

Abstract
Pulmonary embolism is an important clinical entity with considerable mortality despite advances in diagnosis and treatment. In the present article, the authors offer a comprehensive review focused mainly on epidemiology, risk factors, risk stratification, pathophysiological considerations and clinical presentation. Diagnosis based on assessment of clinical likelihood, electrocardiography, chest x-ray, D-dimer levels, markers of myocardial injury and overload, and blood gases is discussed in detail. Special attention is devoted to the clinical use of computed tomography, pulmonary angiography and echocardiography in the setting of pulmonary embolism.
Keywords: Diagnosis, Epidemiology, Pulmonary embolism, Risk stratification
Pulmonary embolism (PE) is a relatively common acute cardiovascular disorder with high early mortality rates that, despite advances in diagnosis and treatment over the past 30 years, have not changed significantly. Due to pulmonary bed obstruction, PE can result in acute right ventricular (RV) failure, a life-threatening condition. Because most patients ultimately die within the first hours of presentation, early diagnosis is of paramount importance. Emergency management is usually highly effective and RV failure is potentially reversible. Depending on PE presentation, initial treatment is primarily focused on restoring adequate blood flow through the pulmonary bed and preventing PE recurrence. Appropriate therapy is best selected using risk stratification primarily by assessing hemodynamic impact as the strongest marker of short-term prognosis, morphological extent of PE, the patient’s cardiovascular and pulmonary system status, the degree of neurohumoral adaptation and potential risks of the therapy instituted.
INCIDENCE AND MORTALITY OF PE
While no exact epidemiological data are available, the incidence of PE is estimated to be approximately 60 to 70 per 100,000, and that of venous thrombosis approximately 124 per 100,000 of the general population (1,2). The European guidelines for the diagnosis and management of PE report annual incidence rates of venous thrombosis and PE of approximately 0.5 to 1.0 per 1000 inhabitants (3). However, the actual figures are likely to be substantially higher because silent PE can develop in up to 40% to 50% of patients with deep vein thrombosis (DVT) (4). In addition, autopsy studies have shown that PE had been diagnosed before death in 30% to 45% of patients (5). After coronary artery disease and stroke, acute PE ranks third among the most common types of cardiovascular diseases. While clinical data indicate that most cases of PE occur at 60 to 70 years of age, autopsy data show the highest incidence among individuals 70 to 80 years of age. If untreated, acute PE is associated with a significant mortality rate (as high as 30%), whereas the death rate of diagnosed and treated PE is 8%. Up to 10% of acute PE patients die suddenly. Two of three patients succumbing to PE die within 2 h after presentation. The mortality rates associated with various types of PE are summarized in (Table 1) (2,3,6).
TABLE 1.
| Clinical presentation of acute pulmonary embolism | Mortality rate |
|---|---|
| Unselected population | 11.4% at 2 weeks, 17.4% at 3 months |
| Massive pulmonary embolism | |
| Overall | 18% to 65% |
| Treated | Approximately 20% |
| With cardiogenic shock | 25% to 30% |
| With resuscitation | 65% |
| Submassive pulmonary embolism | 5% to 25% |
| Pulmonary embolism with mobile thrombi in right-heart chambers | As high as 27% |
| Small pulmonary embolism | Up to 1% |
RISK FACTORS, MEDICAL HISTORY, PATHOPHYSIOLOGICAL CONSIDERATIONS, CLINICAL PRESENTATION AND RISK STRATIFICATION
Due to the variation in presentation, establishing a diagnosis of PE may not be an easy task; therefore, it is imperative that the possibility of a PE be considered. The accuracy of the diagnosis decreases as patient age increases. Diagnosis is difficult in the presence of comorbidities such as bronchopneumonia, chronic obstructive pulmonary disease (COPD), asthma or chronic fibrotizing pulmonary processes. In contrast, PE is readily diagnosed in patients presenting with DVT. The most common sources of PE (up to 85% of cases) include DVT followed by thrombosis of iliac and renal veins, and the inferior vena cava. The upper limbs are not usually identified as a source of major PE (7).
Risk factors
Venous thromboembolism (VTE) is believed to result from an interaction of the individual patient’s risk factors and the setting or circumstances where it occurs. Patient-associated risk factors are usually permanent, whereas the circumstances tend to be transient in nature. Patient risk factors include age, personal history of VTE, active malignancy or another disabling conditions such as heart or respiratory failure, congenital or acquired coagulation disorders, hormone replacement therapy and oral contraception. According to the British Thoracic Society, risk factors are traditionally classified into major and minor categories (Table 2) (8). Critically ill patients receiving intensive care are also considered to be a population at risk for developing VTE.
TABLE 2.
