

Abstract
Objectives
To determine whether glycine receptor α1 subunit-specific autoantibodies (GlyRα1-IgG) occur in a broader spectrum of brainstem and spinal hyperexcitability disorders than the progressive encephalomyelitis with rigidity and myoclonus phenotype recognized to date, and to ascertain disease specificity.
Design
Retrospective, case-control study.
Settings
Mayo Clinic, Rochester, Minnesota, and University of Barcelona, Spain.
Patients
Eighty-one patients with stiff-man syndrome phenotype, 80 neurologic control subjects, and 20 healthy control subjects.
Intervention
Glycine receptor α1–transfected cells to test serum or cerebrospinal fluid from cases and control subjects.
Main Outcome Measures
Frequency of GlyRα1-IgG positivity among stiff-man syndrome phenotype cases and control subjects. Comparison of GlyRα1-IgG seropositive and seronegative cases.
Results
Seropositive cases (12% of cases) included 9 with stiff-man syndrome (4 classic; 5 variant; 66% were glutamic acid decarboxylase 65–IgG positive) and 1 with progressive encephalomyelitis with rigidity and myoclonus. Immunotherapy responses were noted more frequently in GlyRα1-IgG–positive cases (6 of 7 improved) than in seronegative cases (7 of 25 improved; P=.02). The single seropositive control patient had steroid-responsive vision loss and optic atrophy with inflammatory cerebrospinal fluid.
Conclusions
Glycine receptor α1–IgG aids identification of autoimmune brainstem/spinal cord hyperexcitability disorders and may extend to the glycinergic visual system.
Inhibitory synaptic transmission mediated by γ-aminobutyric acid and glycine is critical for regulating motor neuron excitability in the brainstem and spinal cord. Central nervous system hyperexcitability disorders ascribed to loss of this input are characterized by exaggerated startle, stiffness, and spasms of the axis and limbs. Autoimmunity accounts for classic stiff-man syndrome (SMS; also known as stiff-person syndrome), which principally affects the lumbar spine and proximal lower extremities),1,2 variant SMS (limited to the axis or extremities [eg, stiff-leg syndrome]),3 and progressive encephalomyelitis with rigidity and myoclonus (PERM),4 in which hyperexcitability is widespread, severe, and sometimes fatal. Autoantibodies specific for the 65 kDa isoform of the γ-amino-butyric acid–synthesizing enzyme glutamic acid decarboxylase 65 (GAD65)–IgG are detected in 80% of patients with classic SMS.5
Other autoantigens pertinent to SMS phenotype include amphiphysin,6 gephyrin,7 and the α1 subunit of a heteromeric (α1β) glycine-gated chloride channel (GlyRα1)8 that is enriched in the spinal cord and brainstem.9 Published reports have emphasized an association of GlyRα1-IgG with the rare PERM phenotype.8,10–14 However, to our knowledge, this autoantibody has not been investigated systematically in more common forms of brainstem and spinal cord hyperexcitability, including classic and variant SMS.
METHODS
This study was approved by the institutional review boards of Mayo Clinic (Rochester, Minnesota) and the Hospital Clinic (University of Barcelona, Spain).
DETECTION OF GlyRα1 AUTOANTIBODIES (UNIVERSITY OF BARCELONA)
HEK293 cells were transfected, as reported previously,15 with a plasmid (pRK5) containing the human GlyRα1 subunit cDNA, or control plasmid without insert. After culturing for 24 hours posttransfection, the cells were fixed and permeabilized for 10 minutes (4% paraformaldehyde and 0.3% Triton X-100), washed in phosphate-buffered saline, and held overnight at 4°C with patient serum (1:40) or cerebrospinal fluid (CSF; 1:5) plus a monoclonal mouse IgG specific for a noncompeting epitope of GlyRα1 (residues 96–105; Synaptic Systems SYSY; diluted 1:500). Bound human and mouse IgGs were demonstrated after holding 1 additional hour at 37°C with Alexa Fluor–conjugated IgGs specific for human IgG (488) and mouse IgG (594), respectively, both diluted 1:1000 (molecular probes; Invitrogen). Resulting images were visualized by fluorescence microscope and captured using Zeiss Axiovision software (Zeiss).
