches for CAG revealed several previously undescribed long DNA segments of heterologous origin that were located in loci other than tRNA genes [49]. These clusters, most probably the consequences of genetic transfer via conjugative plasmids or other mobile genetic elements, contain 1.6 per cent of the genes in LAL14/1. Biological functions can be predicted for only a small fraction of these genes. For example, the CAGs carry several copies of vapBC genes of toxin/antitoxin systems, some CRISPR-related genes and genes coding for methyl- and glycosyltransferases.
A comparative analysis of the genome structure and composition of three closely related strains (HVE10/4, REY15A and LAL14/1) confirms that they have a very similar genomic pattern with a strong conservation of synteny. Nevertheless, the presence in each of these genomes of multiple local rearrangements raised the issue of the stability of the LAL14/1 genome. All three strains carry many copies of IS elements of various families. The transposition of an IS or transposon is an important source of genome instability and rearrangements in any cell [96]. Such instability is well documented in the case of REY15A, for which a relatively high incidence of the pyrEF-deletion mutants (one from 50 analysed PyrEF-colonies) is observed [7,19].
Sulfolobus islandicus LAL14/1 seems to be genetically more stable as no deletions in the pyrEF locus were detected by PCR analysis of 100 colonies of spontaneous LAL14/1 pyrEF mutants resistant to FOA (C. Jaubert, C. Danioux, G. Sezonov 2013, unpublished data). This could be due to a lower transposition activity in LAL14/1 and consequently lower frequency of genome rearrangements.
Thus, in vivo and in silico indications concerning the stability of the genome of strain LAL14/1 suggest that it would be a useful model for genetic studies. Strain LAL14/1 is the host of the model rudivirus SIRV2 [10,89,97] and has been used to study virus–host interactions in Archaea [11–13]. The availability of the sequence of the LAL14/1 genome makes global genomic analysis of the interaction between viral and host genomes during the infection cycle possible. A genetic approach would facilitate investigations of the role of particular host genes involved in this interaction, as well as the host immune response dependent on the activity of CRISPRs. We successfully inactivated two genetic loci in LAL14/1 of different sizes, pyrEF (2.2 kb) and CRISPR_1/cas (6.6 kb), using allelic replacement and in-frame markerless genetic exchange approaches. We thereby demonstrated the potential of LAL14/1 for genetic experimentation.
More than 90 per cent of the archaeal genomes analysed code for an adaptive immunity system called the CRISPR/cas system [98–100]. In LAL14/1, CRISPRs are represented by five loci associated with three cas (subtype I-A) and two cmr (subtype III-B) gene cassettes. The presence in the CRISPR arrays of LAL14/1 spacers matching extrachromosomal elements (SIRV1 virus, pARN1) make this strain an interesting model for studying the biological and functional role of CRISPRs in Archaea. The presence of two spacers perfectly matching the genome of SIRV1 virus allows speculation about a connexion between the CRISPR composition and the SIRV1-resistance phenotype of S. islandicus LAL14/1. Deletion of the CRISPR_1 carrying one of two SIRV1-specific spacers did not change the phenotype of the obtained mutant; it remained as resistant to SIRV1 as the initial strain. A construction of a double mutant ΔCRISPR_1 ΔCRISPR_5 will help to better characterize the eventual involvement of CRISPR generated immunity in resistance of LAL14/1 to SIRV1.
Recent publications report spacer acquisition under laboratory conditions in several bacterial [84,101–104] and archaeal (S. solfataricus) [105] models. However, analysis of the CRISPR content of 12 independent SIRV2-resistant mutants of LAL14/1 did not detect any new insertions in the CRISPR sequences (data not shown). Consequently, the acquired resistance does not appear to be related to CRISPRs and presumably involves a different mechanism of resistance [106].
