

Abstract
The unregulated activity of inteins during expression and consequent side reactions during work-up limits their widespread use in biotechnology and chemical biology. Therefore, we exploited a mechanism-based approach to regulate intein autocatalysis for biotechnological application. The system, inspired by our previous structural studies, is based on reversible trapping of the intein’s catalytic cysteine residue through a disulfide bond. Using standard mutagenesis, the disulfide trap can be implemented to impart redox control over different inteins and for a variety of applications both in vitro and in Escherichia coli. Thereby, we first enhanced the output for bioconjugation in intein-mediated protein ligation, also referred to as expressed protein ligation, where precursor recovery and product yield were augmented fourfold to sixfold. Second, in bioseparation experiments, the redox trap boosted precursor recovery and product yield twofold. Finally, the disulfide-trap intein technology stimulated development of a novel bacterial redox sensor. This sensor reliably identified hyperoxic E. coli harboring mutations that disrupt the reductive pathways for thioredoxin and glutathione, against a background of wild-type cells.
Keywords: redox regulation, expressed protein ligation, bioseparations, redox sensor
Introduction
Autocatalytic protein domains called inteins occur as insertion elements in proteins that self-splice after expression (Kane et al., 1990). In protein splicing, an intein cleaves itself from adjacent N- and C-terminal protein segments, called exteins, and ligates the newly exposed termini (Paulus, 2000; Perler, 1998; Fig. 1a). Inteins can also self-splice from heterologous proteins. That promiscuity has enabled the development of various intein-based techniques for cellular and chemical biology (Cheriyan and Perler, 2009; Vila-Perello and Muir, 2010). The efficiency of these methods is, however, limited by the continuous catalytic activity of inteins. This can lead to wasteful side reactions during expression in vivo, and during the ensuing workup, which not only depletes the precursor substrate but also complicates analysis of the end-product. Accordingly, imparting control over intein activity has become an important goal in the field. The majority of approaches utilize intein mutagenesis. Recent examples include introducing photo-reactive groups (Binschik et al., 2011; Vila-Perello et al., 2008), or destabilizing mutations (Zeidler et al., 2004), so that splicing occurs in response to exposure to UV light or a temperature shift, respectively. Other proof-of-principle studies have made use of “split” inteins that engage in bimolecular splicing reactions upon conditional reassembly (Tyszkiewicz and Muir, 2008).
Figure 1.

General scheme of protein splicing and intein redox trap. a: Scheme for protein splicing and intein redox trap. Top: splicing occurs spontaneously to generate ligated exteins (blue and green) and to free the intein (red), with the catalytic cysteine at its N terminus. Bottom: a disulfide bond between intein (red) and N-extein (blue) blocks catalytic activity; a reducing agent (e-) breaks the disulfide bond, and self-splicing ensues. b: Extreme stability of the DnaE intein precursor in the absence of added reducing agent (TCEP). Intein precursor is stable in Tris buffer (0.05 M, pH 8) at 22°C in the absence of TCEP (top gel) and labile in the presence of TCEP (bottom gel). The identity of the species that migrates more slowly than the precursor and appears after 48 h is not known. c: Kinetics of precursor disappearance. The plot was constructed using data from panel (b) and shows that a reducing agent (TCEP) is required for efficient precursor processing.
Extein mutagenesis provides an alternative strategy to modulate intein activity (Amitai et al., 2009). Recently, we exploited extein mutagenesis to render the fused Synechocystis PCC6803 (Ssp) DnaE intein responsive to changing redox conditions (Callahan et al., 2011). Redox-responsiveness was imparted by a disulfide bond that we engineered between the intein’s catalytic cysteine and a cysteine in the flanking N-extein sequence (Fig. 1a). The paired cysteines were separated by proline and glycine, mimicking the active site of thioredoxin. This intein flanked by the Cys–Pro–Gly tripeptide, CPGCDnaE, catalyzed self-cleavage at its N-terminal splicing junction at typical rates during expression in the reducing environment of wild-type Escherichia coli and in buffer containing a reducing reagent. However, CPGCDnaE self-cleavage was substantially inhibited when expressed in the “origami” strain of E. coli, which lacks two oxidoreductases (ΔtrxB and ΔgorA) and has an oxidizing cellular environment (Stewart et al., 1998), as well as in buffer lacking a reducing agent. We defined the mode of redox-responsiveness crystallographically, and identified naturally occurring inteins that may function as adaptive post-translational switches, aside from the customary role as molecular parasites (Callahan et al., 2011).