Risk factors of venous thromboembolism, according to the British Thoracic Society, 2003
| Major risk factors (RR = 5 to 20) | Minor risk factors (RR = 2 to 4) |
|---|---|
| Postoperative states: Major abdominal/pelvic surgery, hip/knee joint replacement, postoperative intensive care Obstetrics: Late pregnancy, Caesarian section, puerperium Lower limb affections: Fractures, extensive varicosities Malignancies: Abdominal/pelvic, advanced/metastatic stage Limited mobility: Hospitalization, geriatric care Miscellaneous: History of previous venous thromboembolism |
Cardiovascular: Congenital heart disease, heart failure, hypertension, superficial venous thrombosis, central venous catheter Humoral: Estrogen use: oral contraception, hormone replacement therapy Miscellaneous: Chronic obstructive lung disease, neurological impairment, latent malignancy, thrombotic defects, long-distance travel in the sitting position, obesity Other: Inflammatory bowel disease, nephrotic syndrome, chronic dialysis, myeloproliferative disease, paroxysmal nocturnal hemoglobinuria |
Adapted from reference 8
Personal history
The overwhelming majority of patients with PE (>85%) tend to complain primarily of sudden-onset or worsened resting dyspnea. PE, however, can also present as progressive exercise-induced dyspnea. In addition, more than one-half of patients experience chest pain that is sometimes difficult to distinguish from an angina of ischemic origin. The pain experienced in PE is usually not dull; instead, it is sharp, stinging and occasionally related to respiratory excursions. Other presentations of PE can include cough (approximately 20% of patients), hemoptysis (7% [a consequence of lung infarction]) and syncope (14%). The often reported triad of dyspnea, chest pain and hemoptysis does not actually occur frequently (5% to 7%) (2). Regardless, more than 90% of PE patients present with dyspnea, tachypnea or chest pain (3). In the most serious cases, PE can ultimately result in cardiac arrest, shock or hypotension (see below). Isolated, rapidly progressing dyspnea usually develops in patients with extensive central PE with a major hemodynamic impact. This is in contrast to (no or minimal symptoms associated with) minor embolisms occluding peripheral branches of the pulmonary artery or, possibly, presenting as a lung infarction syndrome. The aforementioned chest pain may also reflect true RV ischemia or infarction in right-heart overload. In patients with chronic heart or lung disease, exacerbation of dyspnea can be the sole symptom of underlying PE. While identification of a patient’s risk factors is, no doubt, crucial in the initial diagnostic process, up to 30% of cases of PE develop idiopathically (ie, without an identifiable risk factor) (3).
Pathophysiological considerations
The severity of acute PE is determined primarily by its hemodynamic impact, presenting as sudden pulmonary hypertension. However, a pivotal role in the ultimate hemodynamic consequence is also played by cardiovascular functional status (‘cardiovascular reserve’), as well as adaptation of pulmonary and neurohumoral systems. In fact, even a morphologically extensive PE can present as a hemodynamically minor one and vice versa (Figure 1). It is traditionally believed that patients without a history of heart or pulmonary disease require pulmonary bed obstruction of 30% to 50% to develop pulmonary hypertension (9). In patients with a heart or lung disease, however, even a minor obstruction in the pulmonary circulation is sufficient to cause pulmonary hypertension. The contribution of reflex and humoral pulmonary vasoconstriction reported in an experimental setting is not considered to be relevant in clinical practice. The acutely developing pulmonary hypertension in PE leads to an increase in RV afterload, morphologically presenting as RV dilation and may eventually cause right-heart failure. Once pulmonary vascular resistance has risen to a level that the RV is unable to tolerate, PE can result in sudden death through pulseless electrical activity (formerly electromechanical dissociation) or asystole (Figure 2). A less sudden drop in RV cardiac output results in decreasing left ventricular (LV) filling, deteriorated diastolic LV function due to RV dilation (ventricular interdependence) and interventricular septal bulging. These events can lead to a fall in blood pressure and present as syncope, hypotension or cardiogenic shock. RV overload and reduction in coronary flow secondary to high RV pressure in the presence of massive PE can lead to subendocardial RV ischemia or infarction, with a potential contribution of coronary atherosclerosis. Patients surviving the initial episode of right-heart failure develop compensatory mechanisms via activation of the sympathetic nervous system. Inotropic and chronotropic stimulation, together with the Frank-Starling mechanism, results in the development of pulmonary hypertension crucial for maintaining pulmonary artery flow and, hence, systemic circulation. Together with systemic vasoconstriction, these mechanisms can preserve systemic blood pressure and organ function. It is believed that the RV of a healthy man exposed to acute overload will not generate more than 40 mmHg of mean pulmonary artery pressure.
Figure 1).
