

Abstract
Terpene synthases catalyze complex reactions, often forming multiple chiral centers in cyclized olefin products from acyclic allylic diphosphate precursors, yet have been suggested to exert little control over the actual reaction, instead largely serving as inert templates. However, recent results highlight stereoelectronic effects exerted by these enzymes. Perhaps not surprisingly, the pyrophosphate co-product released in the initiating and rate-limiting chemical step provides an obvious counter-ion that may drive carbocation migration towards itself. This is emphasized by the striking effects of a recently uncovered single residue switch for diterpene synthase product outcome, whereby substitution of hydroxyl residues for particular aliphatic residues has been shown to be sufficient to “short-circuit” complex cyclization and/or rearrangement reactions, with the converse change further found to be sufficient to increase reaction complexity. The mechanistic hypothesis for the observed effects is hydroxyl dipole stabilization of the specific carbocation formed by initial cyclization, enabling deprotonation of this early intermediate, whereas the lack of such stabilization (i.e. with an aliphatic side chain) leads to carbocation migration towards the pyrophosphate co-product, resulting in a more complex reaction. This is further consistent with the greater synergy exhibited between pyrophosphate and aza-analogs of late, relative to early, stage carbocation intermediates, and crystallographic analysis of the monoterpene cyclase bornyl diphosphate synthase wherein mechanistically non-relevant counter-ion pairing between aza-analogs of early stage carbocation intermediates and pyrophosphate is observed. Thus, (di)terpene synthases seem to mediate specific reaction outcomes, at least in part, by providing stereoelectronic effects to counteract those exerted by the pyrophosphate co-product.
Introduction
Terpenoids are the most structurally diverse class of natural products, with over 50,000 already known.1 This chemical diversity is underlaid by manyfold hydrocarbon backbone structures formed by the cyclization and/or rearragement of acyclic precursors catalyzed by terpene synthases. These precursors arise from coupling of the universal 5-carbon isoprenoid precursors isopentenyl diphophsate and dimethylallyl diphosphate into longer chain polyisoprenoid diphosphates. Of particular interest here are the 10-carbon monoterpene precursor geranyl diphosphate (GPP), 15-carbon sesquiterpene precursor farnesyl diphosphate (FPP), and 20-carbon diterpene precursor geranylgeranyl diphosphate (GGPP). Typically, the allylic diphosphate ester bond of these acyclic precursors is then lysed/ionized by a terpene synthase to initiate electrophilic reactions that transform these into the observed manyfold cyclized and/or rearranged hydrocarbon backone structures.2 Although GGPP may first undergo a separate protonation-initiated bicyclization reaction catalyzed by mechanistically distinct (class II) diterpene cylases, this leaves intact the allylic diphosphate ester bond in the resulting labdane-related diterpenoid biosynthetic intermediate,3 enabling subsequent ionization and further transformation by members of the more typical (class I) terpene synthase enzyme family that are the focus of this review.
Terpene synthase
As suggested by their enzymatic classification (EC 4.2.3.x), terpene synthases use lysis/ionization of the allylic diphosphate ester bond to drive carbon bond formation. Despite exhibiting very little to no overall sequence similarity, crystallographic investigations have revealed that terpene synthases from plants, fungi, and bacteria share protein structure homology.2 Specifically, catalysis occurs in an analogous α-helical bundle tertiary assembly that has been termed the class I terpene cyclase fold.4 Catalysis further relies on a trinuclear cluster of divalent metal ion co-factors (generally magnesium), which are coordinated by two binding motifs that provide the only broadly conserved sequence features between microbial and plant terpene synthases,5, 6 and which are further coordinated to the substrate diphosphate moiety to enable initiating lysis/ionization (Figure 1). Nevertheless, the observed enzymatic structural and mechanistic similarities indicates common origins for all terpene synthases, making the observations noted here broadly applicable.
Figure 1.