Here, we first explore the portability of the redox trap through experiments with a ribonucleotide reductase intein from Methanobacter thermotropicum. We thus show that this simple regulatory mechanism can be applied to control the self-cleavage activity of other inteins. We then explore the potential utility of CPGCDnaE for intein biotechnology. Three features of the redox control mechanism are advantageous for applied uses: (1) the disulfide/dithiol switch is non-disruptive with respect to intein structure, solubility, and activity; (2) installation of the redox-switch involves standard mutagenesis, and is therefore straightforward to accomplish; and (3) the strength of the redox trap can be modulated in living cells through specific mutations and in vitro by redox-active small-molecules. Here, we apply CPGCDnaE to two signature intein-based biotechnologies: protein labeling through intein-mediated protein ligation (expressed protein ligation, EPL) and protein purification. In addition, we capitalize on the redox-responsiveness of CPGCDnaE to develop a novel biosensor of oxidative stress in bacteria.
Results and Discussion
The Redox Trap Can Function With Another Intein
With the Cys-3-to-Cys1 disulfide trap, the DnaE precursor persisted over 2 weeks at 22°C in Tris buffer (0.05 M, pH 8) lacking added reducing agents (Fig. 1b and c). We wished to evaluate the generality of this regulatory mechanism by establishing whether the Cys–Pro–Gly–Cys sequence could function as a redox switch of an intein other than with this prototypic Ssp DnaE intein. Since the trap involves mutagenesis of extein residues only, effects on intein structure and function were predicted to be minimal. For these experiments, we utilized the Methanothermobacter thermautotrophicus ribonucleotide reductase (Mth RnR1) intein. This intein is notable for its small size, at only 134 amino acids, making it the smallest self-splicing domain among annotated intein sequences (Perler, 2002), and giving it a potential advantage from the standpoint of biotechnology. Indeed the Mth RnR1 intein has been utilized as a tool for bioseparations and as a catalyst for the first successful in vitro ligation of large, bacterially expressed proteins (Evans et al., 1999). To explore the possibility of creating a redox-responsive Mth RnR1 intein construct, CPGCRnR1, we replaced the intein’s native N-extein sequence with Cys–Pro–Gly. To inhibit C-terminal cleavage and splicing and to isolate N-cleavage activity, the C-terminal asparagine residue was mutated to alanine (Ia). An identical construct but with an alanine at −3, APGCRnR1, was prepared as a control. The resulting constructs were then inserted into a FRET-active reporter, patterned on a similar strategy for the DnaE intein (Amitai et al., 2009).
The reporter, C-Ia-Y, includes cyan fluorescent protein (CFP) fused to the N terminus of CPGCRnR1 (Ia), and yellow fluorescent protein (YFP) fused to the intein’s C terminus. The intact C-Ia-Y is FRET-active, but loses FRET activity upon N-terminal cleavage (C + Ia-Y). An equivalent construct that was unable to form the intein–extein disulfide, APGCRnR1, served as a FRET-active control.
To study redox sensitivity in vitro, CPGCRnR1 and APGCRnR1 were incubated in reducing and non-reducing Tris buffer (0.05 M, pH 8) at 23°C with added hydroxyl-amine (0.1 M) or DTT (0.02 M) to induce N-terminal cleavage. Activity of the control, APGCRnR1, remained high in all conditions (Fig. 2, lower gels, and blue symbols in plots). In comparison, CPGCRnR1 appeared high in reducing buffer with hydroxylamine or DTT as the cleavage agent, but the precursor remained inert in non-reducing buffer (Fig. 2, upper gels, and red symbols in plots). This effect of redox conditions on the cleavage kinetics of the CPGCRnR1 intein is consistent with the behavior of the CPGCDnaE intein (Callahan et al., 2011).
Figure 2.

Redox trapping of RnR1 intein in vitro and E. coli. a: Effect of redox conditions on in vitro cleavage kinetics of the RnR1 intein precursor. Gels show conversion of precursor, C-Ia-Y into Ia-Y, in Tris buffer containing hydroxylamine (HA), HA and TCEP, or dithiothreitol (DTT). Precursor and product with CPGCRnR1 (top gels, C) and APGCRnR1 (bottom gels, A) were detected by in-gel fluorescence (excitation 488 nm, emission 580 nm). b: Plots of precursor disappearance using representative data of (a), shows that reducing conditions are necessary for efficient processing of CPGCRnR1 precursor. c: Effect of cellular redox conditions on the self-cleavage of CPGCRnR1 in vivo. Activity of CPGCRnR1 and APGCRnR1 inteins was determined by FRET signal in culture lysates prepared at 7 h after induction of protein expression in E. coli DHB4 (left), AD494 (middle) and origami (right). Elevated FRET CPGCRnR1 in origami indicates precursor accumulation. Error bars, SD (n >5).