LAL14/1 also carries a unique pseudo-spacer, SiL-E1, which represents an insertion of an approximately 2 kb region from a pING1-like plasmid into the CRISPR_2 array. To our knowledge, such large spacers have not been previously described in archaeal or bacterial CRISPR loci. The acquisition mechanism of this pseudo-spacer as well as its role in LAL14/1 immunity against conjugative plasmids is unclear. However, the fact that SiL-E1 is flanked by perfect repeats of CRISPR_2 (figure 6c) argues against the possibility of a random integration event. Plausible acquisition scenarios include faulty protospacer processing by the Cas machinery or homologous recombination between the episomal plasmid and the pre-existing CRISPR_2 spacer(s) matching the plasmid. Future studies should provide important additional information regarding spacer acquisition mechanisms and reveal whether such atypical spacers are competent in conferring immunity against mobile genetic elements in Archaea.
Physical isolation of the geothermal hot spring in which S. islandicus thrives makes this species a very valuable model to study microbial speciation and evolution [107]. Such studies were recently conducted on S. islandicus strains isolated from the hot spring located in Russia (‘M’) and USA (‘Y/N’) [9,33], and have already provided important insights into the population dynamics of hyperthermophilic Archaea. Our in-depth comparative genomics analysis clearly indicates divergence of the ‘Icelandic trinity’—LAL14/1, HVE10/4 and REY15A—from the groups ‘M’ and ‘L/Y’, supporting previously suggested biogeographical patterns of differentiation of S. islandicus species. Genetic tools developed in this study and those available for REY15A will help to experimentally tackle questions regarding the evolution and divergence of these Icelandic strains and compare the elucidated patterns with those available for S. islandicus strains isolated from other continents.
6. Acknowledgements
This work was supported by PhD fellowships from the ‘Ministère de l'enseignement supérieur et de la recherche’ (C.J. and C.D.) and by Pasteur-Weizmann (C.J.) allocations. We thank Nuno Peixeiro and Sophie Schbath for helpful discussions.
Supplementary Material
Supplementary Material
Supplementary Material
References
- 1.Brock TD, Brock KM, Belly RT, Weiss RL. 1972. Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch. Mikrobiol. 84, 54–68 10.1007/BF00408082 (doi:10.1007/BF00408082) [DOI] [PubMed] [Google Scholar]
- 2.Redder P, Garrett RA. 2006. Mutations and rearrangements in the genome of Sulfolobus solfataricus P2. J. Bacteriol. 188, 4198–4206 10.1128/JB.00061-06 (doi:10.1128/JB.00061-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciaramella M, Pisani FM, Rossi M. 2002. Molecular biology of extremophiles: recent progress on the hyperthermophilic archaeon Sulfolobus. Antonie Van Leeuwenhoek 81, 85–97 10.1023/A:1020577510469 (doi:10.1023/A:1020577510469) [DOI] [PubMed] [Google Scholar]
- 4.She Q, et al. 2001. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc. Natl Acad. Sci. USA 98, 7835–7840 10.1073/pnas.141222098 (doi:10.1073/pnas.141222098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawarabayasi Y, et al. 2001. Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain7. DNA Res 8, 123–140 10.1093/dnares/8.4.123 (doi:10.1093/dnares/8.4.123) [DOI] [PubMed] [Google Scholar]
- 6.Chen L, et al. 2005. The genome of Sulfolobus acidocaldarius, a model organism of the Crenarchaeota. J. Bacteriol. 187, 4992–4999 10.1128/JB.187.14.4992-4999.2005 (doi:10.1128/JB.187.14.4992-4999.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo L, et al. 2011. Genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus–host interaction studies. J. Bacteriol. 193, 1672–1680 10.1128/JB.01487-10 (doi:10.1128/JB.01487-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zillig W, et al. 1998. Genetic elements in the extremely thermophilic archaeon Sulfolobus. Extremophiles 2, 131–140 10.1007/s007920050052 (doi:10.1007/s007920050052) [DOI] [PubMed] [Google Scholar]
