

Abstract
The actin severing protein cofilin is essential for directed cell migration and chemotaxis, in many cell types and is also important for tumor cell invasion during metastasis. Through its severing activity, cofilin increases the number of free barbed ends to initiate actin polymerization for actin-based protrusion in two distinct subcellular compartments in invasive tumor cells: lamellipodia and invadopodia. Cofilin severing activity is tightly regulated and multiple mechanisms are utilized to regulate cofilin activity. In this prospect, we have grouped the primary on/off regulation into two broad categories, both of which are important for inhibiting cofilin from binding to F-actin or G-actin: (1) Blocking cofilin activity by the binding of cofilin to either PI(4,5)P2 at lamellipodia, or cortactin at invadopodia. (2) Blocking cofilin's ability to bind to actin via serine phosphorylation. Although the literature suggests that these cofilin regulatory mechanisms may be cell-type dependent, we propose the existence of a common cofilin activity cycle in which both operate. In this common cycle, the mechanism used to initiate cofilin activity is determined by the starting point in the cycle in a given subcellular compartment.
Keywords: Arp2/3 COMPLEX, ACTIN POLYMERIZATION, CHEMOTAXIS, CORTACTIN, PIP2
Directed cell migration is essential for many normal physiological processes beginning with the migration of embryonic cells during development throughout adult life when cells of the immune system, such as neutrophils and macrophages, chemotax toward pathogens [Soon, 2007]. In addition, chemotaxis based directed cell migration is a hallmark of several disease processes including the invasion of tumor cells into the surrounding tissue stroma towards the endothelium to enter the bloodstream. This coordinated migration process leads to tumor metastasis, which is the spread of tumor cells from the primary site to a distant organ. Thus, it is important to understand the underlying mechanisms that allow for directed cell migration.
The initiation of directed cell migration requires actin polymerization from free barbed ends. In response to the stimulation by growth factors, such as epidermal growth factor (EGF), a cell begins to polymerize actin filaments near the plasma membrane producing force to push the plasma membrane forward resulting in a protrusion of the membrane [Mogilner and Oster, 2003; DesMarais et al., 2005]. There are multiple mechanisms by which cells initiate actin polymerization including cofilin-induced severing to produce free actin filament barbed ends [DesMarais et al., 2005], Arp2/3 complex initiated dendritic nucleation of daughter filaments from pre-existing mother filaments [Pollard, 2007], and formin family nucleation of new filaments [Higgs, 2005]. These mechanisms are coordinated to produce free barbed ends for efficient cell migration [DesMarais et al., 2005; Wang et al., 2007a; Sarmiento et al., 2008].
Increasing the number of free barbed ends at a specific location within a cell is used not only to initiate a protrusion during cell motility, but also to define the location of protrusion [Ghosh et al., 2004]. This results in directed cell migration toward the extracellular source that is triggering actin polymerization, a process known as chemotaxis. There is substantial evidence showing that the actin severing protein cofilin is required for determining the direction of the protrusion by initiating the formation of free barbed ends used for actin polymerization [Ghosh et al., 2004; Mouneimne et al., 2004; Mouneimne et al., 2006; Sidani et al., 2007]. The severing activity of cofilin is sufficient to set the direction of cell migration [Sidani et al., 2007] and initiates actin-based protrusions by increasing the number of free barbed ends for actin polymerization [Mouneimne et al., 2004]. In this review, we will discuss the function of cofilin activity during cell motility and how cofilin activity is regulated in different cell types and in different subcellular compartments. Although there is substantial evidence concluding that cofilin is regulated by multiple mechanisms and that the primary mechanism utilized may be cell-type dependent, we propose the existence of a common cofilin activity cycle.
THE COFILIN/ADF FAMILY
Cofilin, a 19 kDa protein, is in the cofilin/actin-depolymerizing factor (ADF) family of actin-binding proteins. There are several isoforms in the cofilin/ADF family of proteins including cofilin-1 (ubiquitous expressed cofilin isoform), cofilin-2 (the muscle isoform of cofilin), and ADF. Knockout studies in mice have shown that cofilin-1 [Gurniak et al., 2005], but not ADF [Ikeda et al., 2003], is essential for survival during embyogenesis past embryonic day 9.5. In contrast, ADF −/− mice have normal survival phenotypes during embryonic development [Ikeda et al., 2003]. In this review, we will focus on cofilin-1 (referred to hereafter as cofilin)—the ubiquitously expressed isoform required for cell motility.
FUNCTION OF COFILIN ACTIVITY IN VITRO
There is a general agreement that cofilin activity is essential for regulation of actin dynamics during cell motility [Nagata-Ohashi et al., 2004; Sidani et al., 2007; Sun et al., 2007]. Studies have shown that cofilin has two general biochemical functions: (1) To depolymerize actin filaments to supply a pool of actin monomers for steady state actin polymerization [Carlier et al., 1997]. (2) To sever actin filaments to create free barbed ends used for actin polymerization [Ichetovkin et al., 2002]. Thus, there are two leading models to explain the function of cofilin activity during cell motility: (1) The enhanced dissociation model claims that cofilin activity leads to increased rates of dissociation of actin monomers from the pointed end of actin filaments and as a result cofilin activity is utilized by cells to depolymerize and recycle actin to supply a pool of actin monomers for subsequent actin polymerization [Carlier et al., 1997; Kiuchi et al., 2007]. (2) The severing activity model suggests that cofilin activity results in the severing of actin filaments creating new barbed ends for actin polymerization. Cofilin activity has a direct role in actin polymerization in the severing model, but not in the enhanced dissociation model, where cofilin participates indirectly in actin polymerization by recycling actin monomers. These two functions for cofilin activity during actin assembly and disassembly are not mutually exclusive and which function predominates depends on the supply of G-actin monomers available for actin polymerization [DesMarais et al., 2005]. The studies that support either the enhanced dissociation model or the severing model have been done in different cell types, using different extracellular stimuli, at different sub-cellular locations. A recent study demonstrated that the effect of cofilin activity on actin filaments is dependent on the concentration of free cofilin [Andrianantoandro and Pollard, 2006] and hence, the effect of cofilin activity on actin dynamics may differ among cell types and in different subcellular compartments. In this study, low concentrations of free cofilin were optimal for cofilin severing activity. As the cofilin concentration was increased, nucleation of actin filaments was observed. They also demonstrated that cofilin activity can result in the depolymerization of actin filaments without a change in off rate at the pointed end, in support of previous work [Ichetovkin et al., 2000]. This study supports the severing model for cofilin activity and argues against the enhanced dissociation model. It also suggests that cofilin primarily functions during actin-based motility to directly increase actin polymerization via its severing activity or direct nucleation activity. Furthermore, it suggests that the precise function for cofilin activity during cell motility may be determined by the concentration of free cofilin and G-actin in a specific subcellular compartment.