We next tested CPGCRnR1 for redox-regulated processing in living cells. For these experiments, we compared processing CPGCRnR1 and APGCRnR1 in the oxidizing E. coli origami strain (ΔtrxB and ΔgorA), and in the isogenic parental strains, DHB4 and AD494 (ΔtrxB). FRET signal from cultures expressing CPGCRnR1 and APGCRnR1 precursor were nearly identical in AD494 and DHB4, indicating similar cleavage activity (Fig. 2c). However, the FRET signal from intact CPGCRnR1 precursor increased to 1.1 in the hyperoxic origami strain from FRET values of ~0.65 in AD494 and DHB4, and twice that of the FRET signal of APGCRnR1 precursor expressed in parallel (Fig. 2c, green). Together with the in vitro experiments, the inhibition of CPGCRnR1 activity in the origami strain indicates redox responsiveness and suggests that engineering a Cys-3-to-Cys1 disulfide bond is a portable strategy for intein regulation.
Intein Redox Trap Supports Enhanced EPL
EPL attracts substantial interest because of its application to labeling and semi-synthesis of proteins, irrespective of their molecular weight (Evans et al., 1998, 1999; Muir et al., 1998). EPL practitioners have leveraged knowledge of organic chemistry and intein mechanism to prepare protein derivatives equipped with myriad labels and probes (Clark et al., 2010; Muir, 2008; Seyedsayamdost et al., 2007; Smith et al., 2011). One pitfall with EPL, however, is that a significant fraction of the reactive intein precursor required for in vitro EPL reactions is consumed by spontaneous hydrolysis during bacterial expression. Noting that the hyperoxic origami E. coli strain can restrict activity of trapped precursors (CPGCDnaE and CPGCRnR1) during expression, without compromising activity in vitro, we suspected that trapped inteins could serve as efficient EPL biocatalysts.
We therefore prepared a potential EPL precursor by fusing the C terminus of a target protein (TP) to the N terminus of CPGCDnaE intein, TP-CPGCDnaE (Fig. 3a). Eglin C was chosen as the TP because of its small size (7 kDa), which facilitates separation of native and EPL-modified species by using SDS–PAGE. We overexpressed this construct in origami as a soluble protein, and in accord with previous results (Callahan et al., 2011), we observed minimal processing. Thus, >80% of the bacterially expressed TP-CPGCDnaE was recovered as precursor, which is a substantial improvement over the <15% precursor recovered with a non-redox responsive control construct, TP-APGCDnaE, expressed in parallel (Fig. 3b).
Figure 3.

Expressed protein ligation with DnaE intein redox trap. a: EPL scheme. EPL between TP (gray) and synthetic peptide (green) catalyzed by a reductively activated intein (red). b: Redox-trapping enhances precursor (PC) accumulation during bacterial expression in the oxidizing origami strain. Percent precursor remaining with redox-trapped (+) and non-redox trapped (−) intein are shown below the Coomassie-stained gel. c: Time course of EPL product formation catalyzed by reductively activated intein (green squares). Inset: Formation of fluorescently labeled TP in the presence (+) and absence (−) of TCEP. d: Near quantitative labeling of TP. SDS–PAGE of EPL starting material (lane 1) and product mixture (lane 2) visualized by Coomassie staining.
Figure 3a shows the general scheme for the standard “one-pot” EPL where the TP-intein precursor reacts with a fluorescently labeled synthetic peptide bearing a free cysteine at its N terminus. A nucleophilic catalyst, 2-mercaptoethane sulfonate (MESNA), is added to displace the thioester at the intein active site, and then act as a leaving group for the peptide’s N-terminal cysteine.