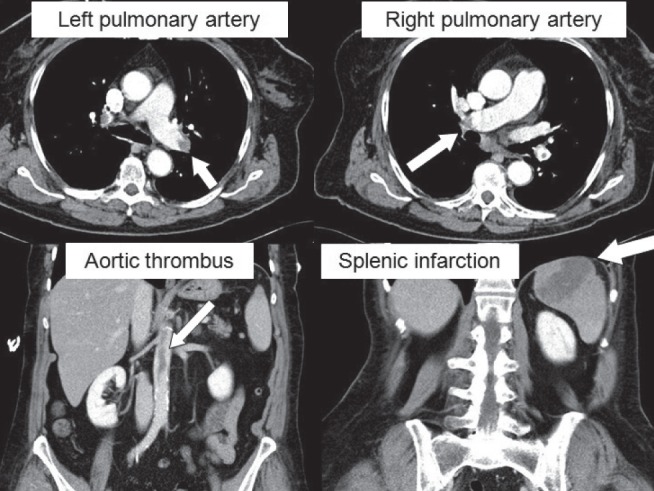
A female patient with clinically small (ie, hemodynamically not significant), yet morphologically extensive, pulmonary embolism (arrows point to large emboli in the left and right pulmonary artery branches on computed tomography angiography). This clinical case is additionally complicated by embolization across a patent foramen ovale to the aorta and spleen. Patient’s clinical course was, however, favourable. Images were reproduced with permission from archives of the Department of Diagnostic Radiology, General University Hospital and 1st Medical School, Charles University in Prague, Prague, Czech Republic
Figure 2).
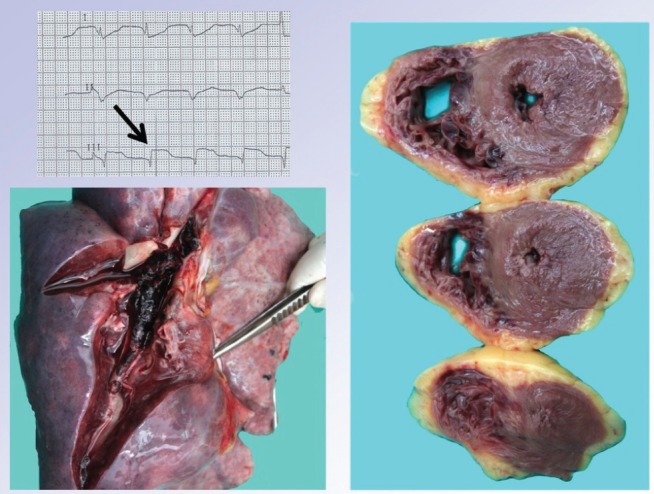
Massive pulmonary embolism in a 35-year-old woman presenting with cardiac arrest and asystole. The patient was brought in by emergency medical services under continuous resuscitation; transient resumption of electrical activity was followed by clear ST-segment elevations in lead III (upper left panel). The patient was then transferred to the catheter laboratory and connected to veno-arterial extracorporeal membrane oxygenation, where pulmonary embolism diagnosis was confirmed by pulmonary angiography detecting virtually complete pulmonary bed obstruction. Thrombolytic therapy and catheter-based mechanical thrombus fragmentation failed to restore adequate flow through the pulmonary bed. The lower panel clearly shows the macroscopic autopsy finding of the pulmonary bed filled up with fresh thrombi. The right panel displays a myocardial section (documenting massive right ventricular wall effusion) indicating critical right-heart overload and possible mimicking of the above electocardiographic finding. Intraresuscitation selective coronary angiography ruled out coronary artery obstruction. (Images reproduced with permission from archives of the Department of Pathology, General University Hospital and 1st Medical School, Charles University in Prague, Prague, Czech Republic)
An additional typical feature of PE is respiratory insufficiency (in approximately 10% of patients) due to several mechanisms. A low cardiac output causes mixed venous blood desaturation with subsequent reverse blood flow into the pulmonary capillaries. A crucial role in this is also played by the ventilation-perfusion mismatch. Up to one in three patients can develop a right to left shunt by the opening of a patent foramen ovale (PFO) in the presence of inverse pressures in the right and left atria, thus contributing to the risk of paradoxical embolization.
The pathophysiological basis for nonthrombotic PE is different and is briefly discussed in a separate paragraph at the end of the section.
Clinical presentation
The definition of the clinical presentation and course of PE depends on the classification system used. The Czech guidelines on the diagnosis, management and prevention of pulmonary embolism (version 2007) of the Czech Society of Cardiology (2) uses the traditional division into acute massive, acute submassive, acute small, subacute massive PE and chronic thromboembolic pulmonary hypertension. The 2008 European guidelines on the diagnosis and management of pulmonary embolism of the European Society of Cardiology (3) use a different system of classification based on assessment of the individual risk of early mortality associated with PE. This classification divides patients into groups at high, intermediate and low risk based on the presence of risk stratification markers (Tables 3 and 4). A comparison of both classification systems is presented in (Table 5). It is evident from Table 5 that both classification systems are similar because the critical factor affecting the prognosis of patients with PE is its hemodynamic impact (ie, presence or absence of RV dysfunction) (2,3). This criterion can be used to divide PE patients into those with symptomatic RV dysfunction (patients with massive/high-risk PE); asymptomatic dysfunction and/or RV overload (patients with submassive/intermediate-risk PE); and normal RV function (small/low-risk PE). A complementary role in this approach is inevitably played by the anatomical extent of PE assessed with a morphological investigation using computed tomography (CT), angiography or a ventilation-perfusion scan, in absolute (ie, the ‘mass’ of thrombi present) and relative (a morphologically small PE in a patient with a reduced cardiovascular reserve) terms (Figure 1). It is important to note that the value of syncope as a sign of massive PE has yet to be conclusively determined, particularly in cases in which the syncope is not followed by prolonged hemodynamic instability; nevertheless, this presentation should always be considered to be serious. Another important note, while the presence of asymptomatic RV dysfunction in submassive PE adequately identifies at-risk patients with presumably higher mortality rates, there is only scanty evidence that it also identifies patients eligible for therapy associated with risk (ie, thrombolytic therapy). The Management Strategies and Prognosis of Pulmonary Embolism (MAPPET-3) study, the only prospective randomized trial to date (10), compared heparin in combination with alteplase versus heparin alone in submassive PE. Although designed to specifically explore the above issue, the trial uncovered controversies rather than clear-cut answers (refer to Bĕlohlávek et al [11], ‘Pulmonary embolism, part II: Management’, in the current issue of Experimental and Clinical Cardiology [pages 139–147]).