PATIENTS
Medical records were reviewed at Mayo Clinic. Coded specimens of serum and CSF were tested for GlyRα1-IgG. Interpreting serologists at the University of Barcelona were blinded to clinical diagnosis. Patients from the following groups were tested.
SMS PHENOTYPE
Serum samples collected during 25 years (Mayo Clinic, 1984–2011; Figure 1) were from 81 patients of a previously described cohort with classic SMS and phenotypically similar disorders5 for whom at least 1 sample was available from the time of diagnosis (80 serum samples and 14 CSF specimens). Mayo Clinic's computerized diagnostic index was searched for patient diagnoses (1984–2011) where neurologic hyperexcitability was the predominant phenotype: stiff-man syndrome; stiff-limb syndrome; stiff-person syndrome; and progressive encephalomyelitis, rigidity, myoclonus, and hyperekplexia. The diagnosis of SMS or PERM was made by the treating physician. Patients were diagnosed as having PERM if they had a rapidly progressive encephalomyelopathy accompanied by whole-body stiffness, spasms, and myoclonus. We retrospectively classified SMS as classic or variant. Patients were classified as having classic SMS if lower extremity and lumbar stiffness and spasms were present, and variant SMS if symptoms were restricted to either axial or extremity muscles or upper body muscles. Patients with hyperekplexia had isolated exaggerated startle in response to tactile or auditory stimuli.
Figure 1.
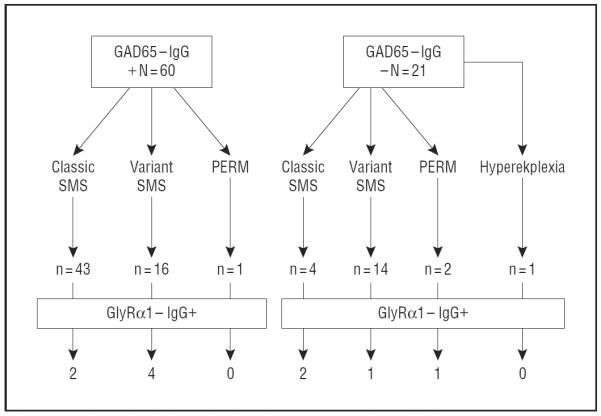
Graph showing that 81 patients with stiff-man syndrome (SMS) phenotype (classic, variant, progressive encephalomyelitis with rigidity and myoclonus [PERM], and hyperekplexia) were tested for glycine receptor α1 (GlyRα1)-IgG.
Clinical, serologic, CSF, neurophysiological, radiologic, treatment, and outcome data were recorded from the medical records of all patients. Electrophysiological studies consisted of multichannel surface electromyography recording over the right orbicularis oculi, sternocleidomastoid, biceps, abductor pollicis brevis, thoracic paraspinals, lumbar paraspinals, anterior tibialis, gastrocnemius, and soleus muscles. Auditory startle reflexes (pattern and habituation of motor responses) were evaluated using binaural 105 dB intensity stimuli 1 minute apart during 3 to 5 trials.16 Exteroceptive responses were determined by electrically stimulating the medial plantar nerve.17 Concentric needle studies were performed in some patients on the lumbar paraspinal or limb muscles.
CONTROL GROUPS
A total of 100 patients (81 serum samples and 46 CSF specimens) represented 5 groups: (1) other autoimmune neurologic disorders (n=13 [4 with encephalopathy, 3 with seizure disorder, 2 with neuropathy, 1 with cerebellar ataxia, 1 with brainstem disorder, 1 with myelopathy, and 1 with neuromyelitis optica]; 6 were GAD65-IgG positive); (2) idiopathic demyelinating diseases (n=12 [10 with multiple sclerosis and 2 with monophasic optic neuritis]); (3) miscellaneous neurologic disorders (n=15 [2 with myelopathy; 2 with fibromyalgia; and 1 each with indeterminate spells, dementia, ataxia, seizure disorder, optic atrophy, central nervous system sarcoidosis, neuropathy, hereditary hyperekplexia, progressive multifocal leukoencephalopathy, Huntington disease, and viral meningoencephalitis]); (4) paraneoplastic or idiopathic autoimmune encephalitis (n=40) and the following antibodies: N-methyl-D-aspartate receptor antibody (n=10), γ-aminobutyric acid B receptor antibody (n=5), leucine-rich, glioma-inactivated 1 (n=10), and ANNA types 1 (anti-Hu; n=10) and 2 (anti-Ri); and (5) serum samples from 20 healthy subjects also were tested.