FUNCTION OF COFILIN ACTIVITY DURING CELL MOTILITY IN VIVO
As described above, initiation of actin polymerization requires the amplification of free barbed ends. In invasive tumor cells, dictyostelium discoideum, and neutrophils, the amplification of free barbed ends occurs in two temporal transients, an early and a late transient [Soon, 2007]. In mammary carcinoma cells, signaling pathways leading to the severing activity of cofilin are required for the early barbed end transient and pathways leading to Arp2/3 complex activation are required for the late barbed end transient [Mouneimne et al., 2004]. In neutrophils, cofilin-generated barbed ends are critical for Arp2/3-dependent actin polymerization and cell migration [Sun et al., 2007]. Interestingly, there is little productive protrusion resulting from the cofilin-generated barbed ends during the first transient [Mouneimne et al., 2004]. The cofilin activity during the first transient has two primary functions: (1) Establish the asymmetry of actin polymerization required to set the direction of cell migration [Ghosh et al., 2004; Sidani et al., 2007] and (2) to supply the mother filaments for dendritic nucleation by Arp2/3 complex resulting in the productive protrusion of the plasma membrane [DesMarais et al., 2004]. Cofilin and the Arp2/3 complex have been shown to function synergistically both in vitro [Ichetovkin et al., 2002] and in vivo [DesMarais et al., 2004] and this synergy is required to produce protrusion of a lamellipodium on the side of the cell facing the chemotactic gradient [Sidani et al., 2007]. When cofilin severs actin filaments to create free barbed ends, new filaments elongating from these barbed ends are preferred sites for Arp2/3 binding [Ichetovkin et al., 2002]. Thus, the result is a synergy between cofilin severing activity and Arp2/3-generated dendritic nucleation resulting in the formation of a branched actin network.
The cooperation between cofilin and the Arp2/3 complex during actin polymerization and depolymerization has been investigated in vitro with purified proteins [Ichetovkin et al., 2002; Chan et al., 2009]. Both studies support a synergistic interplay between cofilin and the Arp2/3 complex. At the optimum concentration of cofilin for severing, around 9 nM [Ichetovkin et al., 2002; Chan et al., 2009], cofilin severs actin filaments to produce mother filaments preferred for dendritic nucleation by Arp2/3 complex. At these concentrations of cofilin, the Arp2/3-generated branches are stable, and productive pushing force can result from polymerization. At higher concentrations of cofilin, a cofilin-dependent cooperative inhibition of Arp2/3 binding to the sides of mother filaments occurs, which increases the debranching rate. This occurs in part through a cofilin-dependent structural change propagated in the mother actin filament resulting in the dissociation of the Arp2/3 complex from the branch site [Chan et al., 2009]. Since cofilin does not bind to newly polymerized ADP-Pi filaments, the stable Arp2/3 branches are biased to the newly polymerized mother filaments generated by cofilin severing [Ichetovkin et al., 2002]. These results emphasize the importance of precisely regulating the concentration of active cofilin in vivo to achieve the balance between the polymerization and depolymerization activities intrinsic to cofilin–a balance needed for chemotaxis and cell migration [Wang et al., 2007a].
Arp2/3 branch stability may also be regulated in vivo by coronin-1B [Cai et al., 2007] and may involve cortactin [Cai et al., 2008] at lamellipodia. Coronin-1B can simultaneously bind to slingshot and the Arp2/3 complex resulting in the inhibition of Arp2/3 activity and the activation of slingshot resulting in cofilin dephosphorylation and activation [Cai et al., 2007]. The net result is the debranching of actin filaments. Together, these studies provide strong evidence that the activities of cofilin and the Arp2/3 are highly regulated and cooperate both during actin assembly resulting in the efficient generation of a branched actin network [Ichetovkin et al., 2002; DesMarais et al., 2004] and during disassembly resulting in actin filament debranching [Cai et al., 2007; Chan et al., 2009]. Together, these mechanisms, underlying chemotaxis and the formation of lamellipodia in tumor cells, are important for the formation of productive protrusions allowing motile cells to sense a gradient and migrate toward that gradient.
THE REGULATION OF COFILIN ACTIVITY
Precise regulatory control of cofilin activity is critical to maintain the normal physiology of the cell since the mis-regulation of cofilin activity can lead to disease states including tumor metastasis [Wang et al., 2007a] and Alzheimer's disease [Bamburg and Wiggan, 2002]. Many studies have now demonstrated, both in vitro and in vivo, that cofilin activity can be regulated by multiple mechanisms [Arber et al., 1998; van Rheenen et al., 2007]. We have grouped the primary on/off regulation into two broad categories, both of which are important for inhibiting cofilin from binding to F-actin or G-actin: (1) Blocking cofilin activity by the binding of cofilin to either PI(4,5)P2 [Gorbatyuk et al., 2006] or cortactin [Oser et al., 2009]. (2) Blocking cofilin's ability to bind to actin via serine phosphorylation at residue 3 [Arber et al., 1998] (Fig. 1). Apart from these two primary on/off mechanisms used to regulate cofilin activity, other mechanisms have been shown to contribute to the amplitude of cofilin activity resulting from the turning on of cofilin including: regulation by pH [Frantz et al., 2008] and by scaffolding activators of cofilin including cyclase-associated protein (CAP), Aip1, β-arrestin [Zoudilova et al., 2007], Memo [Meira et al., 2009], and coronin [Cai et al., 2007; Marshall et al., 2009]. These mechanisms primarily function to fine tune the primary on/off regulatory mechanisms involving PLCγ1-mediated PI(4,5)P2 hydrolysis, cortactin, and cofilin dephosphorylation.
Fig. 1.
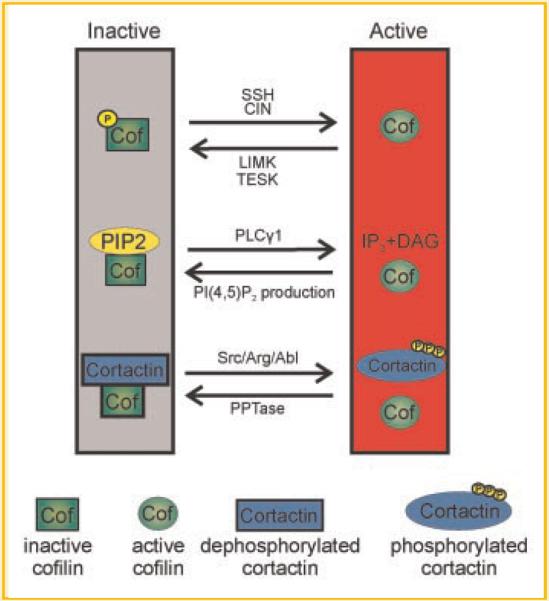
Primary on/off mechanisms that regulate cofilin activity. Left: In resting cells, cofilin remains inactive due to phosphorylation of serine residue 3 via Lim or TES Kinases, by binding to PI(4,5)P2 at the plasma membrane, or by binding to dephosphorylated cortactin in invadopodia. Right: The initiation of cofilin activity can be achieved by dephosphorylation of serine 3 via slingshot (SSH) or chronophin (CIN), and other general phosphatases, PI(4,5)P2-hydrolysis via PLCγ1, or tyrosine phosphorylation of cortactin via either Abl or Src-family kinases.