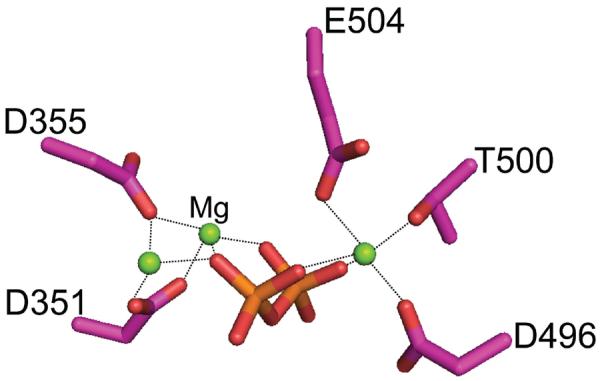
Binding of trinuclear cluster of divalent metal ions by conserved residues and pyrophosphate in bornyl diphosphate synthase.17
Basic catalytic mechanism
Terpene synthases typically catalyze cyclization reactions, which are mediated by intramolecular carbon-carbon double bond addition to carbocation intermediates. Lysis/ionization of the diphosphate ester generates an allylic carbocation that undergoes concerted addition to another π bond within the same substrate molecule, forming a carbocation at a different carbon center, in an SN′ reaction. Such initial cyclization can be followed by further cyclization and/or rearrangements mediated by proton, hydride and/or methyl shifts with correlated carbocation migration. The resulting series of carbocations is then terminated by quenching of the final such intermediate, most typically by deptonation to yield an olefin, although this also can occur after addition of water to yield a hydroxylated or cyclic ether containing product, or via recapture of the ionized pyrophosphate to generate a diphosphate.7
Substrate folding – the template enzyme model
Implicit in this SN′ reaction mechanism is the necessary folding of the substrate by the enzyme to bring together the incipient allylic carbocation and carbon-carbon double bond to be joined (Figure 2).8 In addition, the proximity of this π bond presumably provides anchimeric assistance for the initiating lysis/ionization of the allylic diphosphate ester bond, which is viewed as the rate limiting chemical step, although enzymatic turnover seems to be limited by product release.9, 10 This has led to the view that terpene synthases may dictate product outcome in large part by simply providing a product-like template into which their substrate is folded prior to triggering the relevant carbocation cascade by ionization.11 Indeed, it has been observed that the active site of terpene synthases in the “closed” conformation, but in the absence of any substrate or reaction intermediate analog, are nevertheless distinctly “product-like” in at least some cases.12
Figure 2.

Cyclization of FPP to germacrene A.
Kinetic rather than thermodynamic control
Notably, multiple examples exist of terpene synthase crystal structures wherein substrate or reaction intermediate analogs are bound in non-catalytically relevant conformations in the active site,13–19 although examples also exist wherein relevent conformations are observed, even of the same enzymes with different analogs.16–19 The relevance of the observed substrate/intermediate analog configuration to catalytic mechanisms seems to depend in large part on how closely the analog mimics the reaction product.2
For example, monoterpene cyclases are considered to proceed via initial rearrangement of GPP to the tertiary linalyl diphosphate (LPP), and co-crystal structures of limonene synthase with the inert 2-fluoro analogs of each reveal that exogenous 2F-LPP, but not 2F-GPP (even though it is enzymatically converted to 2F-LPP), is bound in a catalytically relevant conformation.18 Particularly informative is the series of tertiary crystal structures of bornyl diphosphate synthase (BPS) complexed with inorganic pyrophosphate and/or aza-analogs, mimicking various carbocations of the BPS catalyzed reaction (Figure 3).17 In both cases where there is a separate pyrophosphate group, the aza moiety was ion-paired with the pyrophosphate, whose position was essentially invariant (as fixed by its interactions with the enzyme and bound divalent metal ion co-factors). Only in the case of the aza-bornane analog of the final carbocation does its conformation appear to be catalytically relevant. Indeed, the 7-aza-limonene analog is clearly bound “backwards” to enable aza-pyrophosphate ion-pairing, indicating that this is the thermodyamically favored binding mode for such a carbocation. Thus, it seems clear that terpene synthases exert kinetic, rather than thermodynamic, control over the catalyzed reactions,14 which is consistent with the template model for terpene synthase control of product outcome.11
Figure 3.
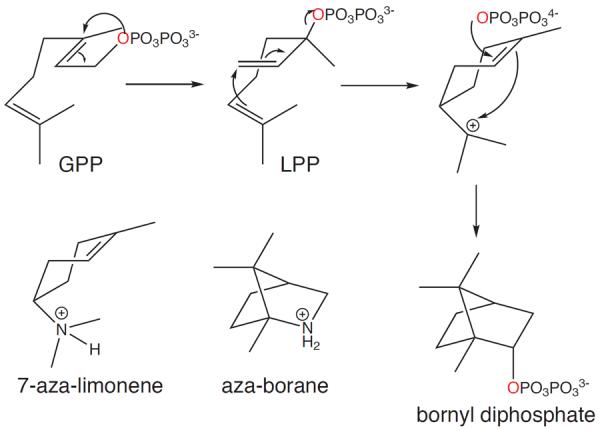
BPS catalyzed cyclization of GPP to bornyl diphosphate, with aza-analogs of early and late carbocation intermediates.17
A role for the pyrophosphate anion co-product
The role of the substrate diphosphate moiety extends beyond that of a simple leaving group. It has been demonstrated that the first step in mono- and many sesqui-terpene cyclization reactions is diphosphate migration from C-1 to the tertiary C-3 position, with concurrent double bond shift to enable C-1 to C-6 bond formation, which is otherwise prevented by the cis conformation of the trans configuration of the original C-2,3 double bond (Figure 3).