- 9.Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. 2009. Biogeography of the Sulfolobus islandicus pan-genome. Proc. Natl Acad. Sci. USA 106, 8605–8610 10.1073/pnas.0808945106 (doi:10.1073/pnas.0808945106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prangishvili D, Arnold HP, Gotz D, Ziese U, Holz I, Kristjansson JK, Zillig W. 1999. A novel virus family, the Rudiviridae: structure, virus–host interactions and genome variability of the Sulfolobus viruses SIRV1 and SIRV2. Genetics 152, 1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bize A, et al. 2009. A unique virus release mechanism in the Archaea. Proc. Natl Acad. Sci. USA 106, 11 306–11 311 10.1073/pnas.0901238106 (doi:10.1073/pnas.0901238106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quax TE, Krupovic M, Lucas S, Forterre P, Prangishvili D. 2010. The Sulfolobus rod-shaped virus 2 encodes a prominent structural component of the unique virion release system in Archaea. Virology 404, 1–4 10.1016/j.virol.2010.04.020 (doi:10.1016/j.virol.2010.04.020) [DOI] [PubMed] [Google Scholar]
- 13.Quax TE, Lucas S, Reimann J, Pehau-Arnaudet G, Prevost MC, Forterre P, Albers SV, Prangishvili D. 2011. Simple and elegant design of a virion egress structure in Archaea. Proc. Natl Acad. Sci. USA 108, 3354–3359 10.1073/pnas.1018052108 (doi:10.1073/pnas.1018052108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allers T, Mevarech M. 2005. Archaeal genetics: the third way. Nat. Rev. Genet. 6, 58–73 10.1038/nrg1504 (doi:10.1038/nrg1504) [DOI] [PubMed] [Google Scholar]
- 15.Jonuscheit M, Martusewitsch E, Stedman KM, Schleper C. 2003. A reporter gene system for the hyperthermophilic archaeon Sulfolobus solfataricus based on a selectable and integrative shuttle vector. Mol. Microbiol. 48, 1241–1252 10.1046/j.1365-2958.2003.03509.x (doi:10.1046/j.1365-2958.2003.03509.x) [DOI] [PubMed] [Google Scholar]
- 16.Worthington P, Hoang V, Perez-Pomares F, Blum P. 2003. Targeted disruption of the alpha-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 185, 482–488 10.1128/JB.185.2.482-488.2003 (doi:10.1128/JB.185.2.482-488.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner M, Berkner S, Ajon M, Driessen AJ, Lipps G, Albers SV. 2009. Expanding and understanding the genetic toolbox of the hyperthermophilic genus Sulfolobus. Biochem. Soc. Trans. 37, 97–101 10.1042/BST0370097 (doi:10.1042/BST0370097) [DOI] [PubMed] [Google Scholar]
- 18.Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV. 2012. Versatile Genetic Tool Box for the Crenarchaeote Sulfolobus acidocaldarius. Front Microbiol. 3, 214. 10.3389/fmicb.2012.00214 (doi:10.3389/fmicb.2012.00214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L, Zhu H, Chen Z, Liang YX, She Q. 2009. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13, 735–746 10.1007/s00792-009-0254-2 (doi:10.1007/s00792-009-0254-2) [DOI] [PubMed] [Google Scholar]
- 20.Leigh JA, Albers SV, Atomi H, Allers T. 2011. Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 35, 577–608 10.1111/j.1574-6976.2011.00265.x (doi:10.1111/j.1574-6976.2011.00265.x) [DOI] [PubMed] [Google Scholar]
- 21.She Q, Zhang C, Deng L, Peng N, Chen Z, Liang YX. 2009. Genetic analyses in the hyperthermophilic archaeon Sulfolobus islandicus. Biochem. Soc. Trans. 37, 92–96 10.1042/BST0370092 (doi:10.1042/BST0370092) [DOI] [PubMed] [Google Scholar]
- 22.Zhang C, Whitaker RJ. 2012. A broadly applicable gene knockout system for the thermoacidophilic archaeon Sulfolobus islandicus based on simvastatin selection. Microbiology 158, 1513–1522 10.1099/mic.0.058289-0 (doi:10.1099/mic.0.058289-0) [DOI] [PubMed] [Google Scholar]
- 23.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 10.1101/gr.074492.107 (doi:10.1101/gr.074492.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewing B, Green P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 [PubMed] [Google Scholar]
- 25.Ewing B, Hillier L, Wendl MC, Green P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 [DOI] [PubMed] [Google Scholar]
- 26.Gordon D, Abajian C, Green P. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202 [DOI] [PubMed] [Google Scholar]
- 27.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okonechnikov K, Golosova O, Fursov M. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 10.1093/bioinformatics/bts091 (doi:10.1093/bioinformatics/bts091) [DOI] [PubMed] [Google Scholar]
- 29.Schbath S. a. H. M. 2011. R'MES: a tool to find motifs with a significantly unexpected frequency in biological sequences. In Advances in genomic sequence analysis and pattern discovery, vol. 7 (eds Elnitski L, Piontkivska O, Welch L.). Science, Engineering, and Biology Informatics Singapore: World Scientific [Google Scholar]
- 30.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57 10.1093/nar/gkm360 (doi:10.1093/nar/gkm360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oberto J, Forterre P. In preparation. Comparative genomics of Thermococcales.