STATISTICAL ANALYSIS
We compared GlyRα1-IgG–positive and GlyRα1-IgG–negative patients with SMS phenotype with respect to age at onset, distribution of stiffness and spasms, and coexisting neurologic disorders using Fisher exact test or Wilcoxon rank sum test where appropriate (JMP version 8.0; SAS Institute).
RESULTS
Ten of the 81 patients with SMS phenotype (12%) were GlyRα1-IgG positive: 5 had variant SMS, 4 had classic SMS, and 1 had PERM (Figure 1 and Figure 2). Glycine receptor α1–IgG was detected in 6 of 80 available serum samples (7.5%) and in 6 of 14 available CSF specimens (43%). Glycine receptor α1–IgG was detected in 6 of 60 serum samples that were GAD65-IgG positive (10%) and in 4 of 21 serum samples that were GAD65-IgG negative (19%). Glycine receptor α1–IgG was not detected in any amphiphysin-IgG–positive serum. The frequency of GlyRα1-IgG was not significantly greater among patients with classic SMS (4/45), variant SMS, or PERM (6/35; P=.32). One (patient 11) of the 80 neurologic control patients' serum samples (1.2%) was positive for GlyRα1-IgG; CSF was negative. None of the 20 healthy subjects' serum samples was GlyRα1-IgG positive.
Figure 2.

Glycine receptor α1 (GlyRα1)–IgG demonstrated in HEK293 cells expressing the human GlyRα1 subunit. The reactivity of serum from 2 representative patients with HEK293 cells expressing the human GlyRα1 subunit is shown in panels A and D. G, Serum from a control patient lacks reactivity with transfected cells. Panels B, E, and H show the reactivity with a commercial GlyRα1 monoclonal antibody. The immunostaining colocalizes with that of patient antibodies (C and F, but not I). Scale bar=20 μm.
GlyRα1-IgG–POSITIVE PATIENTS
The 11 patients in whom GlyRα1-IgG was detected (10 cases, 1 control subject) are described in Table 1 and Table 2 (6 were women). The median age at symptom onset was 43 years (range, 5–69 years). Ethnicities were white (n=9); African American (n=1), and unknown (n=1).
Table 1.
Demographic, Clinical, Electrophysiological, and Radiologic Findings in GlyRα1-IgG–Positive Patients
| Patient No./Sex/Age at Symptom Onset, y | Symptom Duration at Evaluation, y | Symptoms | Distribution of Stiffness | Other Neurologic Signs | Neurophysiologic Findings | Head/Spine MRI | Phenotype |
|---|---|---|---|---|---|---|---|
| 1/M/29 | 13 | Jerking right foot; spasms in lower back; severe startle; fear of falling | RLE; LLE; lumbar spine | Exaggerated acoustic startle response | Normal | Classic SMS | |
| 2/F/44 | 23 | Spasms in lower back; pelvic thrusting; walking difficulties; falls | RUE; LUE; RLE; LLE; lumbar spine | Brisk DTRs; stiff-legged gait | Exaggerated acoustic startle response | Normal | Classic SMS |
| 3/F/5 | 50 | Toe walking; thoracolumbar spasms; pain and scoliosis; gait freezing; had to give up work | RLE; LLE; lumbar spine | Thoracic-lumbar scoliosis | Normal (taking diazepam, 60 mg/d) | Normal | Classic SMS |
| 4/M/62 | 1.5 | Acute upper limb jerking followed fall on ice; rapid progression to stiffness and jerks of all limbs | Severe stiffness and spasms of all extremities; severe flexed posture on standing | Brisk DTRs; myoclonus UEs; impaired upgaze | Not done | Moderate generalized cerebral and cerebellar atrophy | PERM |
| 5/M/37 | 10 | Progressive stiffness in right arm and left leg; intermittent hemiparetic posturing; had to give up playing basketball and golf | RUE; LLE | Brisk DTRs, R Babinski+ | Increased insertional activity and poor relaxation of triceps, tibialis anterior, and medial gastrocnemius | Normal | Variant SMS |