INACTIVATION OF COFILIN BY SERINE PHOSPHORYLATION: ROLE OF SPECIFIC KINASES
Cofilin activity is blocked upon phosphorylation on serine 3 and restored when cofilin is dephosphorylated [Bamburg and Wiggan, 2002]. Phosphorylation of cofilin at serine 3 inhibits cofilin's ability to bind to actin blocking cofilin's actin severing and depolymerization activities [Arber et al., 1998] (Figs. 1 and 2). Many studies have now identified specific kinases and phosphatases involved in phosphorylating and dephosphorylating cofilin. The kinases include the Lim family kinases (Lim 1 and 2) [Arber et al., 1998] and the Tes family kinases (Tes 1 and Tes 2). The phosphatases that dephosphorylate and activate cofilin include slingshot [Nagata-Ohashi et al., 2004] and chronophin [Gohla et al., 2005], and general phosphatases such as PP1, PP2A, and PP2B.
Fig. 2.

The common cofilin activity cycle at lamellipodia. Cofilin cycles through three compartments near lamellipodia: the cytosol, F-actin, and the plasma membrane (PM) compartments. Cofilin remains inactive at the PM by binding to PI(4,5)P2, and in the cytosol when it is serine phosphorylated. When activated by either PLCγ1-mediated PI(4,5)P2 hydrolysis or dephosphorylation by SSH, cofilin translocates to the F-actin compartment where it binds and severs actin filaments resulting in the generation of free barbed ends and the formation of cofilin–G-actin complexes. These free barbed ends are amplified by WAVE2-dependent Arp2/3 activation resulting in efficient actin polymerization and the formation of cellular protrusions in lamellipodia. The release of cofilin from PI(4,5)P2 at the PM is amplified by an increase in pH (mediated by the Na+–H+ exchanger NHE1), which reduces the affinity of cofilin for PI(4,5)P2, Cofilin is then phosphorylated by Lim kinase to inactivate cofilin. The cycle repeats when cofilin is dephosphorylated by SSH to either recycle cofilin to the PM or directly initiate actin filament severing by cofilin. `+' Indicates pH increase. White arrows indicate primary pathways that regulate cofilin activity, yellow arrows indicate indirect pathways, and blue arrows indicate pathways downstream of cofilin severing activity. Modified from Figure 2 of van Rheenen et al. [2009].
Lim kinase phosphorylates cofilin on serine 3 blocking cofilin's severing and actin depolymerization activity (for a review on pathways that regulate Lim kinase-dependent activation of cofilin see Scott and Olson [2007]). Although other substrates for Lim kinase exist, cofilin and ADF are probably the most abundant substrates for Lim kinase. Lim kinases are regulated and activated by phosphorylation at serine 505/508 by upstream pathways involving Rho-GTPases. Specifically Rac1 or Cdc42 can activate Pak, which in turn phosphorylates and activates Lim kinase. Alternatively, Rho-GTPases can activate ROCK, which phosphorylates Lim kinase. Thus, activation of Rho, Rac, or Cdc42 can result in the phosphorylation and activation of Lim kinase leading to the inhibition of cofilin activity.
These pathways allow for precise temporal and spatial control of cofilin activity within a narrow window as required to balance cofilin's intrinsic polymerization and depolymerization activities, which is essential for chemotaxis [Mouneimne et al., 2006]. In this regard, different studies have reported that overexpression of Lim kinase can either enhance cell motility and tumor metastasis [Bagheri-Yarmand et al., 2006] or inhibit cell motility [Hotulainen et al., 2005] and tumor metastasis [Wang et al., 2006]. Such conflicting results occur because overexpression of Lim kinase may decrease or increase the output of the cofilin pathway, depending on the relative level of cofilin expression and activity in the cell type used for study. Lim kinase 1 is overexpressed in the invasive subpopulation of metastatic carcinoma cells, but cofilin expression or activity is also increased in these cells [Wang et al., 2004, 2007b]. Increased Lim kinase activity results in increased cofilin phosphorylation and, in the presence of increased cofilin activity, this is sufficient to sharpen the cell's response to chemoattractants [Mouneimne et al., 2006]. Overall, during tumor cell invasion, both Lim kinase expression and cofilin activity are simultaneously increased. These findings demonstrate that, to understand the effect of altering Lim kinase expression on cell motility processes, the output of the cofilin pathway needs to be measured directly, and not inferred indirectly.
ACTIVATION OF COFILIN ACTIVITY BY DEPHOSPHORYLATION: ROLE OF SPECIFIC PHOSPHATASES
Dephosphorylation of cofilin at serine 3 by specific phosphatases, including slingshot and chronophin, can result in the activation of cofilin (for a review see Huang et al. [2006]; Fig. 2). In many cell types, such as neutrophils, cofilin phosphorylation levels are high in resting cells and dramatically decrease upon growth factor stimulation as a result of phosphatase activation [Sun et al., 2007]. Neuregulin stimulation in MCF-7 cells results in slingshot activation also leading to dramatic decreases in phosphorylated cofilin upon growth factor stimulation [Nagata-Ohashi et al., 2004]. In these cell types under resting conditions, the cellular pool of cofilin consists mostly of phosphorylated cofilin. As a result, cofilin is initially dephosphorylated leading to its activation (Fig. 2).
Slingshot is a family of phosphatases that selectively dephosphorylate cofilin downstream of growth factor stimulation [Nagata-Ohashi et al., 2004]. Apart from functioning to dephosphorylate cofilin, slingshot also dephosphorylates and inactivates Lim kinase [Soosairajah et al., 2005] resulting in increased control of cofilin activation. Slingshot is known to be activated by high cellular F-actin levels, which increases cofilin activity [Nagata-Ohashi et al., 2004] suggesting that slingshot-mediated cofilin activation controls the intracellular pool of G-actin. PKD1 was recently identified as the upstream kinase that phosphorylates and inactivates slingshot [Eiseler et al., 2009b]. Activated PKD1 increases the phosphorylation and de-activation of slingshot resulting in increased cofilin phosphorylation and decreased cofilin activity. PKD1 activity inhibits total barbed end formation through increasing phosphorylated cofilin and thus decreasing the output of the cofilin pathway. Increasing Lim kinase expression decreases the output of the cofilin pathway and results in decreased tumor cell invasion, intravasation, and metastasis [Wang et al., 2006]. Similarly, PKD1 expression can suppress the invasiveness of mammary carcinoma cell lines [Eiseler et al., 2009a]. It would be interesting to determine whether PKD1-mediated decreases in cofilin activity [Eiseler et al., 2009b] can block tumor cell metastasis in vivo. In summary, PKD1 and slingshot have opposing roles leading to either an overall decrease or increase in cofilin activity [Soosairajah et al., 2005; Eiseler et al., 2009b].