- 32.Lerat E, Daubin V, Moran NA. 2003. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-Proteobacteria. PLoS Biol. 1, E19. 10.1371/journal.pbio.0000019 (doi:10.1371/journal.pbio.0000019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, Krause DJ, Whitaker RJ. 2012. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol. 10, e1001265. 10.1371/journal.pbio.1001265 (doi:10.1371/journal.pbio.1001265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng X, Kessler A, Phan H, Garrett RA, Prangishvili D. 2004. Multiple variants of the archaeal DNA rudivirus SIRV1 in a single host and a novel mechanism of genomic variation. Mol. Microbiol. 54, 366–375 10.1111/j.1365-2958.2004.04287.x (doi:10.1111/j.1365-2958.2004.04287.x) [DOI] [PubMed] [Google Scholar]
- 35.Blum H, Zillig W, Mallok S, Domdey H, Prangishvili D. 2001. The genome of the archaeal virus SIRV1 has features in common with genomes of eukaryal viruses. Virology 281, 6–9 10.1006/viro.2000.0776 (doi:10.1006/viro.2000.0776) [DOI] [PubMed] [Google Scholar]
- 36.Lillestol RK, Shah SA, Brugger K, Redder P, Phan H, Christiansen J, Garrett RA. 2009. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol. Microbiol. 72, 259–272 10.1111/j.1365-2958.2009.06641.x (doi:10.1111/j.1365-2958.2009.06641.x) [DOI] [PubMed] [Google Scholar]
- 37.Shah SA, Garrett RA. 2011. CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res. Microbiol. 162, 27–38 10.1016/j.resmic.2010.09.001 (doi:10.1016/j.resmic.2010.09.001) [DOI] [PubMed] [Google Scholar]
- 38.Touzain F, Petit MA, Schbath S, El Karoui M. 2011. DNA motifs that sculpt the bacterial chromosome. Nat. Rev. Microbiol. 9, 15–26 10.1038/nrmicro2477 (doi:10.1038/nrmicro2477) [DOI] [PubMed] [Google Scholar]
- 39.Bush EC, Lahn BT. 2006. The evolution of word composition in metazoan promoter sequence. PLoS Comput. Biol. 2, e150. 10.1371/journal.pcbi.0020150 (doi:10.1371/journal.pcbi.0020150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rocha EP, Danchin A, Viari A. 2001. Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res. 11, 946–958 10.1101/gr.GR-1531RR (doi:10.1101/gr.GR-1531RR) [DOI] [PubMed] [Google Scholar]
- 41.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147. 10.1371/journal.pone.0011147 (doi:10.1371/journal.pone.0011147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.She Q, Peng X, Zillig W, Garrett RA. 2001. Gene capture in archaeal chromosomes. Nature 409, 478. 10.1038/35054138 (doi:10.1038/35054138) [DOI] [PubMed] [Google Scholar]
- 43.She Q, Shen B, Chen L. 2004. Archaeal integrases and mechanisms of gene capture. Biochem. Soc. Trans. 32, 222–226 10.1042/BST0320222 (doi:10.1042/BST0320222) [DOI] [PubMed] [Google Scholar]
- 44.Muskhelishvili G, Palm P, Zillig W. 1993. SSV1-encoded site-specific recombination system in Sulfolobus shibatae. Mol. Gen. Genet. 237, 334–342 [DOI] [PubMed] [Google Scholar]
- 45.Held NL, Whitaker RJ. 2009. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ. Microbiol. 11, 457–466 10.1111/j.1462-2920.2008.01784.x (doi:10.1111/j.1462-2920.2008.01784.x) [DOI] [PubMed] [Google Scholar]
- 46.Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. 2011. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 75, 610–635 10.1128/MMBR.00011-11 (doi:10.1128/MMBR.00011-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mochizuki T, Sako Y, Prangishvili D. 2011. Provirus induction in hyperthermophilic archaea: characterization of Aeropyrum pernix spindle-shaped virus 1 and Aeropyrum pernix ovoid virus 1. J. Bacteriol. 193, 5412–5419 10.1128/JB.05101-11 (doi:10.1128/JB.05101-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.She Q, Phan H, Garrett RA, Albers SV, Stedman KM, Zillig W. 1998. Genetic profile of pNOB8 from Sulfolobus: the first conjugative plasmid from an archaeon. Extremophiles 2, 417–425 10.1007/s007920050087 (doi:10.1007/s007920050087) [DOI] [PubMed] [Google Scholar]