| 6/M/14 | 3 | Spasms and pain in thoracic and lumbar regions, intermittent at first and then continuous; athlete (football), had to give up all physical activity | Thoracic and lumbar spine only | Thoracic-lumbar scoliosis | Continuous motor unit activity at the midthoracic level | Normal | Variant SMS |
| 7/F/45 | 1 | Stiffness and spasms in right leg; mild symptoms | RLE | Bilateral extensor plantars | Exaggerated acoustic startle response and exteroceptive reflexes | Normal | Variant SMS |
| 8/F/69 | 4 | Stiffness and spasms in right leg, severe balance difficulties as a result; required a walker to ambulate | RLE | Hammer toes; high arch R foot | Exaggerated exteroceptive response in R leg only | Normal | Variant SMS |
| 9/F/43 | 22 | Spasms in whole body, lower back, and lower extremities; during attacks of spasms, could not do any activities of daily living | Lumbar spine | Brisk DTRs | Continuous motor unit activity left medial and lateral gastrocnemius only (when receiving clonazepam, 3 mg/d, and baclofen, 15 mg/d) | Normal | Classic SMS |
| 10/F/34 | 2 | Stiffness in all limbs; spasms in both legs; legs became stiff and difficult to move; leg spasms while driving | RUE; LUE; RLE; LLE | Brisk DTRs | Not done | Normal | Variant SMS |
| 11/M/42 | 1 | Progressive vision loss; had to stop driving (bus driver by profession) | None | Not done | Nonenhancing T2-signal abnormalities in the bilateral superior colliculi, superior cerebellar peduncle and left brachium pontis, and bilateral occipital white matter | Optic atrophy |
Abbreviations: DTRs, deep tendon reflexes; F female; GlyRαi, glycine receptor α1; LEs, lower extremities; LLE, left lower extremity; LUE, left upper extremity; M, male; MRI, magnetic resonance imaging; PERM, progressive encephalomyelitis with rigidity and myoclonus; R, right; RLE, right lower extremity; RUE, right upper extremity; SMS, stiff-man syndrome; UEs, upper extremities.
Table 2.
Oncologic, Immunologic, Treatment, and Outcome Data for 11 GlyRα1-IgG–Positive Patients
| Patient No. | Cancer Detected | Other Autoimmune History | GAD65-IgG, nmol/La | CSFb,c | GlyRα1-IgG Serum, 1:40 | GlyRα1-IgG CSF, 1:5 | Treatment | Outcome | Follow-up Period, mo |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Hodgkin lymphoma | 0.00 (serum) | Not tested | + | NA | Chemotherapy;d IV methylprednisolone; IVIg, 0.4 g/kg, twice weekly for 6 wk | Improved considerably with all treatments; IVIg most beneficial; mild residual spasms in back and lower extremities | 12 | |
| 2 | Addison disease; type 1 DM; hypothy-roidism | 544 (serum); 29.1 (CSF) | Normal | − | + | Baclofen, 10 mg, 3 times daily | Moderate improvements; residual low back spasms (further dose escalation of baclofen and diazepam trial recommended) | 48 | |
| 3 | Thyroid disorder | 12.6 (serum); 0.15 (CSF) | Normal | − | + | IVIg, 0.4g/kg, daily for 3 d, weekly for 6 wk, then gradually tapered over 1 y; azathioprine, 2.5 mg/kg/d, initiated simultaneously and continued as monotherapy after IVIg discontinued | Near normal after treatment for 1 y; no symptoms; mildly exaggerated lumbar lordosis | 24 | |
| 4 | 0.00 (serum) | Not tested | + | NA | IV methylprednisolone daily for 5 d | Substantial improvements after treatment for 5 d | 3 | ||
| 5 | 260 (serum); 27 (CSF) | Protein level, 67 mg/dL; WBC count, 1 /μL; OCBs; IgG index/synthesis rate normal | − | + | Prednisone, 60 mg/d, tapered after 3 mo; azathioprine, 150 mg/d; baclofen, 10 mg, 3 times daily | No symptoms or signs after treatment for 10 mo | 10 | ||