Besides slingshot, the phosphatase chronophin (CIN) is a specific phosphatase for cofilin [Gohla et al., 2005]. CIN dephosphorylates cofilin leading to increased cofilin activity. By increasing the dephosphorylation of cofilin, CIN decreases total cellular F-actin levels [Gohla et al., 2005]. In contrast to slingshot, CIN does not dephosphorylate and inactivate Lim kinase [Huang et al., 2006]. CIN is important for the initial activation of cofilin in neutrophils leading to the formation of free barbed ends and polymerization of mother filaments used for subsequent Arp2/3 activation [Sun et al., 2007].
REGULATION OF COFILIN ACTIVITY BY PI(4,5)P2 BINDING
Recent evidence shows that the initial activation of cofilin activity in mammary carcinoma cells is regulated in vivo by pathways involving the release of cofilin from inhibitory binding interactions [van Rheenen et al., 2007; Oser et al., 2009]. PI(4,5)P2 controls the initial activation of cofilin at the leading edge [van Rheenen et al., 2007] and cortactin controls the initial activation of cofilin in invadopodia-organelles that mediates focal degradation of extracellular matrix (ECM) in metastatic carcinoma cells using matrix metalloproteinase (MMP) activity [Oser et al., 2009]. The binding of either PI(4,5)P2 or cortactin to cofilin blocks cofilin's activity even when cofilin is dephosphorylated [Gorbatyuk et al., 2006; Oser et al., 2009]. First, we will describe what is known about PI(4,5)P2's ability to regulate cofilin and then compare and contrast it with cortactin's direct regulation of cofilin severing activity. Furthermore, we will speculate whether these two forms of regulation are utilized throughout the cell or are unique to invadopodia and lamellipodia.
In vitro binding experiments demonstrated that cofilin binds to membrane lipids, one of which is PI(4,5)P2, and this inhibits cofilin's severing activity [Gorbatyuk et al., 2006]. Cofilin binds to PI(4,5)P2 whether or not it is phosphorylated in vitro [Moriyama et al., 1996]. However, dephosphorylated, but not phosphorylated, cofilin is enriched in the plasma membrane in mammary carcinoma cells [Song et al., 2006] suggesting that, in vivo, the PI(4,5)P2-bound cofilin fraction is dephosphorylated. When cofilin is bound to PI(4,5)P2, its actin binding activity and hence severing activity, is inhibited [Gorbatyuk et al., 2006]. Loss of cofilin binding to PI(4,5)P2 is the mechanism for activating cofilin at the leading edge of mammary carcinoma cells and in muscle cells [Song et al., 2006; Hosoda et al., 2007; van Rheenen et al., 2007]. Knocking down cofilin or PLCγ1 with siRNA demonstrated that both cofilin and PLCγ1 are required for chemotaxis and for the generation of the first barbed end transient that occurs after EGF stimulation in mammary carcinoma cells [Mouneimne et al., 2004, 2006; Meira et al., 2009].
The precise mechanism by which cofilin is regulated by PI(4,5)P2 binding at the plasma membrane was elucidated using FRET and FRAP approaches with live mammary carcinoma cells [van Rheenen et al., 2007] (Fig. 2). In resting cells, cofilin is directly bound to PI(4,5)P2 at the plasma membrane. Upon stimulation with EGF, cofilin is released from the membrane via PLCγ1-mediated PI(4,5)P2 hydrolysis and binds to actin. Cofilin then severs actin filaments to create free barbed ends for actin polymerization. The amount of serine phosphorylated cofilin increases after EGF stimulation [Song et al., 2006] excluding dephosphorylation of cofilin as the mechanism for initial cofilin activation in this cell type. Recently, it was also shown that Memo, a scaffolding protein with no enzymatic activity, increases both the depolymerization and severing activity of cofilin in vitro, binds to a cofilin/PLCγ1/ErbB2 complex in vivo, and thus increases the output of the PLCγ1/cofilin pathway [Meira et al., 2009]. This suggests that local activation of cofilin at the leading edge via release from PI(4,5)P2 by PLCγ1 can be amplified by the presence of the scaffolding protein Memo.
For some time it has been known that cofilin severing and depolymerization activities are increased at elevated physiological pHs. A recent study demonstrated that the binding of cofilin to PI(4,5)P2 is weakened by increases in pH that occur after activation of the Na+/H+ exchanger [Frantz et al., 2008]. Upon growth factor stimulation, the Na+/H+ exchanger is activated leading to local increases in pH resulting in decreased cofilin-PI(4,5)P2 binding at the membrane and increased cofilin activity (Fig. 2). Thus, increasing pH functions to promote cofilin activity by decreasing its affinity for the endogenous activity inhibitor, PI(4,5)P2, and thus promoting cofilin-actin binding.
All of these regulatory steps have been assembled into a cofilin activity cycle for the leading edge of the lamellipodium, which explains cofilin's role in protrusion and chemotaxis (Fig. 2) [van Rheenen et al., 2009].
DIRECT REGULATION OF COFILIN ACTIVITY BY CORTACTIN IN INVADOPODIA
In carcinoma cells, cofilin localizes both at the leading edge [Chan et al., 2000; Mouneimne et al., 2004] and at invadopodia [Yamaguchi et al., 2005]. It has been shown that cofilin is localized to invadopodia and its presence is required for the stability of the invadopodium and matrix degradation activity [Yamaguchi et al., 2005]. Similar to the function of cofilin activity at the leading edge, cofilin is important for the formation of free barbed ends in invadopodia [Oser et al., 2009]. Interestingly, the mechanisms used to regulate cofilin activity at the plasma membrane and invadopodia are different in the same mammary carcinoma cell type [van Rheenen et al., 2007; Oser et al., 2009]. In invadopodia, cofilin is primarily regulated by cortactin (Fig. 3) [Oser et al., 2009], a multi-domain scaffolding protein that is known to activate the Arp2/3 complex and bind to the branch points of actin filaments and stabilize them [Weaver et al., 2001]. In vitro, cofilin and cortactin bind directly and cortactin inhibits cofilin's severing activity. Upon tyrosine phosphorylation of cortactin, after EGF stimulation, the interaction between cortactin and cofilin decreases thereby releasing cofilin's actin binding and severing activities to create free barbed ends for actin polymerization. Within minutes, cortactin is dephosphorylated and the cofilin–cortactin interaction is restored inhibiting further cofilin activity. Similar to the PI(4,5)P2 mechanism that regulates the initial activation of cofilin at the plasma membrane (Fig. 2), cofilin is bound and released from cortactin in invadopodia resulting in cofilin severing activity (Fig. 3). Cortactin and cofilin are both involved in many other cellular processes involving actin polymerization (for reviews see Ammer and Weed [2008] and Van Troys et al. [2008]). It will be interesting to determine whether cortactin regulates cofilin during other actin-based motile processes as it does in the invadopodium.