- 49.Cortez D, Forterre P, Gribaldo S. 2009. A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 10, R65. 10.1186/gb-2009-10-6-r65 (doi:10.1186/gb-2009-10-6-r65) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang CT, Zhang R, Ou HY. 2003. The Z curve database: a graphic representation of genome sequences. Bioinformatics 19, 593–599 10.1093/bioinformatics/btg041 (doi:10.1093/bioinformatics/btg041) [DOI] [PubMed] [Google Scholar]
- 51.Zhang R, Zhang CT. 2005. Identification of replication origins in archaeal genomes based on the Z-curve method. Archaea 1, 335–346 10.1155/2005/509646 (doi:10.1155/2005/509646) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortez D, Quevillon-Cheruel S, Gribaldo S, Desnoues N, Sezonov G, Forterre P, Serre MC. 2010. Evidence for a Xer/dif system for chromosome resolution in archaea. PLoS Genet. 6, e1001166. 10.1371/journal.pgen.1001166 (doi:10.1371/journal.pgen.1001166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flynn KM, Vohr SH, Hatcher PJ, Cooper VS 2010. Evolutionary rates and gene dispensability associate with replication timing in the archaeon Sulfolobus islandicus. Genome Biol. Evol. 2, 859–869 10.1093/gbe/evq068 (doi:10.1093/gbe/evq068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dueber EC, Costa A, Corn JE, Bell SD, Berger JM. 2011. Molecular determinants of origin discrimination by Orc1 initiators in archaea. Nucleic Acids Res. 39, 3621–3631 10.1093/nar/gkq1308 (doi:10.1093/nar/gkq1308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lundgren M, Andersson A, Chen L, Nilsson P, Bernander R. 2004. Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc. Natl Acad. Sci. USA 101, 7046–7051 10.1073/pnas.0400656101 (doi:10.1073/pnas.0400656101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson NP, Dionne I, Lundgren M, Marsh VL, Bernander R, Bell SD. 2004. Identification of two origins of replication in the single chromosome of the archaeon Sulfolobus solfataricus. Cell 116, 25–38 10.1016/S0092-8674(03)01034-1 (doi:10.1016/S0092-8674(03)01034-1) [DOI] [PubMed] [Google Scholar]
- 57.Robinson NP, Bell SD. 2007. Extrachromosomal element capture and the evolution of multiple replication origins in archaeal chromosomes. Proc. Natl Acad. Sci. USA 104, 5806–5811 10.1073/pnas.0700206104 (doi:10.1073/pnas.0700206104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976 10.1093/nar/gki201 (doi:10.1093/nar/gki201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 4, 19. 10.1186/1745-6150-4-19 (doi:10.1186/1745-6150-4-19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grynberg M, Erlandsen H, Godzik A. 2003. HEPN: a common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem. Sci. 28, 224–226 10.1016/S0968-0004(03)00060-4 (doi:10.1016/S0968-0004(03)00060-4) [DOI] [PubMed] [Google Scholar]
- 61.Ellen AF, Rohulya OV, Fusetti F, Wagner M, Albers SV, Driessen AJ. 2011. The sulfolobicin genes of Sulfolobus acidocaldarius encode novel antimicrobial proteins. J. Bacteriol. 193, 4380–4387 10.1128/JB.05028-11 (doi:10.1128/JB.05028-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O'Connor EM, Shand RF. 2002. Halocins and sulfolobicins: the emerging story of archaeal protein and peptide antibiotics. J. Ind. Microbiol. Biotechnol. 28, 23–31 [DOI] [PubMed] [Google Scholar]