To evaluate CPGCDnaE as a biocatalyst for protein labeling, we subjected bacterially expressed precursor to EPL conditions with a fluorescein-modified peptide, FLU-P1 (NEB Biolabs, Ipswich, MA). Through pilot experiments we discovered that the reducing power of the standard EPL buffer is insufficient to activate CPGCDnaE, despite the presence of MESNA, so that the precursor remained inert even after overnight incubation. We therefore conducted subsequent experiments with added Tris(2-carboxyethyl)phosphine (TCEP; 5 × 10−3 M, final). When the precursor (2 × 10−5 M) and the FLU-P1 (3 × 10−3 M) were incubated under these conditions at 23°C, we observed rapid accumulation of fluorescently labeled TP (Fig. 3c). Plots of the fluorescence intensity over time show that the majority of covalent ligation of the TP, in the presence of TCEP is complete in the first 5 h (Fig. 3c, green symbols). In comparison, no labeled product was detectable in reactions lacking TCEP (Fig. 3c, blue symbols). Analysis of reaction mixtures incubated overnight with TCEP, showed near total consumption of the precursor and quantitative conversion of TP into the peptide labeled product (Fig. 3d). Compared to its non-redox controlled counterpart, APGCDnaE, CPGCDnaE enhanced yield from EPL by at least fourfold. Collectively, these results indicate that CPGCDnaE is an effective catalyst for EPL showing minimal precursor loss during bacterial expression followed by efficient labeling in vitro.
Precursor Stabilization for Bioseparations
Next, we investigated CPGCDnaE as a tool for bioseparations. By functioning as “self-cleaving affinity tags,” inteins have facilitated economical, single-step purification of a wide range of bacterially expressed proteins (Chong et al., 1997; Wood et al., 1999; Xu et al., 2000). Early intein-based protein purification systems were based on intein mutants that catalyzed self-cleavage at their N or C terminus in response to shifts in temperature or pH or low molecular weight thiol reagents, like DTT. A substantial cost advantage has been reported for bioseparations using self-cleaving inteins, as compared with His-tag binding, because intein purification schemes obviate the need for costly proteases (Banki and Wood, 2005).
Thus, we set out to test CPGCDnaE as a tool for bioseparations. We prepared a tri-part precursor, C-I-CBD, composed of CFP fused to the N terminus of CPGCDnaE, which was joined to the chitin-binding domain (CBD; Fig. 4a). Along with this potential redox-responsive protein purification construct, we prepared a control construct in which the cysteine in the flanking N-extein was replaced with alanine. Both constructs were expressed as soluble proteins in origami. Before chromatography, samples of cell extract were analyzed to assess the extent of precursor processing that occurred during expression. Extracts were not boiled prior to SDS–PAGE, to maintain functional CFP for densitometric analysis. As shown in Figure 4b, we again observed elevated levels (2.2-fold) of precursor in the redox-responsive precursor compared to the control construct, with precursor/product ratios in this case being double for the redox-trapped intein. The soluble tri-part precursor was then bound via its CBD to chitin resin, followed by successive washes to remove unbound material. CFP was collected along with detectable amounts of a higher molecular species following incubation of the column in reducing buffer containing DTT or hydroxyl-amine. Samples from the purification of CFP using CPGCDnaE are shown in Figure 4c. Comparison of these results with those obtained using the non-redox responsive control precursor, showed that the yield of isolated of CFP more than doubled.
Figure 4.
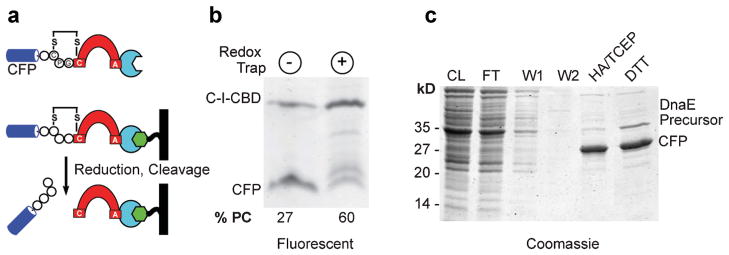
Protein purification with DnaE intein redox trap. a: Tri-part precursor, C-I-CBD, consisting of CFP (blue) fused to intein (red) followed by a CBD (turquoise). Bacterially expressed C-I-CBD is immobilized on a chitin column (black/green), reductively activated, and treated with nucleophile to release CFP. b: Redox-trapping enhances accumulation of C-I-CBD precursor. SDS–PAGE showing intact and processed C-I-CBD with control (lane 1) and redox-trapped intein (lane 2) following bacterial expression in origami, as visualized by in gel fluorescence (excitation 457 nm, emission 526 nm). Numbers below gel reflect precursor/product ratio. c: Purification of CFP following reductive activation and on-bead cleavage of C-I-CBD precursor. Proteins were separated by SDS–PAGE and visualized by Coomassie staining. Lanes: CL (cleared lysate); FT (flow through); W1 (wash 1); W2 (wash 2); cleaved CFP using hydroxlamine (HA/TCEP) or dithiothreitol (DTT).