Furthermore, inorganic pyrophosphate, corresponding to the anionic co-product released by initiating ionization, is able to drive terpene synthase active site closure.12, 14, 17, 20 Given that terepene synthase enzymatic turnover is limited by product release,9, 10 this indicates that the pyrophosphate co-product is retained in the active site during the electrophilic reaction cycle. Indeed, it has been suggested that the pyrophosphate co-product is tightly bound and may serve as a general acid/base during terpene synthase reactions without becoming reattached itself.12, 21 Further consistent with such tight binding is the stereospecificity of bornyl diphosphate formation by BPS, which reattaches the bornyl cation to the same oxygen of the diphosphate involved in the original diphosphate ester bond of the GPP substrate, indicating that the pyrophosphate anion remains in a fixed orientation during the catalyzed reaction.22
Obviously, the ionized pyrophosphate and carbocation intermediates of the relased olefin must be separated to prevent recapture (except for the bornyl cation in the case of the BPS catalyzed reaction). Nevertheless, we have suggested that the pyrophosphate co-product also may affect the reaction outcome by providing a counter-ion that directs carbocation migration. This was first based on our extensive studies of the model diterpene cyclase, abietadiene synthase (AS).23–29 Specifically, despite the inability of inorganic pyrophosphate to inhibit AS, which is unlike mono- and sesequi- terpene synthases,24 pyrophosphate strongly potentiates inhibition of AS by an aza-analog of a late stage intermediate in the catalyzed reaction, increasing affinity by over three orders of magnitude.26 This is indicative of strong ion-pairing, and is a much stronger effect than observed with other such carbocation mimic analogs,30–32 including earlier stage reaction intermediate analogs with AS (R.J.P., H.-J. Lee, R.B. Croteau, and R.M. Coates; unpublished results). Further evidence for the ability of the pyrophosphate anion co-product to influence to diterpene product outcome is presented here below.
A single residue switch for diterpene synthase product outcome
It has long been supposed that terpene synthases would exhibit some ability to stabilize at least certain carbocation reaction intermediates. However, while it has been suggested that terpene synthase exert such stereoelectronic effects, via both carbocation-quadrupole interactions with the ring π electrons of aromatic side chains33 and/or carbocation interactions with fixed and protected dipholes in the enzyme active site,2 until recently there was no direct evidence for such interactions. Below we present recent studies from our group that provide strong evidence for a stereoelectronic role of a hydroxyl dipole in a wide range of diterpene synthase catalyzed reactions, and further support a role for the pyrophosphate anion co-product in directing the olefin cyclization-rearrangement reaction.
Labdane-related diterpene synthases
As noted in the Introduction, in diterpene biosynthesis GGPP is often first cyclized in a separate protonation-initiated reaction to a bicyclic intermediate such as labdadienyl/copalyl diphosphate (CPP, Figure 4). This biosynthetic intermediate is then further cyclized and/or rearranged by a more typical (class I) terpene synthase. The derived large group of natural products (~7,000 known) has been termed the labdane-related diterpenoids.3 Notably, the relevant (labdane-related class I) diterpene synthases are generally specific for such bicyclic diphosphates, exhibiting much less reactivity with more typical acyclic substrates such as GGPP. The presence of the bicycle in these imparts rigidity relative to acyclic substrates such as GGPP, so that accommodating changes in substate conformation necessary for altered product outcome requires significant remodelling of the active site. Hence, we have hypothesized that these labdane-related diterpene synthases can then serve as a model system for analysis of terpene synthase substrate and product specificity.34–36
Figure 4.
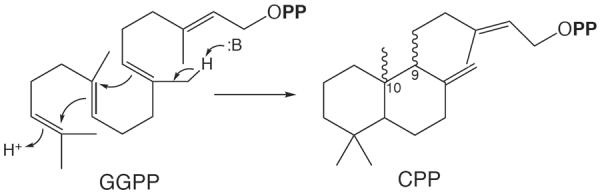
Protonation-initiated cyclization of GGPP to CPP stereoisomeric intermediates in labdane-related diterpenoid biosynthesis.3
Discovery
Because terpene synthases are generally conserved by taxonomic origin rather than biochemical function,37 we undertook a functional genomics investigation of rice (Oryza sativa). In particular, because rice was known to make a number of labdane-related diterpenoid natural products, the corresponding multiple diterpene synthase activities had been demonstrated,38 and the agronomic importance of this staple cereal crop plant had led to early sequencing of its genome.39–41 This led to parallel investigations by our own group and a consortium in Japan. The Japanese studies were focused on the rice subspecies japonica (cv. Nipponbare),42–45 while we largely worked with the indica subspecies (cv. IR24).34–36 Intriguingly, one functional difference between the labdane-related diterpene synthase arsenal of these subspecies became apparent,36 which proved to be extremely informative.