- 63.Prangishvili D, Holz I, Stieger E, Nickell S, Kristjansson JK, Zillig W. 2000. Sulfolobicins, specific proteinaceous toxins produced by strains of the extremely thermophilic archaeal genus Sulfolobus. J. Bacteriol. 182, 2985–2988 10.1128/JB.182.10.2985-2988.2000 (doi:10.1128/JB.182.10.2985-2988.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ajon M, Frols S, van Wolferen M, Stoecker K, Teichmann D, Driessen AJ, Grogan DW, Albers SV, Schleper C. 2011. UV-inducible DNA exchange in hyperthermophilic archaea mediated by type IV pili. Mol. Microbiol. 82, 807–817 10.1111/j.1365-2958.2011.07861.x (doi:10.1111/j.1365-2958.2011.07861.x) [DOI] [PubMed] [Google Scholar]
- 65.Frols S, et al. 2008. UV-inducible cellular aggregation of the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by pili formation. Mol. Microbiol. 70, 938–952 10.1111/j.1365-2958.2008.06459.x (doi:10.1111/j.1365-2958.2008.06459.x) [DOI] [PubMed] [Google Scholar]
- 66.Brugger K, Redder P, She Q, Confalonieri F, Zivanovic Y, Garrett RA. 2002. Mobile elements in archaeal genomes. FEMS Microbiol. Lett. 206, 131–141 10.1016/S0378-1097(01)00504-3 (doi:10.1016/S0378-1097(01)00504-3) [DOI] [PubMed] [Google Scholar]
- 67.Filee J, Siguier P, Chandler M. 2007. Insertion sequence diversity in archaea. Microbiol. Mol. Biol. Rev. 71, 121–157 10.1128/MMBR.00031-06 (doi:10.1128/MMBR.00031-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Redder P, She Q, Garrett RA. 2001. Non-autonomous mobile elements in the crenarchaeon Sulfolobus solfataricus. J. Mol. Biol. 306, 1–6 10.1006/jmbi.2000.4377 (doi:10.1006/jmbi.2000.4377) [DOI] [PubMed] [Google Scholar]
- 69.Feschotte C, Mouches C. 2000. Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol. Biol. Evol. 17, 730–737 10.1093/oxfordjournals.molbev.a026351 (doi:10.1093/oxfordjournals.molbev.a026351) [DOI] [PubMed] [Google Scholar]
- 70.Feschotte C, Swamy L, Wessler SR. 2003. Genome-wide analysis of mariner-like transposable elements in rice reveals complex relationships with stowaway miniature inverted repeat transposable elements (MITEs). Genetics 163, 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang G, Dong J, Chandrasekharan MB, Hall TC. 2001. Kiddo, a new transposable element family closely associated with rice genes. Mol. Genet. Genomics 266, 417–424 10.1007/s004380100530 (doi:10.1007/s004380100530) [DOI] [PubMed] [Google Scholar]
- 72.Yang G, Hall TC. 2003. MDM-1 and MDM-2: two mutator-derived MITE families in rice. J. Mol. Evol. 56, 255–264 10.1007/s00239-002-2397-y (doi:10.1007/s00239-002-2397-y) [DOI] [PubMed] [Google Scholar]
- 73.Zhang Q, Arbuckle J, Wessler SR. 2000. Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc. Natl Acad. Sci. USA 97, 1160–1165 10.1073/pnas.97.3.1160 (doi:10.1073/pnas.97.3.1160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR. 2003. An active DNA transposon family in rice. Nature 421, 163–167 10.1038/nature01214 (doi:10.1038/nature01214) [DOI] [PubMed] [Google Scholar]
- 75.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR. 2009. Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science 325, 1391–1394 10.1126/science.1175688 (doi:10.1126/science.1175688) [DOI] [PubMed] [Google Scholar]
- 76.Berkner S, Lipps G. 2007. An active nonautonomous mobile element in Sulfolobus islandicus REN1H1. J. Bacteriol. 189, 2145–2149 10.1128/JB.01567-06 (doi:10.1128/JB.01567-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blount ZD, Grogan DW. 2005. New insertion sequences of Sulfolobus: functional properties and implications for genome evolution in hyperthermophilic archaea. Mol. Microbiol. 55, 312–325 10.1111/j.1365-2958.2004.04391.x (doi:10.1111/j.1365-2958.2004.04391.x) [DOI] [PubMed] [Google Scholar]