As with other inteins, the chief advantage of using CPGCDnaE for bioseparations is that the TP can be isolated by site-specific cleavage by DTT or hydroxylamine, obviating the need for costly proteases. The specific advantage of CPGCDnaE is the long-term stability of the precursor in an inactive but catalytically competent state. It should be noted that proteins that have been purified (or ligated) using the CPGCDnaE intein will bear a tri-peptide “scar,” Cys–Pro–Gly, at their C terminus. The Pro and Gly residues are inert chemically; cysteine, however, can act as nucleophile so that it could be employed as a handle to attach any of the increasing number of electrophilic reporter groups (Chalker et al., 2009). At the same time, it is conceivable that in some cases the extra residues could prove deleterious to downstream applications. The user must therefore weigh the potential liability of the final product harboring a tripeptide Cys–Pro–Gly against the benefit of enhanced yield.
A Kinetic Biosensor of Oxidizing Environments Based on Intein Cleavage
In addition to in vitro applications, inteins also find use in prokaryotic and eukaryotic cell biology as genetically encoded biosensors (Li et al., 2011; Peck et al., 2011; Skretas and Wood, 2005; Yuen et al., 2006). Engineering of intein biosensors often involves insertion of a receptor into the intein, such that ligand binding and intein self-splicing are coupled (Li et al., 2011; Tyszkiewicz and Muir, 2008). In principle, biosensors could also be developed that are based on intein self-cleavage, rather than self-splicing, although this approach remains largely unexplored.
In previous work and as described above, we observed that self-cleavage of a FRET-active CPGCDnaE precursor, C-Ia-Y, containing an intein flanked by cyan and YFPs, resulted in a loss of FRET (Amitai et al., 2009). Introduction of the Cys–Pro–Gly–Cys-based redox trap into the C-Ia-Y precursor resulted in high FRET values during bacterial expression in origami, relative to FRET values when expressed in the isogenic parental strains, AD494 and DHB4 (Callahan et al., 2011). Based on this observation, we asked whether the FRET-active CPGCDnaE construct (Fig. 5a) could serve as an indicator to identify bacteria with hyperoxic cellular environments.
Figure 5.

FRET-active CPGCDnaE as a redox biosensor. a: FRET-active reporter of intein self-cleavage. Intein (red) is expressed as an internal fusion with FRET-active fluorescent proteins (blue and yellow). b: Elevated FRET of intein reporter when expressed in oxidizing origami. Representative hisotgram of FRET values from cultures of parental (blue, yellow) and oxidizing origami (green) strains of E. coli harboring FRET-active intein reporter. c: Gel analysis correlates with elevated FRET in origami. Precursor (C-Ia-Y) and product (Ia-Y) after expression in AD494 and origami strains were separated by non-reducing SDS–PAGE and detected by in-gel fluorescence (excitation 488 nm, emission 580 nm). Samples with high FRET showed precursor accumulation.
To serve as a useful cellular marker, the FRET-active CPGCDnaE construct should allow identification of hyperoxic mutants against a background of wild-type cells. As a test of this application, we first generated a mixed population of origami, AD494 and DHB4 colonies, harboring the FRET-active CPGCDnaE expression construct (CamR), by spreading the bacteria together on LB agar plus chloramphenicol (Cam). The reducing power of the wild-type DHB4 strain (−259 mV) and AD494 strain (−237 mV) have been determined by using an engineered yellow fluorescent protein (rxYFP) (Ostergaard et al., 2001); the reducing power of origami has not been reported to our knowledge. Colonies from this plate were transferred to LB media plus Cam, grown to mid-log phase, and induced with arabinose to express C-Ia-Y. Following induction, we determined precursor accumulation via FRET. When these FRET values were plotted as a histogram, distinct low FRET and high FRET populations emerged. We next returned to the master plates to unequivocally differentiate origami (Kanr, Tetr), AD494 (Kanr), and DHB4, by using their unique antibiotic resistance profiles. As apparent from Figure 5b, where FRET values have been colored according to the determined host strain, there is a strong correlation between elevated FRET values and expression in origami. Further, and in accord with previous work (Amitai et al., 2009; Callahan et al., 2011), elevated FRET readings were found to correspond to precursor accumulation by gel analysis (Fig. 5c). Finally, we evaluated the FRET data using Z′ factor analysis (Zhang et al., 1999) to assess the reliability of this redox reporter in a high throughput sorting experiment.