TABLE 3.
Markers used for risk stratification according to the guidelines for the diagnosis and management of pulmonary embolism of the European Society of Cardiology (3)
| Clinical markers | Shock |
| Hypotension* | |
| Markers of right ventricular dysfunction | Right ventricular dilation, hypokinesia or echocardiographic signs of pressure overload |
| Right ventricular dilation on spiral CT angiography | |
| Increased BNP or NT-proBNP levels | |
| Increase in right-heart pressures during right-heart catheterization | |
| Markers of myocardial injury | Troponin T or I positivity |
Hypotension is defined as blood pressure below 90 mmHg or a decrease in blood pressure ≥40 mmHg for 15 min, not caused by arrhythmia, hypovolemia or sepsis. BNP Brain natriuretic peptide; CT Computed tomography; NT-proBNP N-terminal proBNP
TABLE 4.
Risk stratification* of acute pulmonary embolism (PE) according to the guidelines for the diagnosis and management of pulmonary embolism of the European Society of Cardiology (3)
| PE-related early-mortality risk |
Risk markers
|
Potential treatment implications | ||
|---|---|---|---|---|
| Clinical (shock or hypotension) | RV dysfunction | Myocardial injury | ||
| High (>15%) | + | (+)† | (+)† | Thrombolysis or embolectomy |
| Nonhigh | ||||
| Intermediate (3% to 15%) | – | + + – |
+ – + |
Hospital admission |
| Low <1% | – | – | – | Early discharge or home-based management |
Risk stratification takes into account anticipated early mortality.
In the presence of shock or hypotension, there is no need to confirm the presence of right ventricular (RV) dysfunction or myocardial injury to classify the condition as high-risk PE
TABLE 5.
Comparison of classification of individual forms of acute pulmonary embolism according to Czech (2) and European (3) guidelines for the diagnosis and management of pulmonary embolism
| Czech Society of Cardiology guidelines | European Society of Cardiology guidelines |
|---|---|
| Massive pulmonary embolism | High-risk pulmonary embolism |
| Submassive pulmonary embolism | Intermediate-risk pulmonary embolism |
| Small pulmonary embolism | Low-risk pulmonary embolism |
PE patients relatively often present with serious conditions such as cardiac arrest, shock and hypotension (Table 6) (12). The authors of the present article analyzed data from their acute PE registry, which included patients with verified acute and subacute PE between the period from 2003 to 2009, and showed that 20% of patients belonged to the group with massive PE, 47% with submassive and the remaining 33% consisted of small PE. Overall, 67% of cases could be classified as hemodynamically severe PE (177 of 266 patients). Total in-hospital patient mortality was 5.6%, involving 24.5% and 1.6% mortality for massive and submassive PE, respectively. It should be noted that the patient group was a relatively at-risk population, with 14 patients requiring resuscitation, 25 diagnosed to have mobile thrombi in the right-heart chambers and four developed embolization into the systemic circulation in the presence of a PFO. Overall, thrombolysis was undertaken in 97 patients, of which seven received rescue thrombolysis, six were treated by catheter embolectomy and four underwent surgical embolectomy. Given the above data, there is absolutely no doubt about the relevance of the initial presentation of PE, although our patient sample clearly differed in the incidence of the individual forms of PE compared with other reported series (13,14). The main reason for this difference is attributable to selection of patients referred to a tertiary cardiac centre.
TABLE 6.