| 6 | 0.06 (serum); 0.00 (CSF) | Normal | − | + | Prednisone, 60 mg/d, and plasma exchange every other day for 10 d (5 treatments) | Substantial improvements in spasms following plasma exchange; symptoms worsened with prednisone alone | 12 | ||
| 7 | Type 1 DM; thyroid disorder | 56.5 | Not tested | + | NA | Diazepam, 5 mg, 3 times daily | Near normal; mild stiff-legged gait | 36 | |
| 8 | Addison disease; type 1 DM; thyroid disease | 39.1 (serum); 3.42 (CSF) | Protein level, 59 mg/dL; WBC count, 5/μL; IgG index/synthesis rate, normal; OCBs, 6 | − | + | IVIg, 1g/kg, twice monthly for 2 mo; diazepam, 10 mg, twice daily | Mild improvements in spasms with either treatment; still required walker to ambulate | 60 | |
| 9 | Vitiligo | 0.00 (serum); 3.42 (CSF) | Normal | + | + | Clonazepam, 1 mg, 3 times daily | Spasms improved substantially in initial 6 mo, then worsened (prior to Mayo Clinic evaluation) | 0 | |
| 10 | 0.00 (serum) | Not tested | + | NA | NA | NA | 0 | ||
| 11 | 0.00 (serum) | Protein level, 47 mg/dL; WBC count, 2 /μL; OCBs, 11; IgG index, 1.38; IgG synthesis rate, 15.11 | + | − | IV methylprednisolone 1 g daily for 5 d followed by weekly for 6 wk | Improved visual acuity (20/400 to 20/150) | 0 |
Abbreviations: CSF, cerebrospinal fluid; DM, diabetes mellitus; GAD65, glutamic acid decarboxylase 65; GlyRα1, glycine receptor α1; IV, intravenous; IVIg, intravenous immune globulin; NA, not available; OCBs, oligoclonal bands (CSF exclusive); WBC, white blood cell.
Normal range for GAD65-IgG, 0.00–0.02 nmol/L.
Protein, cell count, OCBs, IgG index, and synthesis rate evaluated in all who were tested.
Normal ranges: protein level, 0–45 mg/dL; WBC count, ≤5/μL (to convert to ×109 per liter, multiply by 0.0001); IgG index, ≤0.85; IgG synthesis rate, ≤12 mg/24 h; and OCBs (CSF exclusive), <4.
Adriamycin, bleomycin, vinblastine, and dacarbazine therapy.
Among patients with SMS phenotype, age at symptom onset was not significantly different in seropositive and seronegative patients (Wilcoxon rank sum test; P = .35). Median follow-up from diagnosis was 12 months (range, 0–60 months). Response rates to treatment with benzodiazepines were similar for seropositive patients (6 of 7 improved) and seronegative patients (62 of 68 improved). Among patients for whom we had immunotherapy data (treatments were heterogenous), substantial improvements were noted in 5 of 6 seropositive patients compared with 7 of 25 seronegative patients (P = .02).
ILLUSTRATIVE PATIENTS
Patient 1
At age 29 years, this man developed jerking of his right foot that subsequently spread to his lower back. On examination, he had stiffness and superimposed spasms of the lumbar paraspinal muscles and lower extremities. Despite being seronegative for GAD65 Ab, he was diagnosed as having classic SMS because the clinical findings were characteristic. He improved considerably with diazepam therapy. At age 41 years, he was diagnosed as having stage IV Hodgkin lymphoma. Within 1 month of starting adriamycin, bleomycin, vinblastine, and dacarbazine therapy, stiffness worsened and spasms progressed to affect the whole body; he also developed severe startle, fear of falling, and anxiety. Electrophysiological studies were consistent with SMS. Symptoms improved periodically during the ensuing 12 months of treatment with methylprednisolone, prednisone, and intravenous immune globulin. One year later, lymphoma was in remission and the neurologic symptoms had improved considerably.