Fig. 3.

The common cofilin activity cycle in invadopodia. Cofilin cycles through two compartments near invadopodia: the cytosol and the F-actin compartments. Cofilin remains inactive in the F-actin compartment by binding to cortactin, and in the cytosol when it is serine phosphorylated. When cortactin is tyrosine phosphorylated by either Abl or Src-family kinases, cortactin no longer inhibits cofilin's severing activity and cofilin binds and severs actin filaments resulting in the generation of free barbed ends and the formation of cofilin–G-actin complexes. These free barbed ends are amplified by N-WASp-dependent Arp2/3 activation resulting in efficient actin polymerization in invadopodia. In addition, cortactin tyrosine phosphorylation activates Dynamin II's GTPase activity, which remodels actin filaments making them more accessible to cofilin. Cofilin is then phosphorylated by Lim kinase to inactivate cofilin. The cycle repeats when both cofilin and cortactin are dephosphorylated allowing the re-binding of cofilin to cortactin and inhibition of cofilin severing activity. Black arrows indicate primary pathways that regulate cofilin activity, yellow arrows indicate indirect pathways, and blue arrows indicate pathways downstream of cofilin severing activity.
Cortactin phosphorylation not only directly regulates cofilin activity at invadopodia, but regulates the activity of the Arp2/3 complex through a cortactin phosphorylation/Nck1/N-WASp pathway [Oser et al., 2009]. This pathway has been described in vitro [Tehrani et al., 2007] and recently it was demonstrated that invadopodia use this pathway to polymerize actin in vivo [Oser et al., 2009]. As described earlier, the synergy between cofilin and the Arp2/3 complex leads to efficient actin polymerization and filament remodeling both in vitro [Ichetovkin et al., 2002; Chan et al., 2009] and in vivo [DesMarais et al., 2004]. Thus, cortactin may be the scaffolding protein that coordinates this synergy in invadopodia by regulating the severing and debranching activities of cofilin. It will be interesting to determine whether cortactin plays a role in the synergy between cofilin and the Arp2/3 complex at the leading edge [DesMarais et al., 2004].
The kinase that phosphorylates cortactin in invadopodia to regulate actin polymerization is not known. There are many kinases known to phosphorylate cortactin in vitro including Src, Fer, Arg, and Abl [Ammer and Weed, 2008]. Abl family kinases may be the preferred kinases to phosphorylate cortactin both in vitro and in vivo [Boyle et al., 2007]. Arg binds to cortactin at two distinct sites and these Arg–cortactin binding interactions are critical for the formation of cell edge protrusions in response to fibronectin in fibroblasts [Lapetina et al., 2009]. Together, these findings suggest that an Arg–cortactin signaling pathway may regulate cofilin activity in invadopodia. Interestingly, PLCγ1 can activate Abl family kinases [Plattner et al., 2003] and initiate cofilin activity [van Rheenen et al., 2007]. Thus, it is reasonable to hypothesize that PLCγ1-mediated Abl activation leading to cortactin tyrosine phosphorylation may amplify the initial activation of cofilin in both invadopodia and at the leading edge of lamellipodia.
INDIRECT MECHANISMS USED BY CORTACTIN TO REGULATE COFILIN ACTIVITY
In addition to cortactin's direct regulation of cofilin activity by the binding of cofilin, there is evidence that cortactin can indirectly regulate cofilin activity. Cortactin can regulate dynamin II's GTPase activity resulting in increased accessibility of actin filaments to severing by cofilin [Mooren et al., 2009]. In other words, the binding of cortactin to dynamin II increases dynamin II's GTPase activity resulting in increased cofilin severing and barbed end formation for polymerization (Fig. 3). Interestingly, when cortactin is tyrosine phosphorylated, it has increased affinity for dynamin II resulting in increased dynamin II GTPase activity [Zhu et al., 2007] suggesting that phosphorylated cortactin can more effectively promote cofilin severing both directly by release of cofilin [Oser et al., 2009] and indirectly through dynamin II [Mooren et al., 2009]. Invadopodia are enriched in cortactin, cofilin, and dynamin II [Baldassarre et al., 2003] and thus increased phosphorylation of cortactin upon growth factor stimulation can potentially initiate cofilin activity through both direct [Oser et al., 2009] and indirect [Mooren et al., 2009] mechanisms.
In addition, coronin may cooperate with cortactin to regulate cofilin's severing activity [Cai et al., 2007, 2008]. At the leading edge, coronin-1B, cofilin, and slingshot complex can replace cortactin at branch points of actin filaments resulting in debranching of actin filaments. Coronin-1B competes with cortactin for actin filament branches [Cai et al., 2008] and can induce the debranching of actin filaments upon activating cofilin activity via slingshot [Cai et al., 2007]. A recent study demonstrated that coronin-2A, another coronin family member, is important for coordinating the dephosphorylation of cofilin via slingshot at focal adhesions leading to focal adhesion turnover in mammary carcinoma cells [Marshall et al., 2009]. Specifically, coronin-2A knockdown cells showed decreased cofilin activity, increased focal adhesion size, and decreased focal adhesion turnover rates [Marshall et al., 2009]. Cells expressing a cortactin mutant that cannot be tyrosine phosphorylated have decreased cofilin activity [Oser et al., 2009] and also show increased focal adhesion size and decreased rates of focal adhesion turnover [Kruchten et al., 2008]. These defects can be rescued by expressing a cortactin phospho-mimic [Kruchten et al., 2008] that fails to inhibit cofilin's severing activity [Oser et al., 2009]. These studies suggest that coronin may coordinate the dephosphorylation of cofilin via slingshot and the re-binding of cofilin to cortactin during the cofilin activity cycle at the leading edge (Fig. 2) and at invadopodia (Fig. 3). Which specific coronin isoform is involved in coordinating the dephosphorylation of cofilin via slingshot in invadopodia remains to be determined.