Incidence of individual clinical manifestations of acute pulmonary embolism*
| Clinical status | Incidence, % |
|---|---|
| Cardiac arrest or shock | 13 |
| Hypotension without signs of shock | 9 |
| Hemodynamic stability, but echocardiographic signs of right ventricular dysfunction | 31 |
| Hemodynamic stability, without echocardiographic signs of right ventricular dysfunction | 47 |
More than 50% of cases of pulmonary embolism present as a serious condition. Adapted from reference 12
Acute massive PE
The most severe form of PE is an acute massive PE (ie, high risk) with mortality rates exceeding 20% irrespective of treatment. Acute massive PE can ultimately result in sudden death secondary to massive obstruction of the pulmonary bed (approximately 10% of PE cases). Acute massive PE is characterized by hemodynamic instability (ie, manifesting itself as syncope, the prognostic significance of which remains controversial [15] and the Guidelines of the European Society of Cardiology does not include it in the definition of high-risk PE), persistent hypotension and cardiogenic shock (with hypotension defined as a sudden fall in systolic blood pressure to <90 mmHg or more, or by ≥40 mmHg from baseline). Acute massive PE causes acute cor pulmonale, a condition consistent with acute clinically manifesting RV failure (ie, acute RV dilation, hypokinesis, tachycardia, presence of gallop rhythm, with systolic murmur from tricuspid regurgitation and signs of increased central venous pressure [increased jugular vein filling in the semirecumbent position]). There may be evidence of hepatojugular reflux or even hepatomegaly in the presence of hepatic congestion. Simple tachycardia and tachypnea (≥24 beats/min) are not considered to be signs of circulatory instability. Circulatory ‘instability’ requires the use of inotropic or vasopressor support to maintain adequate organ perfusion.
Acute submassive PE
From the perspective of macrohemodynamics (ie, in terms of systemic blood pressure and organ perfusion), a patient with acute submassive PE is hemodynamically stable, but can present with tachycardia and tachypnea. The condition is primarily characterized by the presence of signs of RV dysfunction, documented either by echocardiography or CT angiography (CTA) and/or right-heart catheterization. Patients with acute submassive PE comprise a heterogeneous population with mortality rates in the range of 5% to 25% (2,3,12) (ie, approximately double that of stable patients).
Acute small PE
Acute small PE has a good prognosis, with a three-month mortality rate <1%. It can be asymptomatic or accompanied only by tachypnea (≥24 breaths/min) and tachycardia (≥100 beats/min), or it can manifest itself as only a mild elevation of body temperature. Silent PE is not a rare occurrence; a study published by Meignan et al (4) in 2000 involving 622 patients with proximal DVT reported silent PE diagnosed by routine lung perfusion scans in 40% to 50% of the patients.
Pulmonary infarction
Pulmonary infarction is caused by small, distally embolizing thrombi with no hemodynamic consequences but can cause alveolar hemorrhages and pleuritis or small pleural exudate (often hemorrhagic). The most common clinical presentations of lung infarction include pleural pain, irritated cough, fever, hemoptysis, signs of lung consolidation and even friction rub. X-ray or CT can show peripheral or, occasionally, triangle-shaped infiltrate (Figure 3). It is sometimes difficult to distinguish pulmonary infarction from acute pneumonia. Lung infarction is more likely to occur in cardiac patients with chronic heart failure complicated by PE.
Figure 3).

Computed tomography image of a minor pulmonary infarction (arrow) in a patient with submassive pulmonary embolism. (Image reproduced with permission from archives of the Department of Diagnostic Radiology, General University Hospital and Charles University Medical School 1, Prague, Czech Republic)
Chronic thromboembolic pulmonary hypertension
Chronic thromboembolic pulmonary hypertension (CTEPH) is defined as mean pulmonary artery pressure ≥25 mmHg with normal capillary wedge pressure persisting six months after acute PE; and angiographic signs of typical pulmonary bed obstruction. Its prevalence is reported to be approximately 4% after PE. Pulmonary artery systolic pressure >50 mmHg during acute attack of PE is considered to be a risk factor. However, most patients with CTEPH have no history of acute PE (16). CTEPH is a serious condition with a dismal prognosis. Clinical features of CTEPH include progressive exertional dyspnea, syncope and right-heart failure. Physical examination reveals signs of RV hypertrophy (precordial or epigastric pulsation), accentuated second sound over the pulmonary artery, typical electrocardiography (ECG) picture and echocardiography-documented hypertrophy of the RV free wall. In indicated cases, the condition is manageable surgically by pulmonary artery endarterectomy (see below).
Subacute massive PE
Subacute massive PE (the term ‘successive PE’ is no longer used) is caused by numerous small emboli. Pulmonary bed obstruction takes longer to develop (approximately one to two weeks). It presents particularly by increasing exertional dyspnea and fatigue. The possibility of subacute massive PE should be considered in all patients experiencing progressive exertional dyspnea over a period of one to two weeks.
A relatively frequent ‘complication’ following an episode of PE can be the finding of a malignancy. However, in these cases PE should be viewed as the first presentation of the procoagulation state associated with malignant disease. Similarly, the long-term mortality rates of PE patients are essentially dependent on the incidence of malignancies.
DIAGNOSIS OF ACUTE PE
The diagnosis of PE is based on assessment of clinical likelihood, ECG, chest x-ray, laboratory investigations (primarily D-dimers and markers of cardiac injury and overload), imaging techniques (most commonly CTA or ventilation-perfusion scintigraphy) and echocardiography.