Patient 3
A 55-year-old woman with autoimmune thyroid disease reported that scoliosis and toe walking were noted at age 5 years. From teen years onwards, she experienced gait-freezing spells. While in her 20s, she noted anxiety and easy startling. Stiffness and spasms in the low back and lower extremities began when she was in her 50s. Examination revealed exaggerated lumbar lordosis, stiffness, and spasms of the lumbar region and lower extremities. She was diagnosed as having classic SMS. She was GAD65-IgG positive. After treatment for 1 year with azathioprine and prednisone, examination revealed mildly exaggerated lumbar lordosis.
Patient 6
A 17-year-old boy reported that pain and spasms restricted to the thoracic and lumbar regions began at age 14 years. Examination revealed slight scoliosis and involuntary spasms of the thoracic paraspinal muscles. Electromyograph demonstrated continuous motor unit activity in those muscles. He was diagnosed as having variant SMS, with restricted involvement of the back muscles. He was GAD65-IgG low positive. Mild improvement was observed with diazepam therapy. Immunotherapy conferred significant improvement but relapse was rapid after discontinuing plasma exchange.
Patient 11
A 42-year-old man experienced bilateral blurring of vision, which worsened progressively during 1 year then stabilized. He experienced no stiffness, spasms, or other motor symptoms. Eye examination 2 years after symptom onset revealed bilateral optic atrophy without observable retinal abnormalities; visual acuity was 20/400 bilaterally. Head magnetic resonance imaging demonstrated nonenhancing T2-signal abnormalities in the superior colliculi, superior cerebellar peduncles, left brachium pontis, and bilateral occipital white matter. After a 6-week trial of corticosteroid therapy, the patient reported improved vision; visual acuity was 20/200 (right eye) and 20/150 (left eye).
COMMENT
The current study revealed that 12% of patients with an acquired SMS phenotype, with and without GAD65-IgG, were seropositive for GlyRα1-IgG. Phenotypes included disorders indistinguishable clinically and electrophysiologically from classic SMS, variant SMS, and PERM. Thus, in clinical practice, serologic tests for GlyRα1-IgG complement tests for GAD65-IgG and amphiphysin-IgG in aiding identification of autoimmune brainstem/spinal cord hyperexcitability disorders that are potentially immunotherapy responsive. Cerebrospinal fluid testing for GlyRα1-IgG was more sensitive than serum testing. Possible explanations for this finding include intrathecal synthesis of antibody, but there are also differences in testing dilutions for serum and CSF specimens; CSF is tested diluted 1:5, while serum is diluted 1:40 before testing, to reduce nonorgan specific antibody binding to cells. Patients who were GlyRα1-IgG positive in both serum and CSF did not differ clinically from patients with GlyRα1-IgG detectable in CSF only. As recommended for N-methyl-D-aspartate receptor–IgG, it would seem prudent in clinical evaluation to test serum and CSF simultaneously for GlyRα1-IgG.18
A neoplasm (Hodgkin lymphoma) was identified in 1 of 10 GlyRα1-IgG–positive patients. Cancers reported to date in GlyRα1-IgG–positive patients include thymoma13 and lung cancer.19 Similar to a previously reported seropositive patient who had thymoma,13 our patient with Hodgkin lymphoma improved neurologically after cancer treatment. The frequency of cancer accompanying GlyRα1-IgG remains to be determined.
The greater frequency of substantial clinical improvement in GlyRα1-IgG–seropositive SMS phenotype patients suggests that GlyRα1-IgG may predict immunotherapy responsiveness. However, the heterogeneity of immunotherapy regimens used and the retrospective ascertainment of responses from medical record review preclude definitive conclusions. Prospective studies could clarify this question. Although the pathogenicity of GlyRα1-IgG is yet unproven, immunotherapy responsiveness has been documented for other neurologic disorders characterized serologically by IgGs targeting neuronal cell surface proteins.15 In contrast, GAD65-IgG recognizes a cytoplasmic-facing synaptic vesicle protein.2 The IgGs that are specific for cytoplasmic and nuclear autoantigens are considered surrogate markers of inflammatory organ-specific autoimmune disorders mediated by cytotoxic T cells, a hypothesis supported by autopsy findings in some patients with SMS.20 Immunotherapy responses in GAD65-IgG–positive patients with SMS phenotype are variable and posttherapy normalization is uncommon.5,21
As recognized in early reports,8,10–14 GlyRα1-IgG was highly specific for brainstem/spinal cord hyperexcitability disorders. However, our detection among 80 disease specificity (neurologic) control samples of a single seropositive patient with immunotherapy-responsive progressive vision loss and inflammatory CSF, in the absence of SMS phenotype, suggests that the spectrum of GlyRα1 receptor autoimmunity extends beyond the spinal cord and brainstem. Of pertinence, glycine and γ-aminobutyric acid are the major inhibitory neurotransmitters of human retina.22 Consistent with the clinical disorders we encountered with GlyRα1-IgG, in situ hybridization studies have revealed that GlyRα1 messenger RNA is largely restricted to the retina, brainstem, and spinal cord.22 Systematic investigation of the neuroophthalmologic spectrum of glycine receptor autoimmunity is needed.