SCAFFOLDING ADAPTORS THAT MODULATE COFILIN ACTIVITY
Apart from the primary mechanisms used to regulate cofilin activity, several scaffolding activators have been identified that function to either enhance or inhibit the output of the cofilin pathways regulated by dephosphorylation and PI(4,5)P2. 14-3-3 Proteins bind to phosphorylated slingshot and inhibit the activity of slingshot [Nagata-Ohashi et al., 2004] thereby decreasing the output of the cofilin pathway. Downstream of G-protein coupled receptors, β-arrestin's 1 and 2, form a complex with cofilin, CIN, and Lim kinase, promoting the phosphatase activity of CIN and inhibiting Lim kinase activity resulting in efficient cofilin dephosphorylation in MDA-MB-468 cells [Zoudilova et al., 2007]. Thus, β-arrestin functions as a scaffolding activator of pathways that control cofilin via dephosphorylation. As described earlier, Memo, a scaffolding protein downstream of the ErbB2 receptor in T47D cells, may be the scaffolding activator involved in the PLCg1/PI(4,5)P2/cofilin pathway [Meira et al., 2009]. In summary, the coordination of signaling pathways that regulate cofilin activity via dephosphorylation and PI(4,5)P2 binding involve scaffolding activators to increase the output of the pathway resulting in efficient activation of cofilin.
COORDINATION OF COFILIN ACTIVATION BY PLCγ1/PI(4,5)P2 AND INACTIVATION BY LIM KINASE-THE LEGI MODEL
Many studies investigating cofilin regulation have focused solely on one mechanism and thus little is known about whether multiple mechanisms of cofilin regulation are simultaneously utilized to regulate cofilin within a single subcellular compartment. Gene expression profiling studies using microarrays have shown that multiple pathways leading to cofilin activation are simultaneously upregulated within invasive tumor cells [Wang et al., 2004; Wang et al., 2007b]. Thus, it is likely that individual pathways controlling cofilin activation cooperate to result in increased control of cofilin activity. Given that all forms of regulation may be utilized and important for regulating cofilin activity in a single cell, it is misleading to analyze one form of regulation, such as the phosphorylation status of cofilin, and draw a conclusion about the overall activity of the cofilin pathway in that cell. To deduce the activity status of cofilin, one must look at the output of cofilin activity—the ability to sever actin filaments to create free barbed ends for actin polymerization.
For example, in mammary carcinoma cells, the phosphorylation of cofilin, and cofilin activity, simultaneously increase upon EGF stimulation in mammary carcinoma cells [Song et al., 2006]. As described earlier, the initial activation of cofilin requires PLCγ1/PI(4,5)P2 pathway [van Rheenen et al., 2007]. In addition, the global increase in cofilin phosphorylation is also important for spatially restricting cofilin's activity, which is necessary for chemotaxis [Mouneimne et al., 2006; Song et al., 2006]. That is, increasing total cofilin phosphorylation generates a region of focal dephosphorylated cofilin with high cofilin activity at the front of the cell facing the chemoattractant gradient, which is required for localizing protrusion toward the source of chemoattractant [Mouneimne et al., 2006]. Thus, activation of both PLCγ1 (leading to initial cofilin activation) and Lim kinase (leading to global increases in cofilin phosphorylation) pathways are activated in response to EGF and are both necessary for proper chemotaxis of tumor cells. These findings support a local excitation/global inhibition (LEGI) model for cofilin activation. LEGI models have been used to explain how cells respond to chemoattractants in other eukaryotic model organisms, such as dictyostelium discoideum [Devreotes and Janetopoulos, 2003]. We propose that the LEGI model can be used to explain cofilin activity in other cell types where the mechanisms that support the local excitation and global inhibition of cofilin are determined by the starting point in the cofilin activity cycle (Figs. 2 and 3).
A COMMON COFILIN ACTIVITY CYCLE
The specific mechanisms that regulate the initial activation of cofilin depend on the starting point in the cofilin cycle from which cofilin is activated during stimulation. This appears to vary with cell type [van Rheenen et al., 2009]. All pathways known to regulate cofilin activity can be connected to generate a common cofilin activity cycle (Fig. 4A). The mechanisms responsible for regulating cofilin activity in a specific subcellular compartment in a specific cell type depends on the starting point of cofilin activity and the composition of molecules in the specific subcellular compartment. For example, in some cell types before stimulation, the majority of the cellular pool of cofilin is phosphorylated cofilin [Kanamori et al., 1995; Sun et al., 2007]. As a result, the initial activation of cofilin requires dephosphorylation by phosphatases. In contrast, in unstimulated tumor cells, the vast majority of the cellular pool of cofilin is dephosphorylated cofilin [Zebda et al., 2000; Song et al., 2006], but cofilin remains inactive [Chan et al., 2000] in part due to the binding of cofilin to PI(4,5)P2 at the plasma membrane. As a result, the initial activation of cofilin requires PLCγ1-dependent hydrolysis of PI(4,5)P2 [van Rheenen et al., 2007]. Thus, the starting point of the cycle determines the mechanisms required for the initial activation of cofilin.
Fig. 4.

A: The common cofilin activity cycle in both lamellipodia and invadopodia. The PM at lamellipodia is enriched with PI(4,5)P2 and the on/off binding of cofilin to PI(4,5)P2 is the primary mechanism used to regulate cofilin activity. In contrast, as shown in B, the PM on the ventral cell surface where invadopodia form is depleted for PI(4,5)P2. Invadopodia form in PI(3,4)P2-enriched PM areas through a Tks5-PI(3,4)P2 binding interaction. In invadopodia, the on/off binding of cofilin to cortactin is the primary mechanism used to regulate cofilin activity. White and black arrows indicate pathways used to regulate cofilin activity in lamellipodia and invadopodia, respectively. B: (Top) X–Y and (bottom) X–Z images of an MTLn3 cell stained with antibodies against PI(4,5)P2 (green) and cofilin (red) showing that PI(4,5)P2 and cofilin co-localize in the PM where lamellipodia form (yellow), but PI(4,5)P2 is depleted from the ventral cell surface where invadopodia form (Courtesy of Dr. Robert Eddy). The leading edge appears yellow as a result of the co-localization of cofilin and PI(4,5)P2. In contrast, the ventral cell surface is red due to the presence of cofilin, but absence of PI(4,5)P2.
CROSS-TALK AMONG COFILIN REGULATORY MECHANISMS
Little is known about the interdependence of the primary on/off mechanisms that control cofilin activity. It has been demonstrated that phosphorylated cofilin can bind to PI(4,5)P2 [Moriyama et al., 1996] and thus it is conceivable that cofilin activation by dephosphorylation and PI(4,5)P2 hydrolysis can simultaneously be achieved in a single compartment within a cell downstream of growth factor stimulation. In support of this hypothesis, inhibition of PLCγ1 activity with a chemical inhibitor blocked cofilin dephosphorylation in neutrophils [Zhan et al., 2003] and macrophages [Matsui et al., 2001] suggesting that the activities of PLCγ1 and cofilin-specific phosphatases are tightly coupled downstream of growth factor stimulation. Recently, it has also been demonstrated that phospho-cofilin is present in the complex with PLCγ1/Memo/ErbB2 at the plasma membrane [Meira et al., 2009]. Thus, it is possible that a fraction of the cofilin bound to PI(4,5)P2 is phosphorylated, and after the initial activation of cofilin by PLCγ1, a population of phospho-cofilin is released from the plasma membrane, and quickly dephosphorylated by cofilin-specific phosphatases. Future studies will determine whether there is a redundancy of regulatory mechanisms that control the initial activation of cofilin within a single subcellular compartment.