In particular, depending on subspecies origin (indica or japonica), orthologs of one of the rice diterpene synthases produced either tetracyclic ent-isokaur-15-ene or tricylic ent-pimara-8(14),15-diene, respectively. Production of ent-pimaradiene represents deprotonation of the presume ent-pimar-15-en-8-yl+ intermediate in the cyclization of ent-CPP to kaurane type diterpenes (Figure 5). This pair of functionally distinct subspecies orthologs share 98% identity at the amino acid (aa) sequence level, and there are only three differences in the active site.36 It was then possible to map their functional difference to a single residue, alteration of which was sufficient to essentially dictate product outcome, with the presence of a Thr at this position leading to production of ent-pimaradiene while an Ile leads to ent-isokaurene production,46 with similar ent-isokaurene production resulting from the presence of Val, which is more closely isosteric to Thr.47
Figure 5.
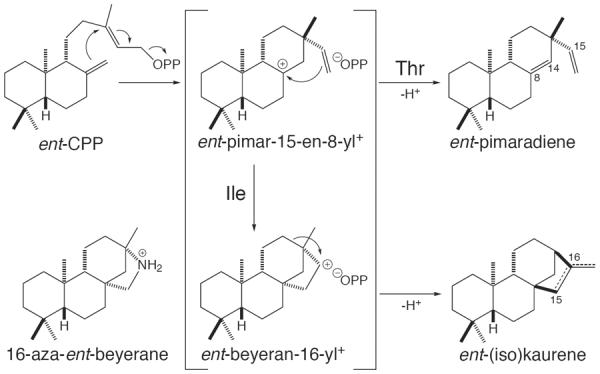
Cyclization-rearrangement of ent-CPP to ent-(iso)kaurene can be short-circuited to produce ent-pimaradiene by substitution of a specific Ile with Thr.46
Furthermore, rice contains another, closely related ent-isokaurene synthase that shares 89% aa sequence identity with the functionally divergent orthologs, and also contains a Ile at this position. Similarly, Thr substitution then converts this enzyme to the production of ent-pimaradiene as well. Strikingly, this position is conserved as Ile in the ent-kaur-16-ene synthases found in all higher plants for gibberellin phytohormone biosynthesis and, despite sharing only 41–52% aa sequence identity with the rice ent-isokaurene synthases, Thr substitution had a very similar effect in two disparate ent-kaurene synthases, “short-circuiting” the more complex cyclization of ent-CPP to ent-kaurene to production of the “simpler” ent-pimaradiene.46
The ability of the inert aliphatic Ile to enable a more “complex” reaction mechanism (i.e. further cyclization and rearrangement), while introduction of a hydroxyl dipole short-circuits this, seems counter-intuitive. Specifically, this implies that there must be some other effect promoting further cyclization of the ent-pimarenyl+ intermediate, despite the accompanying, energetically unfavorable tertiary to secondary carbocation transition. Notably, studies with an aza-analog of the beyeran-16-yl secondary carbocation intermediate initially formed upon tetracyclization demonstrated that inhibition of ent-kaurane synthase by this 16-aza-ent-beyerane was strongly potentiated by inorganic pyrophosphate, suggesting strong ent-beyeranyl+/pyrophosphate ion pairing in this cyclization-rearrangement reaction. However, this does not appear to lead to covalent bond formation, as the corresponding ent-beyeranyl diphosphate is not a substrate for ent-kaurene synthase, although it can be shown to bind in the active site as a competitive inhibitor.48 From these results, we hypothesized that the side chain of this “switch” residue is adjacent to the ent-pimaren-8-yl carbocation, such that the presence of a hydroxyl dipole stabilizes this intermediate long enough for deprotonation to occur, while the lack of such stabilization (i.e. in the presence of an aliphatic residue) enables the tightly bound pyrophosphate anion co-product to drive carbocation migration towards itself. In particular, to form the ent-beyeranyl+/pyrophosphate ion pair, with subsequent rearrangement to the more stable tertiary ent-kauran-16-yl+, which is still adjacent to the pyrophosphate counter-ion, prior to concluding deprotonation.