Assessing clinical likelihood
There are several scoring systems and models of clinical likelihood of PE, which are included in both currently available guidelines for the diagnosis and management of PE (2,3) (eg, one developed by Wells, or the revised Geneva score). However, the scores are not commonly used in clinical practice. The most recent meta-analysis failed to demonstrate their adequacy in final exclusion of PE (17). As mentioned above, what actually matters is the possibility of PE; after this, it is usually straghtforward to confirm or rule out the diagnosis using other laboratory tests. However, stand-alone laboratory investigations and clinical imaging, particularly with D-dimers or CTA, may not have clinical relevance, and may be economically inefficient and/or detrimental to the patient (18–20).
Electrocardiography and chest x-ray in PE
ECG signs of right-heart overload (Table 7), primarily T-wave inversion in V1–V4 leads, QR in V1 lead, classical S1Q3 picture, and incomplete or complete right bundle branch block can be helpful in the diagnosis of PE, in particular if the PE occurred very recently. As a caveat, ST-segment elevation (most often seen only in V1 lead) in PE can mimic an acute ST-segment elevation myocardial infarction and lead to an incorrect decision in the initial therapeutic strategy (21,22).
TABLE 7.
The most common electrocardiographic changes of acute right-heart overload/failure
| Sinus tachycardia |
| T-wave inversion in leads V1–V4, III and aVF |
| Incomplete or complete right bundle branch block |
| Cardiac axis tilt >90° |
| S-wave in lead I and Q wave in lead III |
| Atrial fibrillation |
| P pulmonale in leads II and III |
| ST-segment elevation in leads V1–V2 |
Adapted from reference 2
In PE, chest x-ray plays a crucial role, particularly in ruling out other causes of acute dyspnea and chest pain. However, chest x-ray alone will not be helpful in confirming or excluding PE. Potential radiographic signs of acute PE include atelectasia, diaphragm elevation on the affected side, prominence of the hilus and pulmonary artery, pleural exudate; even oligemia in certain areas of the lungs (Westermark sign) can be found. Lung infarction can be detected in only a small proportion (10% to 20% at most) of PE patients.
Clearly, PE cannot be confirmed or conclusively ruled out based on the above clinical signs, ECG or chest x-ray; however, they all can reinforce the clinical suspicion and be part of the comprehensive assessment of patients with suspected PE.
Laboratory tests used in PE
D-dimers:
Plasma D-dimers are the end product of plasmin-mediated fibrin degradation. Plasma D-dimer levels are elevated in the presence of an acute blood clot because activation of the coagulation system is associated with simultaneous fibrinolysis activation. To explain, normal D-dimer levels should be interpreted as PE or DVT being unlikely; hence, the negative predictive value of D-dimers is satisfactory. Although D-dimers are fibrin specific, fibrin specificity for VTE is not high because fibrin is also produced under several other circumstances such as malignancies, inflammation, infection, necrosis or aortic dissection. The implication is that D-dimers have a low positive predictive value and, are thus, unsuitable markers for PE to be confirmed. D-dimer specificity is also limited in elderly patients, pregnant women and hospitalized individuals. D-dimer levels should be determined using ELISA or line immuno assay because agglutination tests do not have adequate sensitivity. Thus, negative D-dimer levels analyzed using adequate techniques in patients with low or intermediate clinical likelihood of VTE rule out PE and DVT. In these patients, the three-month risk of VTE is significantly less than 1%. D-dimer analysis is not required in cases with highly suspected PE. In patients with already proven thrombembolism with persistently elevated D-dimer levels in long-term follow-up, especially after termination of anticoagulant therapy, the risk of disease recurrence is increased (23).
Markers of myocardial injury and overload:
Markers of myocardial injury include cardiac troponins (troponins T and I) and the overload markers natriuretic peptides (brain natriuretic peptide [BNP]; and N-terminal prohormone BNP [NT-proBNP]). These markers reflect the biochemical response to PE; the mechanism of their release remains poorly understood. In PE, any increase in cardiac troponin levels is ‘non-coronary related’, with the implication that it is not caused by coronary artery occlusion or preocclusion as such (Table 8). In PE, elevation of cardiac troponin levels has been suggested to reflect its severity and is primarily used for risk stratification in hemodynamically stable normotensive patients. Some studies have shown that elevation of troponin level is associated with increased mortality in PE patients, both those with and without coronary artery disease. A large meta-analysis investigating troponin levels in almost 2000 patients with various forms of PE determined that any increase was associated with up to a five-fold increase in the risk of death (24). Other meta-analyses have confirmed the finding with a four- to eightfold increase in the risk of early death (25,26). However, the value of these findings is complicated by the fact that no cut-off value of increase in troponin levels has yet been defined to identify those at higher or lower risk. Thus, an increase in troponin levels per se only confirms that the patient is at risk without allowing any decisions about their future management (use of thrombolysis or early discharge and, possibly, management of PE on an outpatient basis). Similar to the cut-off value of individual markers, the dynamics of their release have not yet been elucidated. The above holds true only for patients with overt RV dysfunction as the most reliable hallmark of a poor prognosis in PE. Troponin levels should be determined on admission and at 6 h to 8 h thereafter; troponin analysis is of no value in patients admitted later than 72 h after the onset of symptoms.