CONCLUDING REMARKS: WHY PI(4,5)P2 AND CORTACTIN IN DIFFERENT SUBCELLULAR COMPARTMENTS?
So why have these two distinct cofilin regulatory mechanisms, PI(4,5)P2 and cortactin, evolved in different subcellular compartments? One can speculate that these divergent forms of cofilin regulation may depend on the requirements for the biogenesis of the specific organelle. Cortactin is absolutely essential for invadopodium formation and function in many cancer cell types (for a review on cortactin see Weaver [2008]), but is not essential for lamellipodium formation [Bryce et al., 2005; Desmarais et al., 2009]. Invadopodia are invasive structures found in metastatic, but not non-metastatic carcinoma cells [Yamaguchi et al., 2005], and thus it is conceivable that the regulation of signaling pathways leading to actin polymerization are specific to invadopodia. In addition, cortactin is highly overexpressed in many human cancers and the evidence suggests that cortactin is important for metastasis, but not for growth of the primary tumor. Thus, the high protein expression of cortactin in invasive tumor cells may help to explain why cortactin is essential for regulating many processes in invadopodia.
Lamellipodia form in regions of the plasma membrane enriched for PI(4,5)P2, where PI(4,5)P2 inhibits cofilin activity at the plasma membrane [van Rheenen et al., 2007]. However, PI(4,5)P2 is not enriched in the plasma membrane on the ventral cell surface where invadopodia form (Fig. 4B), or in podosomes [Oikawa et al., 2008]—a related structure to invadopodia found in myelocytic cells. Interestingly, Tks5, a scaffolding protein enriched in invadopodia [Oser et al., 2009] and required for invadopodium formation [Stylli et al., 2009], binds selectively to PI(3,4)P2 [Abram et al., 2003], and the accumulation of Tks5 at PI(3,4)P2-enriched membrane locations is essential for podosome formation [Oikawa et al., 2008]. In addition, Tks5 recruits cortactin to podosomes [Crimaldi et al., 2009]. Thus, one can speculate that cortactin regulates cofilin activity in subcellular locations where the membrane is not enriched with PI(4,5)P2, such as invadopodia. This implies that the biological composition of invadopodia and lamellipodia may determine the primary mechanisms used to regulate cofilin activity. It will be interesting to determine whether cortactin is also involved in regulating cofilin activity in other subcellular compartments, such as in lamellipodia.
ACKNOWLEDGMENTS
We thank Dr. Rob Eddy for providing the image in Figure 4B, Dr. Jacco van Rheenen for providing Figure 2, and The Journal of Cell Science for granting copyright permission for Figure 2 (from Fig. 2 of van Rheenen et al., 2009). This work was funded by CA100324, CA126511, GM38511 (John Condeelis), and GM064346 (Jacco Van Rheenen).
REFERENCES
- Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–16851. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- Ammer AG, Weed SA. Cortactin branches out: Roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65:687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Mazumdar A, Sahin AA, Kumar R. LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int J Cancer. 2006;118:2703–2710. doi: 10.1002/ijc.21650. [DOI] [PubMed] [Google Scholar]
- Baldassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, Luini A, Buccione R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell. 2003;14:1074–1084. doi: 10.1091/mbc.E02-05-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12:598–605. doi: 10.1016/s0962-8924(02)02404-2. [DOI] [PubMed] [Google Scholar]
- Boyle SN, Michaud GA, Schweitzer B, Predki PF, Koleske AJ. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation. Curr Biol. 2007;17:445–451. doi: 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15:1276–1285. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–929. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Makhov AM, Schafer DA, Bear JE. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134:828–842. doi: 10.1016/j.cell.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Beltzner CC, Pollard TD. Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr Biol. 2009;19:537–545. doi: 10.1016/j.cub.2009.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimaldi L, Courtneidge SA, Gimona M. Tks5 recruits AFAP-110, p190RhoGAP, and cortactin for podosome formation. Exp Cell Res. 2009;315:2581–2592. doi: 10.1016/j.yexcr.2009.06.012. [DOI] [PubMed] [Google Scholar]
- DesMarais V, Macaluso F, Condeelis J, Bailly M. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J Cell Sci. 2004;117:3499–3510. doi: 10.1242/jcs.01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarais V, Ghosh M, Eddy R, Condeelis J. Cofilin takes the lead. J Cell Sci. 2005;118:19–26. doi: 10.1242/jcs.01631. [DOI] [PubMed] [Google Scholar]
- Desmarais V, Yamaguchi H, Oser M, Soon L, Mouneimne G, Sarmiento C, Eddy R, Condeelis J. N-WASP and cortactin are involved in invadopodium-dependent chemotaxis to EGF in breast tumor cells. Cell Motil Cytoskeleton. 2009;66:303–316. doi: 10.1002/cm.20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: Distinctions between directional sensing and polarization. J Biol Chem. 2003;278:20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009a;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009b;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C, Barreiro G, Dominguez L, Chen X, Eddy R, Condeelis J, Kelly MJ, Jacobson MP, Barber DL. Cofilin is a pH sensor for actin free barbed end formation: Role of phosphoinositide binding. J Cell Biol. 2008;183:865–879. doi: 10.1083/jcb.200804161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk VY, Nosworthy NJ, Robson SA, Bains NP, Maciejewski MW, Dos Remedios CG, King GF. Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol Cell. 2006;24:511–522. doi: 10.1016/j.molcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Gurniak CB, Perlas E, Witke W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol. 2005;278:231–241. doi: 10.1016/j.ydbio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Higgs HN. Formin proteins: A domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Hosoda A, Sato N, Nagaoka R, Abe H, Obinata T. Activity of cofilin can be regulated by a mechanism other than phosphorylation/dephosphorylation in muscle cells in culture. J Muscle Res Cell Motil. 2007;28:183–194. doi: 10.1007/s10974-007-9117-6. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–664. doi: 10.1091/mbc.E04-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Han J, Pang KM, Knecht DA, Condeelis JS. Actin filaments are severed by both native and recombinant dictyostelium cofilin but to different extents. Cell Motil Cytoskeleton. 2000;45:293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. doi: 10.1016/s0960-9822(01)00629-7. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Cunningham LA, Boggess D, Hawes N, Hobson CD, Sundberg JP, Naggert JK, Smith RS, Nishina PM. Aberrant actin cytoskeleton leads to accelerated proliferation of corneal epithelial cells in mice deficient for destrin (actin depolymerizing factor) Hum Mol Genet. 2003;12:1029–1037. doi: 10.1093/hmg/ddg112. [DOI] [PubMed] [Google Scholar]