Extension
The hypothesized role for the pyrophosphate anion co-product in driving ent-kaurene synthase product formation is reminiscent of that we had previously advanced for abietadiene synthase (AS).26 In the AS catalyzed class I reaction, cyclization of CPP to isopimar-15-en-8-yl+ is followed by an intramolecular 1,4-proton transfer,23, 26, 49 which forms a secondary isopimar-8(14)-en-15-yl+ that then undergoes a 1,2-methyl shift to the tertiary abieta-8(14)-en-13-yl+ prior to deprotonation, with the carbocation in the later two intermediates closer to the pyrophosphate anion co-product than that in the initial isopimar-15-en-8-yl+ (Figure 6). As noted above, our initial suggestion that the pyrophosphate anion co-product has a role in driving production of the rearranged abietadiene tricycles arose from the strong potentiating effect of inorganic pyrophosphate on binding of the 15-aza-isopimarene analog of the secondary isopimar-8-en-15-yl+ intermediate to AS,26 while 14-aza-isopimarene, mimicking the initial tertiary isopimar-15-en-8-yl+, exhibits much less synergy with pyrophosphate (R.J.P., H.-J. Lee, R.B. Croteau, and R.M. Coates; unpublished results). While the previously discovered product outcome switch position was conserved as aliphatic residues in closely related abietadiene and isopimara-7,15-diene synthases, we noted a similar hydroxyl/aliphatic conservation pattern four residues prior, with this residue also located in the active site (Figure 7), one turn of the helix away. Gratifyingly, substitution of the corresponding Ala in AS with Ser led to “short-circuiting” of the usual AS catalyzed cyclization-rearrangement reaction, with the resulting mutant enzyme producing essentially only isopimara-7,15-diene.47 This is analogous to the results with ent-(iso)kaurene synthases and resulting hypothetical mechanism described above, as well as our previously advanced hypothesis for the role of the pyrophosphate anion co-product in determining AS catalyzed product outcome. Specifically, in the absence of any counteracting stabilizing influence (i.e. interaction with a hydroxyl dipole), the tightly bound pyrophosphate anion co-product drives carbocation migration towards itself, here via intramolecular 1,4-proton shift to create the secondary isopimar-8(14)-en-15-yl+ that is strongly ion paired to the pyrophosphate, albeit this intermediate is quickly rearranged to the more stable tertiary abiet-8(14)-en-13-yl+ that is still adjacent to the pyrophosphate counter-ion, prior to concluding deprotonation. Notably, the shift in active site location of this switch residue presumably reflects the difference in configuration between the enantiomeric (ent-CPP) substrate of the ent-(iso)kaurene synthases, relative to that (CPP) for AS.
Figure 6.
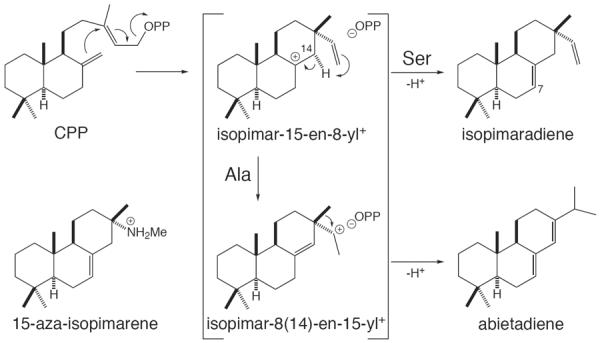
Cyclization-rearrangement of CPP to abietadiene can be short-circuited to produce isopimaradiene by substitution of a specific Ala with Ser.47
Figure 7.
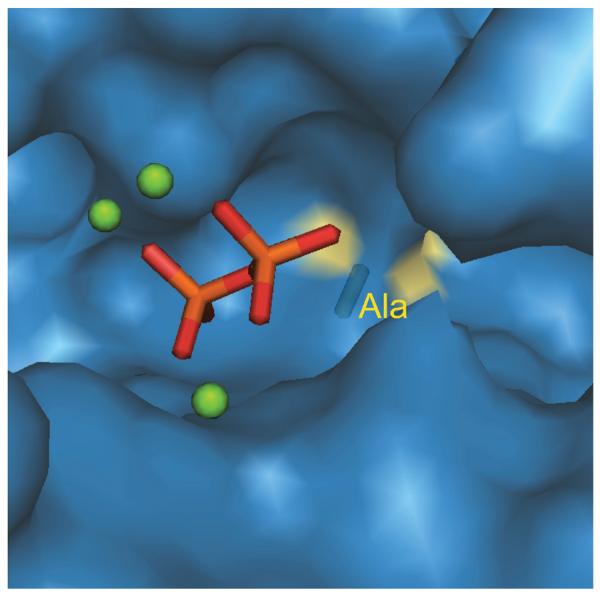
Location of switch residue (Ala) in model of abietadiene synthase, under trinuclear cluster and pyrophosphate shown to define relative substrate orientation.
Application
Implicit in the mechanistic hypothesis presented above is that the identity of the residue at this position acts as a true “switch”, including the ability to increase reaction complexity in addition to short-circuiting more complex reactions. This was investigated with a syn-pimara-7,15-diene synthase from rice, whose encoding gene location in a diterpenoid biosynthetic gene cluster34 dedicated to production of the derived momilactones50 indicates a long standing role in straight-forward tricycle production, and which contains a Thr at the previously identified ent-(iso)kaurene synthase product outcome switch position, consistent with its relatively simple reaction mechanism (i.e. deprotonation of the syn-pimar-15-en-8-yl+ intermediate formed by initial cyclization of syn-CPP). Upon substitution of this Thr with Ile, we found that the resulting mutant enzyme now largely produced a novel diterpene that was identified as syn-aphidicol-15-ene, whose production requires further cyclization and rearrangement of the initially formed syn-pimar-15-en-8-yl+ (Figure 8). Notably, previous biomimetic studies with syn-copalol had demonstrated that similar such extended cyclization and rearrangements can occur in organic solvent,51 consistent with our observations that the presence of an aliphatic residue allows more complex reactions. However, it should be noted that the mutant enzyme more specifically catalyzes such extended cyclization-rearrangement, which presumably reflects, in part, carbocation migration towards the retained pyrophosphate anion co-product, as well as enabling substrate folding. Accordingly, our results demonstrate that the identity of the residue at this position can act as a true switch for diterpene synthase product outcome, and are consistent with our hypothesis for the underlying mechanism.52 In addition, the ability of substitution at the same position as in ent-(iso)kaurene synthases to alter product outcome in this syn-CPP reactive diterpene synthase suggests that the determinant for the location of the switch residue must be the C-9 stereochemistry shared between ent- and syn-CPP (c.f. Figures 5 and 6).