The formula above yields a Z′ factor of 0.7 using FRET values in origami as “high” readings, and DHB4 (or AD494) as “low” readings. A Z′ factor between 0.5 and 1.0 is generally interpreted to mean an excellent assay. The result suggests that FRET-active CPGCDnaE can serve as a reliable indicator of hyperoxia in vivo.
Other biosensors of redox conditions are known. One example is the GFP-based reporter (roGFP) that produces spectral changes upon disulfide–dithiol exchange of an engineered pair of cysteine residues (Hanson et al., 2004; Lohman and Remington, 2008). The present system operates in a somewhat different way than roGFP, in that it couples disulfide–dithiol exchange and spectral changes to peptide bond cleavage. Thus, the intein biosensor reports on the rate of a kinetic process, rather than on the position of an equilibrium. Because peptide bond cleavage is essentially irreversible, an intein-based reporter provides the opportunity to validate changes in redox status by corroborating spectral data with chemical reactivity.
Summary
Here, we showed that disulfide trapping could bring the M. thermautotrophicus Mth RnR1 intein under redox control, indicating the generality of our approach. As in other engineered disulfide traps (Matsumura and Matthews, 1989), the intein is catalytically competent in the oxidized form, but unreactive until stimulated by a reducing agent. Using the DnaE intein, we demonstrated the compatibility of redox-responsive activity with two prominent intein-based technologies: protein modification and protein purification. Finally, we established the practicality of the CPGCDnaE intein as an indicator of hyperoxic conditions in living cells.
Methods
Reagents and Strains
Taq polymerase, restriction enzymes, and the modified peptide, FluP1, were purchased from New England Biolabs, Ipswich, MA. Primers, as listed in Supplementary Table SI, were obtained from Integrated DNA Technologies, Coralville, IA. All other chemicals were purchased from Sigma (St. Louis, MO) or GoldBio (St. Louis, MO) unless otherwise noted. The origami strain of E. coli was obtained from Novagen (Darmstadt, Germany), and its isogenic parental strains, AD494 and DHB4, were provided by Professor Jon Beckwith (Harvard University, Cambridge, MA).
N-Cleavage Activity of DnaE and RnR Inteins
FRET-active reporters of N-extein cleavage activity for the Mth RnR intein were generated as described (Callahan et al., 2011). Primers introduced the desired extein sequence, the Asn to Ala point mutation, and a 5′ XhoI site and 3′ SbfI site which were used to ligate the amplified fragment between the CFP and YFP coding sequences in the plasmid C-Ia-Y (Amitai et al., 2009). The N-extein cleavage activity of the DnaE and RnR inteins in vivo during bacterial expression was assessed by FRET (Amitai et al., 2009). FRET signal was determined 7 or 18 h after induction of protein expression in E. coli DHB4, AD494, and origami. To assay N-extein cleavage activity in vitro, we followed the conversion of precursor, C-Ia-Y into Ia-Y by in-gel YFP fluorescence. Reactions were initiated by adding DTT (0.02 M) or hydroxylamine (0.1 M) to precursor in TE buffer (Tris 0.05 M pH 8, 0.01 M EDTA). Hydroxyaminolysis was conducted +/− the reducing agent TCEP (0.02 M). At selected intervals aliquots of the reaction were quenched with SDS–PAGE loading buffer and frozen at −80°C. Samples were separated by SDS–PAGE; the gels were then scanned with a Typhoon 9400 imager by excitation at 488 nm and emission at 580 nm. The relative abundance of precursor to cleavage product was determined by fluorescence intensity (Callahan et al., 2011).
Bioseparation
Constructs harboring CPGCDnaE and APGCDnaE inteins were generated by replacing the coding sequence of YFP in the respective C-Ia-Y plasmid (Amitai et al., 2009) with that of the Bacillus circulans CBD (Chong et al., 1997). The resulting tri-part precursor, C-I-CBD, was expressed in E. coli origami from the arabinose-inducible promoter in pBAD33 (Guzman et al., 1995). Bacterially expressed precursor was extracted using B-PER reagent (Pierce, Rockford, IL), mixed with six volumes of ice-cold chitin column buffer (0.2 M Tris pH 8, 0.5 M NaCl, 0.1 mM EDTA) and bound to chitin beads (NEB) according to the manufacturer’s instructions. CFP was released from the beads using three volumes of buffer containing DTT or hydroxylamine/TCEP.