- 78.Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 1, e60. 10.1371/journal.pcbi.0010060 (doi:10.1371/journal.pcbi.0010060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Makarova KS, et al. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477 10.1038/nrmicro2577 (doi:10.1038/nrmicro2577) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zillig W, Prangishvilli D, Schleper C, Elferink M, Holz I, Albers S, Janekovic D, Gotz D. 1996. Viruses, plasmids and other genetic elements of thermophilic and hyperthermophilic Archaea. FEMS Microbiol. Rev. 18, 225–236 10.1111/j.1574-6976.1996.tb00239.x (doi:10.1111/j.1574-6976.1996.tb00239.x) [DOI] [PubMed] [Google Scholar]
- 81.Greve B, Jensen S, Brugger K, Zillig W, Garrett RA. 2004. Genomic comparison of archaeal conjugative plasmids from Sulfolobus. Archaea 1, 231–239 10.1155/2004/151926 (doi:10.1155/2004/151926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prangishvili D, et al. 1998. Conjugation in archaea: frequent occurrence of conjugative plasmids in Sulfolobus. Plasmid 40, 190–202 10.1006/plas.1998.1363 (doi:10.1006/plas.1998.1363) [DOI] [PubMed] [Google Scholar]
- 83.Stedman KM, She Q, Phan H, Holz I, Singh H, Prangishvili D, Garrett R, Zillig W. 2000. pING family of conjugative plasmids from the extremely thermophilic archaeon Sulfolobus islandicus: insights into recombination and conjugation in Crenarchaeota. J. Bacteriol. 182, 7014–7020 10.1128/JB.182.24.7014-7020.2000 (doi:10.1128/JB.182.24.7014-7020.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. 2012. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3, 945. 10.1038/ncomms1937 (doi:10.1038/ncomms1937) [DOI] [PubMed] [Google Scholar]
- 85.Marchfelder A, et al. 2012. Small RNAs for defence and regulation in archaea. Extremophiles 16, 685–696 10.1007/s00792-012-0469-5 (doi:10.1007/s00792-012-0469-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11, 181–190 10.1038/nrg2749 (doi:10.1038/nrg2749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571 10.1038/nature08703 (doi:10.1038/nature08703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gudbergsdottir S, Deng L, Chen Z, Jensen JV, Jensen LR, She Q, Garrett RA. 2011. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 79, 35–49 10.1111/j.1365-2958.2010.07452.x (doi:10.1111/j.1365-2958.2010.07452.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prangishvili D, Koonin EV, Krupovic M. 2013. Genomics and biology of Rudiviruses, a model for the study of virus–host interactions in Archaea. Biochem. Soc. Trans. 41, 443–450 10.1042/BST20120313 (doi:10.1042/BST20120313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prangishvili D, Quax TE. 2011. Exceptional virion release mechanism: one more surprise from archaeal viruses. Curr. Opin. Microbiol. 14, 315–320 10.1016/j.mib.2011.04.006 (doi:10.1016/j.mib.2011.04.006) [DOI] [PubMed] [Google Scholar]
- 91.Palm P, Schleper C, Grampp B, Yeats S, McWilliam P, Reiter WD, Zillig W. 1991. Complete nucleotide sequence of the virus SSV1 of the archaebacterium Sulfolobus shibatae. Virology 185, 242–250 10.1016/0042-6822(91)90771-3 (doi:10.1016/0042-6822(91)90771-3) [DOI] [PubMed] [Google Scholar]