Figure 8.
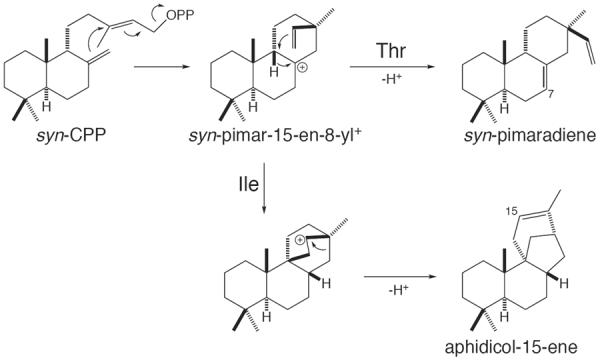
Cyclization-rearrangement of syn-CPP to syn-aphidicolene catalyzed by syn-pimaradiene synthase upon substitution of a particular Thr with Ile.52
However, it must be noted that our results with the rice syn-pimaradiene synthase appear to have been somewhat fortuitious. In particular, it is clear that other, presumably secondary changes can mask the ability of the pyrophosphate anion co-product to drive carbocation migration, as it takes additional active site residue subsitutions to convert isopimaradiene synthase to the production of abietadienes.53
Stereospecificity
As explicitly stated in our mechanistic hypothesis, the switch residue should be proximal to the initially formed pimar-15-en-8-yl carbocation, with its location varying with substrate stereochemistry. Consistent with this supposition, subsitution of the ent-(iso)kaurene synthase switch residue in the normal CPP specific AS (Thr for Val) does not affect product outcome. More direct proof for proximity of the switch residue to the pimar-15-en-8-yl carbocation may be found in our recent observation that the introduction of Cys at the relevant switch position in AS results in a self-inactivating mutant enzyme. While further characterization is required, it is tempting to speculate that this derives from alkylation of the Cys side chain by pimar-15-en-8-yl+, which would directly demonstrate the hypothesized proximity.
Conclusions
Here we have discussed the recent findings from our group demonstrating the presence of a single residue switch for product outcome in the diterpene synthases relevant to labdane-related diterpenoid natural products biosynthesis, which is the central focus of our studies. While no directly analogous results have been reported with other terpene synthases, we suggest that our hypothesis for the underlying mechanism is more widely applicable. In particular, that the retained pyrophosphate anion co-product in the active site will drive carbocation migration towards itself in the absence of other counter-acting stereoelectronic effects. The example arising from our results is the ability of a proximal hydroxyl dipole to stabilize the pimar-15-en-8-yl carbocation formed by initial cyclization of CPP, enabling terminating deprotonation, while the lack of such stabilizing interaction (i.e. when an aliphatic side chain is present instead) allows extended, more complex cyclization-rearrangement reactions, which seems to be driven by carbocation migration towards the pyrophosphate anion co-product. Such favorable ion pairing alleviates the accompanying, energetically unfavorable tertiary to secondary carbocation transition, as indicated by the strong potentiating affect of inorganic pyrophosphate on the corresponding aza-analogs. In addition, in each case the relevant secondary carbocation undergoes rearrangement to a tertiary carbocation that remains proximal to the pyrophosphate anion co-product, retaining some ion pairing potential. From consideration of the generalized terpene cyclization reaction, with intramolecular SN′ addition from a distal double bond on the incipient allylic carbocation arising from lysis/ionization of the diphosphate ester, the resulting pyrophosphate anion co-product will then be proximal to the original allylic double bond, but distal from the initally formed carbocation. Hence, secondary addition from the pyrophosphate proximal double bond will form a carbocation that will be stabilized by ion pairing with the pyrophosphate anion. The counter-intuitive implication of this mechanism is that production of more “simply” cyclized terpenes requires stereoelectronic stabilization of the corresponding “final” carbocation by the enzyme, while the production of more “complex” products may not require any specific interaction with the enzyme beyond sterically imposed substrate folding. Consistent with broader applicability of this mechanistic hypothesis is a recent theoretical study indicating a role for the pyrophosphate anion co-product in the reaction catalyzed by the monoterpene cyclase bornyl diphosphate synthase.54
Biographies
Reuben J. Peters received his B.S. in Molecular Biology from U.C. San Diego in 1992, and a Ph.D. in Biochemistry and Biophysics with David Agard from U.C. San Francisco in 1998. After working on terpene synthase structure-function relationships as a Postdoctoral Fellow of the Jane Coffin Childs Memorial Fund for Medical Research with Rodney Croteau (Washington State University), Peters joined the faculty at Iowa State University in 2002, where he is currently an associate professor. His research interests focus on elucidation of the enzymatic mechanisms and metabolic pathways underlying biosynthesis of labdane-related diterpenoid natural products, along with characterization of their physiological and other, potentially useful functions.