Expressed Protein Ligation
Constructs for EPL were generated by fusing eglin C to the N-extein of the CPGC or APGC DnaE intein. The intein coding sequences were amplified and cloned into pET45b (Novagen) as BamHI–HindIII fragments, followed by insertion of the amplified eglin C gene using NcoI and BamHI. The resulting plasmids, pET45b His6–eglinC-C/APGCDnaE, were transformed into origami-DE3 for protein expression as described. Bacterially expressed precursor was extracted from cell paste with B-PER reagent then purified using an NiNTA spin column (Qiagen, Valencia, CA). To effect labeling of eglin C, purified precursor was combined with the FluP1 peptide in EPL buffer (Tris pH 8.5, 0.1 M NaCl) with 10 mM TCEP added as a reducing agent, with 10 mM MESNA added to induce N-extein cleavage.
Redox Biosensor
We used the FRET-active CPGCDnaE construct to distinguish origami against a background of DHB4 and AD494. Expression cultures (1.5 mL) were grown in LB media plus chloramphenicol (20 μg/mL) within deep-well plates. Overnight cultures were seeded 1:50 into fresh LB media, grown for 2 h at 37°C, followed by induction with arabinose to a final concentration of 0.4%. Incubation temperature was reduced to 18°C and growth was continued for 18 h. Cells were pelleted then lysed with 100 μL of B-PER. Analysis of precursor levels in the soluble cell extract by FRET and in-gel fluorescence using YFP fluorescence was carried out as described above.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM39422 and GM44844 to Marlene Belfort.
Footnotes
Author contributions: BPC designed and performed all experiments with technical assistance from MS and under the guidance of MB. MS prepared the figures. BPC and MB wrote the manuscript.
Some of the research was performed while the authors were at Wadsworth Center, NY State Department of Health.
Additional supporting information may be found in the online version of this article.
References
- Amitai G, Callahan BP, Stanger MJ, Belfort G, Belfort M. Modulation of intein activity by its neighboring extein substrates. Proc Natl Acad Sci USA. 2009;106(27):11005–11010. doi: 10.1073/pnas.0904366106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banki MR, Wood DW. Inteins and affinity resin substitutes for protein purification and scale up. Microb Cell Factories. 2005;4:32. doi: 10.1186/1475-2859-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binschik J, Zettler J, Mootz HD. Photocontrol of protein activity mediated by the cleavage reaction of a split intein. Angew Chem. 2011;50(14):3249–3252. doi: 10.1002/anie.201007078. [DOI] [PubMed] [Google Scholar]
- Callahan BP, Topilina NI, Stanger MJ, Van Roey P, Belfort M. Structure of catalytically competent intein caught in a redox trap with functional and evolutionary implications. Nat Struct Mol Biol. 2011;18(5):630–633. doi: 10.1038/nsmb.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker JM, Bernardes GJ, Lin YA, Davis BG. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem Asian J. 2009;4(5):630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- Cheriyan M, Perler FB. Protein splicing: A versatile tool for drug discovery. Adv Drug Del Rev. 2009;61(11):899–907. doi: 10.1016/j.addr.2009.04.021. [DOI] [PubMed] [Google Scholar]
- Chong S, Mersha FB, Comb DG, Scott ME, Landry D, Vence LM, Perler FB, Benner J, Kucera RB, Hirvonen CA, Pelletier JJ, Paulus H, Xu MQ. Single-column purification of free recombinant proteins using a self-cleavage affinity tag derived from a protein splicing element. Gene. 1997;192:271–281. doi: 10.1016/s0378-1119(97)00105-4. [DOI] [PubMed] [Google Scholar]
- Clark KM, Yu Y, Marshall NM, Sieracki NA, Nilges MJ, Blackburn NJ, van der Donk WA, Lu Y. Transforming a blue copper into a red copper protein: Engineering cysteine and homocysteine into the axial position of azurin using site-directed mutagenesis and expressed protein ligation. J Am Chem Soc. 2010;132(29):10093–10101. doi: 10.1021/ja102632p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TC, Jr, Benner J, Xu M-Q. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998;7:2256–2264. doi: 10.1002/pro.5560071103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TCJ, Benner J, Xu MQ. The in vitro ligation of bacterially expressed proteins using an intein from Methanobacterium thermo-autotrophicum. J Biol Chem. 1999;274:3923–3926. doi: 10.1074/jbc.274.7.3923. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279(13):13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH. Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science. 1990;250:651–657. doi: 10.1126/science.2146742. [DOI] [PubMed] [Google Scholar]