Acknowledgments
Financial Disclosures: Dr McKeon has received research support from the Guthy Jackson Charitable Foundation. Dr Martinez-Hernandez received research grant FI08/00285 from Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, Spain. Dr Lancaster has received research support from Talecris Biotherapeutics, Lundbeck Inc, and the Dana Foundation. Dr Harvey received grant G0601585 from the Medical Research Council and grant 1966 from Action Medical Research. Dr Pittock is a named inventor on patents (12/678 350 filed 2010 and 12/573 942 filed 2008) that relate to functional aquaporin-4 (AQP4)/neuromyelitis optica (NMO)–IgG assays and NMO-IgG as a cancer marker, and he receives research support from Alexion Pharmaceuticals Inc, the Guthy-Jackson Charitable Foundation, and the National Institutes of Health. Dr Lennon is a named inventor on a patent (7101679 issued 2006) relating to AQP4 antibodies for the diagnosis of NMO and receives royalties for this technology. She is a named inventor on patents (12/678 350 filed 2010 and 12/573 942 filed 2008) related to functional AQP4/NMO-IgG assays and NMOIgG as a cancer marker and receives research support from the Guthy-Jackson Charitable Foundation and National Institutes of Health. Dr Dalmau is named inventor on patents (6 387 639 and 7 972 796) related to the of Ma2 and NMDAR as autoantibody diagnostic tests and has patents filed for the use of gamma-aminobutyric acid receptor and leucine-rich glioma-inactivated 1 as diagnostic tests. He receives royalties from the editorial board of Up-To-Date and license fee payments from Euroimmun AG. Dr Dalmau receives research support from Euroimmun AG. His current work has been partially funded by the grants RO1NS077851 and RO1MH094741 from the National Institutes of Health and Fundació la Marató TV3 and Fondo de Investigaciones Sanitarias (FIS, PI11/01780)/Fondo Europeo de Desarrollo Regional (FEDER).
Footnotes
Author Contributions: Study concept and design: McKeon, Pittock, Lennon, and Dalmau. Acquisition of data: McKeon, Martinez-Hernandez, Lancaster, Matsumoto, McEvoy, and Dalmau. Analysis and interpretation of data: McKeon, Martinez-Hernandez, Harvey, Pittock, and Dalmau. Drafting of the manuscript: McKeon and Dalmau. Critical revision of the manuscript for important intellectual content: McKeon, Martinez-Hernandez, Lancaster, Matsumoto, Harvey, McEvoy, Pittock, Lennon, and Dalmau. Statistical analysis: McKeon. Obtained funding: Lancaster, Harvey, and Dalmau. Administrative, technical, and material support: Martinez-Hernandez, Harvey, and Dalmau. Study supervision: McKeon and Dalmau.