Ke Zhou received his BA degree in Biochemistry from Nanjing University in 2000, and his MS degree in Biochemistry from Beijing Normal University in 2003, where he worked on the catalytic function of Calcineurin. He then worked at Unilever China Research Center, where his research focused on screening herbal extracts for compounds with anti-inflammation activity. He joined Prof. Reuben Peters' research group at Iowa State University as a PhD candidate in 2006, where his research is focused on structure-function relationships in diterpene synthases.
Footnotes
† This article is part of the `Enzymes and Proteins' web-theme issue for ChemComm.
‡ Financial support for the work from the authors group described here was provided by a grant from the NIH (GM076324) to R.J.P.
Notes and references
- 1.Buckingham J. Dictionary of Natural Products (on-line web edition) Chapman & Hall/CRC Press; 2002. [Google Scholar]
- 2.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 3.Peters RJ. Nat. Prod. Rep. submitted. [Google Scholar]
- 4.Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 5.Zhou K, Peters RJ. Phytochemistry. 2009;70:366–369. doi: 10.1016/j.phytochem.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrone D, Chambers J, Lowry L, Kim G, Anterola A, Bender K, Peters RJ. FEBS Lett. 2009;583:475–480. doi: 10.1016/j.febslet.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 7.Peters RJ, Croteau RB. Arch. Biochem. Biophys. 2003;417:203–211. doi: 10.1016/s0003-9861(03)00347-3. [DOI] [PubMed] [Google Scholar]
- 8.Cane D. Acc. Chem. Res. 1985;18:220–226. [Google Scholar]
- 9.Cane DE, Chiu HT, Liang PH, Anderson KS. Biochemistry. 1997;36:8332–8339. doi: 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
- 10.Mathis JR, Back K, Starks C, Noel J, Poulter CD, Chappell J. Biochemistry. 1997;36:8340–8348. doi: 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
- 11.Christianson DW. Curr Opin Chem Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shishova EY, Costanzo LD, Cane DE, Christianson DW. Biochemistry. 2007;46:1941–1951. doi: 10.1021/bi0622524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 14.Vedula LS, Rynkiewicz MJ, Pyun H-J, Coates RM, Cane DE, Christianson DW. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 15.Shishova EY, Yu F, Miller DJ, Faraldos JA, Zhao Y, Coates RM, Allemann RK, Cane DE, Christianson DW. J Biol Chem. 2008;283:15431–15439. doi: 10.1074/jbc.M800659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesburg CA, Zhai G, Cane DE, Christianson DW. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 17.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc Natl Acad Sci U S A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C. Proc. Natl. Acad. Sci. U.S.A. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noel JP, Dellas N, Faraldos JA, Zhao M, Hess BA, Jr., Smentek L, Coates RM, O'Maille PE. ACS Chem Biol. 2010;5:377–392. doi: 10.1021/cb900295g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rynkiewicz MJ, Cane DE, Christianson DW. Proc Natl Acad Sci U S A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caruthers JM, Kang I, Rynkiewicz MJ, Cane DE, Christianson DW. J. Biol. Chem. 2000;275:25533–25539. doi: 10.1074/jbc.M000433200. [DOI] [PubMed] [Google Scholar]
- 22.Cane DE, Saito A, Croteau R, Shaskus J, Felton M. J. Am. Chem. Soc. 1982;104:5831–5833. [Google Scholar]
- 23.Ravn MM, Coates RM, Flory J, Peters RJ, Croteau R. Org. Lett. 2000;2:573–576. doi: 10.1021/ol991230p. [DOI] [PubMed] [Google Scholar]
- 24.Peters RJ, Flory JE, Jetter R, Ravn MM, Lee H-J, Coates RM, Croteau RB. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- 25.Peters RJ, Ravn MM, Coates RM, Croteau RB. J. Am. Chem. Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- 26.Ravn MM, Peters RJ, Coates RM, Croteau RB. J. Am. Chem. Soc. 2002;124:6998–7006. doi: 10.1021/ja017734b. [DOI] [PubMed] [Google Scholar]
- 27.Peters RJ, Croteau RB. Biochemistry. 2002;41:1836–1842. doi: 10.1021/bi011879d. [DOI] [PubMed] [Google Scholar]