- Li J, Gierach I, Gillies AR, Warden CD, Wood DW. Engineering and optimization of an allosteric biosensor protein for peroxisome proliferator-activated receptor gamma ligands. Biosens Bioelectron. 2011;29(1):132–139. doi: 10.1016/j.bios.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman JR, Remington SJ. Development of a family of redox-sensitive green fluorescent protein indicators for use in relatively oxidizing subcellular environments. Biochemistry. 2008;47(33):8678–8688. doi: 10.1021/bi800498g. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Matthews BW. Control of enzyme activity by an engineered disulfide bond. Science. 1989;243(4892):792–794. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]
- Muir TW. Studying protein structure and function using semisynthesis. Biopolymers. 2008;90(6):743–750. doi: 10.1002/bip.21102. [DOI] [PubMed] [Google Scholar]
- Muir TW, Sondhi D, Cole PA. Expressed protein ligation: A general method for protein engineering. Proc Natl Acad Sci USA. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: Engineering a redox switch in green fluorescent protein. EMBO J. 2001;20(21):5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus H. Protein splicing and related forms of protein autoprocessing. Ann Rev Biochem. 2000;69:447–496. doi: 10.1146/annurev.biochem.69.1.447. [DOI] [PubMed] [Google Scholar]
- Peck SH, Chen I, Liu DR. Directed evolution of a small-molecule-triggered intein with improved splicing properties in mammalian cells. Chem Biol. 2011;18(5):619–630. doi: 10.1016/j.chembiol.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perler FB. Protein splicing of inteins and hedgehog autoproteolysis: Structure, function, and evolution. Cell. 1998;92(1):1–4. doi: 10.1016/s0092-8674(00)80892-2. [DOI] [PubMed] [Google Scholar]
- Perler FB. InBase: The intein database. Nucleic Acids Res. 2002;30:383–384. doi: 10.1093/nar/30.1.383. < http://tools.neb.com/inbase>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost MR, Yee CS, Stubbe J. Site-specific incorporation of fluorotyrosines into the R2 subunit of E. coli ribonucleotide reductase by expressed protein ligation. Nat Protoc. 2007;2(5):1225–1235. doi: 10.1038/nprot.2007.159. [DOI] [PubMed] [Google Scholar]
- Skretas G, Wood DW. A bacterial biosensor of endocrine modulators. J Mol Biol. 2005;349(3):464–474. doi: 10.1016/j.jmb.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Smith JM, Vitali F, Archer SA, Fasan R. Modular assembly of macrocyclic organo-peptide hybrids using synthetic and genetically encoded precursors. Angew Chem. 2011;50(22):5075–5080. doi: 10.1002/anie.201101331. [DOI] [PubMed] [Google Scholar]
- Stewart EJ, Aslund F, Beckwith J. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 1998;17(19):5543–5550. doi: 10.1093/emboj/17.19.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyszkiewicz AB, Muir TW. Activation of protein splicing with light in yeast. Nat Methods. 2008;5(4):303–305. doi: 10.1038/nmeth.1189. [DOI] [PubMed] [Google Scholar]
- Vila-Perello M, Muir TW. Biological applications of protein splicing. Cell. 2010;143(2):191–200. doi: 10.1016/j.cell.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Perello M, Hori Y, Ribo M, Muir TW. Activation of protein splicing by protease- or light-triggered O to N acyl migration. Angew Chem. 2008;47(40):7764–7767. doi: 10.1002/anie.200802502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DW, Wu W, Belfort G, Derbyshire V, Belfort M. A genetic system yields self-cleaving inteins for bioseparations. Nat Biotechnol. 1999;17:889–892. doi: 10.1038/12879. [DOI] [PubMed] [Google Scholar]
- Xu MW, Paulus H, Chong S. Fusions to self-splicing inteins for protein purification. Methods Enzymol. 2000;326:376–418. doi: 10.1016/s0076-6879(00)26066-7. [DOI] [PubMed] [Google Scholar]
- Yuen CM, Rodda SJ, Vokes SA, McMahon AP, Liu DR. Control of transcription factor activity and osteoblast differentiation in mammalian cells using an evolved small-molecule-dependent intein. J Am Chem Soc. 2006;128:8939–8946. doi: 10.1021/ja062980e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler MP, Tan C, Bellaiche Y, Cherry S, Hader S, Gayko U, Perrimon N. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat Biotechnol. 2004;22(7):871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.