TABLE 8.
Potential causes of increased cardiac troponin levels
| Cardiac disease with potential increase in troponin levels | Noncardiac disease with potential increase in troponin levels |
|---|---|
| Pulmonary embolism, pulmonary hypertension | Infiltrative diseases such as sarcoidosis, amyloidosis, scleroderma, hemochromatosis |
| Hypertrophic cardiomyopathy | Acute neurological conditions including stroke and subarachnoidal hemorrhage |
| Aortic dissection Aortic valve disease Inflammatory diseases – myocarditis, pericarditis, endocarditis Cardiac contusion Acute and chronic advanced-stage heart failure Hypertensive crisis Tachyarrhythmia, bradyarrhythmia Cardioversion, cardiac pacing, endomyocardial biopsy, postablation states Takotsubo syndrome |
Acute and chronic renal failure Burns involving more than 30% of body surface area Rhabdomyolysis Toxic effects of drugs – adriamycin, 5-fluorouracil, herceptin, snake toxins Hypothyroidism Critically ill patients with sepsis, respiratory failure |
Adapted from reference 2
An increase in the natriuretic peptides BNP and NT-proBNP in PE reflects RV overload/dysfunction and is also associated with increased early mortality. Similar to the ‘noncoronary and noncardiac’ causes of troponin level elevation, natriuretic peptide levels can also be increased due to causes other than PE. If disregarding left-heart failure as a typical cause, the physician should consider pulmonary arterial hypertension, CTEPH, and pulmonary hypertension in lung and heart diseases. The prognostic value is similar to that of troponin levels. A meta-analysis has shown that increases in BNP and NT-proBNP levels are associated with a ninefold and sixfold increase in the risk of death in normotensive PE patients, respectively (27). Of note, the relevance of isolated increases in cardiac markers in patients with no signs of RV dysfunction has not been defined to date. Furthermore, while a variety of studies and meta-analyses have documented increased risk in patients with elevated levels of cardiac markers, no study has conclusively shown any benefit in patients receiving more aggressive therapy (thrombolysis). It is to be determined whether the ongoing The Pulmonary EmbolIsm THrOmbolysis (PEITHO) trial will provide an answer to this question. Another useful prognostic approach is the monitoring of elevated levels of cardiac markers of overload (with a short half-life), whereby decrease delayed beyond 24 h identifies very high-risk patients (28), justifying rapid aggressive intervention. At any rate, it is useful to determine and monitor cardiac marker levels in PE as surrogate factors in the decision-making process regarding the degree of aggressiveness of future management.
While novel biomarkers have recently been proposed, such as heart-type fatty acid-binding protein, growth differentiation factor-15, highly sensitive troponin, copeptin, adrenomedullin, and proatrial natriuretic factor, their place in the diagnosis and risk stratification of PE has yet to be established.
Blood gases:
Arterial or venous blood sampling for blood gas analysis is a routine laboratory test. A typical picture of acute PE is a combination of hypoxemia and hypocapnia with respiratory alkalosis as a result of compensatory hyperventilation. Rarely, in severe forms of PE, hypercapnia can be present. Blood gas analysis in PE patients enable more accurate risk stratification of an individual patient, primarily in terms of development of organ dysfunction (both current, when establishing the diagnosis, and over time when monitoring the future course of disease and response to therapy). Monitored parameters of organ function primarily include the presence and development of respiratory and renal dysfunction consistent both with the extent of PE (level of ventilation-perfusion mismatch) and its impact on forward cardiac output and potential development of low cardiac output syndrome. However, any blood sampling for blood gas analysis or any cannulation for regular blood drawing should be carefully considered because unnecessary puncture or cannulation before thrombolysis can result not only in inconvenience but also in fatal bleeding.
Assessment of the thrombophilia, blood clotting tests:
Although up to 50% of cases of VTE can occur in individuals without any identifiable clinical or laboratory risk factor, it may be reasonable to obtain blood samples in selected patients to perform blood tests for the presence of thrombophilia (before thrombolysis and/or initiation of vitamin K antagonist administration). This may hold true particularly for patients who develop thrombosis and are <45 years of age; with idiopathic or recurrent thrombosis, in which thrombosis occurs in unusual locations; with a family history of thromboembolic disease; or in women who experienced thrombosis or PE during pregnancy and plan to become pregnant again. The most common risk factors include antithrombin, protein C and S deficiency, factor V Leiden mutation (ie, activated protein C resistance, and antiphospholipid syndrome). Routine blood clotting tests are common in PE and are used primarily to monitor the adequacy of anticoagulation therapy. When deciding whether to perform thrombolysis, it is helpful to have the results of blood clotting tests at hand, although this is often not feasible in emergencies. In cases where coagulopathy can be reasonably anticipated (eg, a history of hemorrhagic diathesis, warfarinization, cirrhotic patients), it may be prudent to administer only a reduced dose of the thrombolytic agent and wait until the results of blood clotting tests become available.