- 28.Peters RJ, Croteau RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:580–584. doi: 10.1073/pnas.022627099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters RJ, Carter OA, Zhang Y, Matthews BW, Croteau RB. Biochemistry. 2003;42:2700–2707. doi: 10.1021/bi020492n. [DOI] [PubMed] [Google Scholar]
- 30.Croteau R, Wheeler CJ, Aksela R, Oehlschlager AC. J Biol Chem. 1986;261:7257–7263. [PubMed] [Google Scholar]
- 31.Cane DE, Yang G, Coates RM, Pyun H-J, Hohn TM. J. Org. Chem. 1992;57:3454–3462. [Google Scholar]
- 32.Faraldos JA, Kariuki B, Allemann RK. J Org Chem. 2010;75:1119–1125. doi: 10.1021/jo902397v. [DOI] [PubMed] [Google Scholar]
- 33.Dougherty DA. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 34.Wilderman PR, Xu M, Jin Y, Coates RM, Peters RJ. Plant Physiol. 2004;135:2098–2105. doi: 10.1104/pp.104.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrone D, Jin Y, Xu M, Choi S-Y, Coates RM, Peters RJ. Arch. Biochem. Biophys. 2006;448:133–140. doi: 10.1016/j.abb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Xu M, Wilderman PR, Morrone D, Xu J, Roy A, Margis-Pinheiro M, Upadhyaya N, Coates RM, Peters RJ. Phytochemistry. 2007;68:312–326. doi: 10.1016/j.phytochem.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 37.Bohlmann J, Meyer-Gauen G, Croteau R. Proc. Natl. Acad. Sci. USA. 1998;95:4126–4133. doi: 10.1073/pnas.95.8.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohan RS, Yee NKN, Coates RM, Ren YY, Stamenkovic P, Mendez I, West CA. Arch. Biochem. Biophys. 1996;330:33–47. doi: 10.1006/abbi.1996.0223. [DOI] [PubMed] [Google Scholar]
- 39.Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H. Science. 2002;296:79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]
- 40.Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S. Science. 2002;296:92–100. doi: 10.1126/science.1068275. [DOI] [PubMed] [Google Scholar]
- 41.International Rice Genome Sequencing Project Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 42.Cho E-M, Okada A, Kenmoku H, Otomo K, Toyomasu T, Mitsuhashi W, Sassa T, Yajima A, Yabuta G, Mori K, Oikawa H, Toshima H, Shibuya N, Nojiri H, Omori T, Nishiyama M, Yamane H. Plant J. 2004;37:1–8. doi: 10.1046/j.1365-313x.2003.01926.x. [DOI] [PubMed] [Google Scholar]
- 43.Nemoto T, Cho E-M, Okada A, Okada K, Otomo K, Kanno Y, Toyomasu T, Mitsuhashi W, Sassa T, Minami E, Shibuya N, Nishiyama M, Nojiri H, Yamane H. FEBS Lett. 2004;571:182–186. doi: 10.1016/j.febslet.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Otomo K, Kanno Y, Motegi A, Kenmoku H, Yamane H, Mitsuhashi W, Oikawa H, Toshima H, Itoh H, Matsuoka M, Sassa T, Toyomasu T. Biosci. Biotechnol. Biochem. 2004;68:2001–2006. doi: 10.1271/bbb.68.2001. [DOI] [PubMed] [Google Scholar]
- 45.Kanno Y, Otomo K, Kenmoku H, Mitsuhashi W, Yamane H, Oikawa H, Toshima H, Matsuoka M, Sassa T, Toyomasu T. Biosci. Biotechnol. Biochem. 2006;70:1702–1710. doi: 10.1271/bbb.60044. [DOI] [PubMed] [Google Scholar]
- 46.Xu M, Wilderman PR, Peters RJ. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilderman PR, Peters RJ. J. Am. Chem. Soc. 2007;129:15736–15737. doi: 10.1021/ja074977g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roy A, Roberts FG, Wilderman PR, Zhou K, Peters RJ, Coates RM. J. Am. Chem. Soc. 2007;129:12453–12460. doi: 10.1021/ja072447e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravn MM, Coates RM, Jetter R, Croteau R. Chem Commun (Camb) 1998;1998:21–22. [Google Scholar]
- 50.Shimura K, Okada A, Okada K, Jikumaru Y, Ko K-W, Toyomasu T, Sassa T, Hasegawa M, Kodama O, Shibuya N, Koga J, Nojiri H, Yamane H. J. Biol. Chem. 2007;282:34013–34018. doi: 10.1074/jbc.M703344200. [DOI] [PubMed] [Google Scholar]
- 51.Oikawa H, Nakamura K, Toshima H, Toyomasu T, Sassa T. J. Am. Chem. Soc. 2002;124:9145–9153. doi: 10.1021/ja025830m. [DOI] [PubMed] [Google Scholar]
- 52.Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. J. Am. Chem. Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]
- 53.Keeling CI, Weisshaar S, Lin RPC, Bohlmann J. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1085–1090. doi: 10.1073/pnas.0709466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weitman M, Major DT. J Am Chem Soc. 2010;132:6349–6360. doi: 10.1021/ja910134x. [DOI] [PubMed] [Google Scholar]