REFERENCES
- 1.Moersch FP. Woltman HW. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome): report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin. 1956;31(15):421–427. [PubMed] [Google Scholar]
- 2.Solimena M, Folli F, Denis-Donini S, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med. 1988;318(16):1012–1020. doi: 10.1056/NEJM198804213181602. [DOI] [PubMed] [Google Scholar]
- 3.Brown P, Rothwell JC, Marsden CD. The stiff leg syndrome. J Neurol Neurosurg Psychiatry. 1997;62(1):31–37. doi: 10.1136/jnnp.62.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whiteley AM, Swash M, Urich H. Progressive encephalomyelitis with rigidity. Brain. 1976;99(1):27–42. doi: 10.1093/brain/99.1.27. [DOI] [PubMed] [Google Scholar]
- 5.McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230–238. doi: 10.1001/archneurol.2011.991. [DOI] [PubMed] [Google Scholar]
- 6.Rosin L, DeCamilli P, Butler M, et al. Stiff-man syndrome in a woman with breast cancer: an uncommon central nervous system paraneoplastic syndrome. Neurology. 1998;50(1):94–98. doi: 10.1212/wnl.50.1.94. [DOI] [PubMed] [Google Scholar]
- 7.Butler MH, Hayashi A, Ohkoshi N, et al. Autoimmunity to gephyrin in stiff-man syndrome. Neuron. 2000;26(2):307–312. doi: 10.1016/s0896-6273(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291–1292. doi: 10.1212/01.wnl.0000327606.50322.f0. [DOI] [PubMed] [Google Scholar]
- 9.Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56(1):303–309. doi: 10.1016/j.neuropharm.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 10.Mas N, Saiz A, Leite MI, et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 2010;82(12):1399–1401. doi: 10.1136/jnnp.2010.229104. [DOI] [PubMed] [Google Scholar]
- 11.Turner MR, Irani SR, Leite MI, Nithi K, Vincent A, Ansorge O. Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology. 2011;77(5):439–443. doi: 10.1212/WNL.0b013e318227b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piotrowicz A, Thümen A, Leite MI, Vincent A, Moser A. A case of glycine-receptor antibody-associated encephalomyelitis with rigidity and myoclonus (PERM): clinical course, treatment and CSF findings. J Neurol. 2011;258(12):2268–2270. doi: 10.1007/s00415-011-6078-x. [DOI] [PubMed] [Google Scholar]
- 13.Clerinx K, Breban T, Schrooten M, et al. Progressive encephalomyelitis with rigidity and myoclonus: resolution after thymectomy. Neurology. 2011;76(3):303–304. doi: 10.1212/WNL.0b013e318207b008. [DOI] [PubMed] [Google Scholar]
- 14.Iizuka T, Leite MI, Lang B, et al. Glycine receptor antibodies are detected in progressive encephalomyelitis with rigidity and myoclonus (PERM) but not in saccadic oscillations. J Neurol. 2012;259(8):1566–1573. doi: 10.1007/s00415-011-6377-2. [DOI] [PubMed] [Google Scholar]
- 15.Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto JY, Caviness JN, McEvoy KM. The acoustic startle reflex in stiff-man syndrome. Neurology. 1994;44(10):1952–1955. doi: 10.1212/wnl.44.10.1952. [DOI] [PubMed] [Google Scholar]
- 17.Meinck HM, Ricker K, Conrad B. The stiff-man syndrome: new pathophysiological aspects from abnormal exteroceptive reflexes and the response to clomipramine, clonidine, and tizanidine. J Neurol Neurosurg Psychiatry. 1984;47(3):280–287. doi: 10.1136/jnnp.47.3.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108–1110. doi: 10.1212/WNL.0b013e318211c379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vincent A, Leite IM, Waters P, Jacobi C, Becker CM, Meinck HM. Glycine receptor antibodies in progressive encephalomyelitis with rigidity, hyperekplexia, and stiff-person syndrome. Ann Neurol. 2009;66:S50. [Google Scholar]
- 20.Holmøy T, Skorstad G, Røste LS, Scheie D, Alvik K. Stiff person syndrome associated with lower motor neuron disease and infiltration of cytotoxic T cells in the spinal cord. Clin Neurol Neurosurg. 2009;111(8):708–712. doi: 10.1016/j.clineuro.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345(26):1870–1876. doi: 10.1056/NEJMoa01167. [DOI] [PubMed] [Google Scholar]
- 22.Eggers ED, Lukasiewicz PD. Multiple pathways of inhibition shape bipolar cell responses in the retina. Vis Neurosci. 2011;28(1):95–108. doi: 10.1017/S0952523810000209. [DOI] [PMC free article] [PubMed] [Google Scholar]