- Kanamori T, Hayakawa T, Suzuki M, Titani K. Identification of two 17-kDa rat parotid gland phosphoproteins, subjects for dephosphorylation upon beta-adrenergic stimulation, as destrin- and cofilin-like proteins. J Biol Chem. 1995;270:8061–8067. doi: 10.1074/jbc.270.14.8061. [DOI] [PubMed] [Google Scholar]
- Kiuchi T, Ohashi K, Kurita S, Mizuno K. Cofilin promotes stimulus-induced lamellipodium formation by generating an abundant supply of actin monomers. J Cell Biol. 2007;177:465–476. doi: 10.1083/jcb.200610005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruchten AE, Krueger EW, Wang Y, McNiven MA. Distinct phosphoforms of cortactin differentially regulate actin polymerization and focal adhesions. Am J Physiol Cell Physiol. 2008;295:C1113–C1122. doi: 10.1152/ajpcell.00238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapetina S, Mader CC, Machida K, Mayer BJ, Koleske AJ. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J Cell Biol. 2009;185:503–519. doi: 10.1083/jcb.200809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall TW, Aloor HL, Bear JE. Coronin 2A regulates a subset of focal-adhesion-turnover events through the cofilin pathway. J Cell Sci. 2009;122:3061–3069. doi: 10.1242/jcs.051482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui S, Adachi R, Kusui K, Yamaguchi T, Kasahara T, Hayakawa T, Suzuki K. U73122 inhibits the dephosphorylation and translocation of cofilin in activated macrophage-like U937 cells. Cell Signal. 2001;13:17–22. doi: 10.1016/s0898-6568(00)00124-8. [DOI] [PubMed] [Google Scholar]
- Meira M, Masson R, Stagljar I, Lienhard S, Maurer F, Boulay A, Hynes NE. Memo is a cofilin-interacting protein that influences PLCgamma1 and cofilin activities, and is essential for maintaining directionality during ErbB2-induced tumor-cell migration. J Cell Sci. 2009;122:787–797. doi: 10.1242/jcs.032094. [DOI] [PubMed] [Google Scholar]
- Mogilner A, Oster G. Force generation by actin polymerization II: The elastic ratchet and tethered filaments. Biophys J. 2003;84:1591–1605. doi: 10.1016/S0006-3495(03)74969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooren OL, Kotova TI, Moore AJ, Schafer DA. Dynamin2 GTPase and cortactin remodel actin filaments. J Biol Chem. 2009;284:23995–24005. doi: 10.1074/jbc.M109.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- Mouneimne G, Soon L, DesMarais V, Sidani M, Song X, Yip SC, Ghosh M, Eddy R, Backer JM, Condeelis J. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J Cell Biol. 2004;166:697–708. doi: 10.1083/jcb.200405156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouneimne G, DesMarais V, Sidani M, Scemes E, Wang W, Song X, Eddy R, Condeelis J. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–2205. doi: 10.1016/j.cub.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, Ohashi K, Kousaka K, Iwamatsu A, Niwa R, Uemura T, Mizuno K. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol. 2004;165:465–471. doi: 10.1083/jcb.200401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH-src cells. J Cell Biol. 2008;182:157–169. doi: 10.1083/jcb.200801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, Desmarais V, van Rheenen J, Koleske AJ, Condeelis J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186:571–587. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- Sarmiento C, Wang W, Dovas A, Yamaguchi H, Sidani M, El-Sibai M, Desmarais V, Holman HA, Kitchen S, Backer JM, Alberts A, Condeelis J. WASP family members and formin proteins coordinate regulation of cell protrusions in carcinoma cells. J Cell Biol. 2008;180:1245–1260. doi: 10.1083/jcb.200708123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RW, Olson MF. LIM kinases: Function, regulation and association with human disease. J Mol Med. 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- Sidani M, Wessels D, Mouneimne G, Ghosh M, Goswami S, Sarmiento C, Wang W, Kuhl S, El-Sibai M, Backer JM, Eddy R, Soll D, Condeelis J. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J Cell Biol. 2007;179:777–791. doi: 10.1083/jcb.200707009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Chen X, Yamaguchi H, Mouneimne G, Condeelis JS, Eddy RJ. Initiation of cofilin activity in response to EGF is uncoupled from cofilin phosphorylation and dephosphorylation in carcinoma cells. J Cell Sci. 2006;119:2871–2881. doi: 10.1242/jcs.03017. [DOI] [PubMed] [Google Scholar]
- Soon LL. A discourse on cancer cell chemotaxis: Where to from here? IUBMB Life. 2007;59:60–67. doi: 10.1080/15216540701201033. [DOI] [PubMed] [Google Scholar]
- Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, Sampath R, Bamburg JR, Bernard O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–2740. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CX, Magalhaes MA, Glogauer M. Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol. 2007;179:239–245. doi: 10.1083/jcb.200705122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehrani S, Tomasevic N, Weed S, Sakowicz R, Cooper JA. Src phosphorylation of cortactin enhances actin assembly. Proc Natl Acad Sci USA. 2007;104:11933–11938. doi: 10.1073/pnas.0701077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Song X, van Roosmalen W, Cammer M, Chen X, Desmarais V, Yip SC, Backer JM, Eddy RJ, Condeelis JS. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol. 2007;179:1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009;122:305–311. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, Singer RH, Segall JE, Condeelis JS. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- Wang W, Mouneimne G, Sidani M, Wyckoff J, Chen X, Makris A, Goswami S, Bresnick AR, Condeelis JS. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;173:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007a;7:429–440. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007b;67:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- Weaver AM. Cortactin in tumor invasiveness. Cancer Lett. 2008;265:157–166. doi: 10.1016/j.canlet.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: The role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1128. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Bamburg JR, Badwey JA. Products of phosphoinositide specific phospholipase C can trigger dephosphorylation of cofilin in chemoattractant stimulated neutrophils. Cell Motil Cytoskeleton. 2003;54:1–15. doi: 10.1002/cm.10079. [DOI] [PubMed] [Google Scholar]
- Zhu J, Yu D, Zeng XC, Zhou K, Zhan X. Receptor-mediated endocytosis involves tyrosine phosphorylation of cortactin. J Biol Chem. 2007;282:16086–16094. doi: 10.1074/jbc.M701997200. [DOI] [PubMed] [Google Scholar]
- Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem. 2007;282:20634–20646. doi: 10.1074/jbc.M701391200. [DOI] [PubMed] [Google Scholar]