- 92.Reiter WD, Palm P, Yeats S, Zillig W. 1987. Gene expression in archaebacteria: physical mapping of constitutive and UV-inducible transcripts from the Sulfolobus virus-like particle SSV1. Mol. Gen. Genet. 209, 270–275 10.1007/BF00329653 (doi:10.1007/BF00329653) [DOI] [PubMed] [Google Scholar]
- 93.Stedman KM, Schleper C, Rumpf E, Zillig W. 1999. Genetic requirements for the function of the archaeal virus SSV1 in Sulfolobus solfataricus: construction and testing of viral shuttle vectors. Genetics 152, 1397–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng T, Huang Q, Zhang C, Ni J, She Q, Shen Y. 2012. Development of a simvastatin selection marker for a hyperthermophilic acidophile, Sulfolobus islandicus. Appl. Environ. Microbiol. 78, 568–574 10.1128/AEM.06095-11 (doi:10.1128/AEM.06095-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kirzinger MW, Stavrinides J. 2012. Host specificity determinants as a genetic continuum. Trends Microbiol. 20, 88–93 10.1016/j.tim.2011.11.006 (doi:10.1016/j.tim.2011.11.006) [DOI] [PubMed] [Google Scholar]
- 96.Bennett PM. 2004. Genome plasticity: insertion sequence elements, transposons and integrons, and DNA rearrangement. Methods Mol. Biol. 266, 71–113 [DOI] [PubMed] [Google Scholar]
- 97.Peng X, Blum H, She Q, Mallok S, Brugger K, Garrett RA, Zillig W, Prangishvili D. 2001. Sequences and replication of genomes of the archaeal rudiviruses SIRV1 and SIRV2: relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology 291, 226–234 10.1006/viro.2001.1190 (doi:10.1006/viro.2001.1190) [DOI] [PubMed] [Google Scholar]
- 98.Deveau H, Garneau JE, Moineau S. 2010. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475–493 10.1146/annurev.micro.112408.134123 (doi:10.1146/annurev.micro.112408.134123) [DOI] [PubMed] [Google Scholar]
- 99.Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170 10.1126/science.1179555 (doi:10.1126/science.1179555) [DOI] [PubMed] [Google Scholar]
- 100.Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845 10.1126/science.1165771 (doi:10.1126/science.1165771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O'Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 194, 5728–5738 10.1128/JB.01184-12 (doi:10.1128/JB.01184-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. 2012. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol. Microbiol. 85, 1057–1071 10.1111/j.1365-2958.2012.08172.x (doi:10.1111/j.1365-2958.2012.08172.x) [DOI] [PubMed] [Google Scholar]
- 103.Swarts DC, Mosterd C, van Passel MW, Brouns SJ. 2012. CRISPR interference directs strand specific spacer acquisition. PLoS ONE 7, e35888. 10.1371/journal.pone.0035888 (doi:10.1371/journal.pone.0035888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yosef I, Goren MG, Qimron U. 2012. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40, 5569–5576 10.1093/nar/gks216 (doi:10.1093/nar/gks216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Erdmann S, Garrett RA. 2012. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol. Microbiol. 85, 1044–1056 10.1111/j.1365-2958.2012.08171.x (doi:10.1111/j.1365-2958.2012.08171.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bikard D, Marraffini LA. 2012. Innate and adaptive immunity in bacteria: mechanisms of programmed genetic variation to fight bacteriophages. Curr. Opin. Immunol. 24, 15–20 10.1016/j.coi.2011.10.005 (doi:10.1016/j.coi.2011.10.005) [DOI] [PubMed] [Google Scholar]
- 107.Zhang C, Krause DJ, Whitaker RJ. 2013. Sulfolobus islandicus: a model system for evolutionary genomics. Biochem. Soc. Trans. 41, 458–462 10.1042/BST20120338 (doi:10.1042/BST20120338) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.