CT pulmonary angiography:
In recent years, spiral CTA has become the gold standard for assessing patients with suspected PE (2,3), particularly as a tool capable of confirming or excluding the presence of thrombi in the pulmonary bed. Advanced multidetector systems require only a short exposure (approximately 10 s) with the patient holding their breath. Still, CTA is technically difficult to perform in patients with resting dyspnea. Another important consideration is to create a good venous access for sufficiently rapid and smooth contrast agent administration. CTA can be performed simultaneously with indirect CT venography of the deep vein system, pelvis and retroperitoneum by means of visualization of the venous system at a later time following bolus administration of the contrast used for pulmonary bed assessment. Another scan covering the area from the diaphragm down to the thigh is obtained 3 min to 4 min later.
Actual CTA is used to visualize the pulmonary bed, showing either a partly bypassed hypodense defect in the contrast-filled artery or its complete occlusion (Figure 1). A CTA scan will also adequately visualize a lung infarction (Figure 3). Spiral CTA is diagnostically most accurate, particularly in the central segments of the pulmonary artery up to the level of segmental arteries, with >95% sensitivity and specificity. The sensitivity and specificity of the technique when assessing the more peripheral segments of arteries largely depend on the type of the scanner and technical quality of the recording. However, the currently used four-, 16-, 64- or 128-slice scanners offer good visualization of the segmental and subsegmental areas. Meta-analyses have shown that a negative CTA scan is associated with very low risk of subsequent PE, thus comparing well with pulmonary angiography. However, the information provided by CTA does not only confirm the presence or absence of pulmonary bed thrombi. In fact, this technique can also substantially improve risk stratification in PE patients. The European guidelines already consider CTA-documented RV dilation as a surrogate marker in risk stratification. Some studies have reported that CTA-documented increases in the right-to-left ventricular dimension ratio correlates with prognosis (a fivefold increase in risk). The 30-day mortality of patients with a right-to-left ventricular dimension ratio >0.9 was 16% versus 8% mortality of patients without RV dilation (29). Other CTA signs of right-heart overload potentially contributing to risk stratification include the shape of the interventricular septum, main pulmonary artery width, pulmonary artery-to-aorta width ratio, decreased width of the left atrium and pulmonary veins, and contrast agent reflux into the hepatic veins and azygos vein or inferior vena cava (30). In addition, there are other tools, such as the obstruction index of Mastora et al (31) and Qanadli et al (32), in which a quantitatively determined degree of obstruction has been shown to correlate with prognosis (values <40% signalling a good prognosis whereas those >40% are consistent with a significant rise in early mortality) (33).
However, the most important contribution of CT-based assessment of a patient with suspected PE is visualization of the entire thoracic cavity and, hence, ruling out of any other potential pathologies as part of the differential diagnosis. These thoracic structures include, first and foremost, lung parenchyma (inflammation, tumour, emphysema and the above lung infarction), mediastinum (tumours, inflammation, pneumomediastinum), pleural cavity (fluidothorax, pneumothorax, tumours), pericardial space (fluidopericardium, pericardial tumours), and the thoracic aorta (aneurysm, aortic dissection, coarctation of the aorta). This assessment is sometimes referred to as a ‘triple rule-out’ approach in patients complaining of acute chest pain excluding acute coronary syndrome using coronary CTA, excluding PE using pulmonary artery angiography and ascending aorta dissection using thoracic aorta aortography.
Ventilation-perfusion scintigraphy:
This technique is used to follow the distribution of an intravenously administered radiolabelled agent. Ventilation-perfusion (V-P) scintigraphy is a highly sensitive yet non-specific technique. It is well suited to quickly rule out PE in patients at low and medium risk. Its negative predictive value is close to 100%. Ideally, it is performed as a combination of V-P assessment (ie, lung perfusion complemented with inhalation of labelled aerosol to rule out perfusion defect encountered with other lung diseases such as atelectasis, pneumonia, COPD or bullous emphysema). Lung scintigrams can be assessed together with chest x-ray films because the latter are also able to detect the above lung conditions. Drawbacks of lung scintigraphy include frequent intermediate findings; most importantly, its unavailability on a 24 h/seven day per week basis, as is the case in most nuclear medicine departments.
Residual perfusion defects can be detected by V-P scintigraphy in approximately 20% patients after six months of the acute PE episode during the follow-up. Prognostic significance of these defects is unclear. In studies examining persistant pulmonary hypertension, elevated levels of D-dimers and BNP have been reported as risk factors for reccurence of PE (34).
Pulmonary angiography:
Pulmonary angiography (PA) has been long considered to be the gold diagnostic standard for PE. The quality of CTA is currently so high that this technique has virtually replaced it. Nevertheless, PA is still performed in rare cases. Advantages of PA scintigraphy include both the possibility to perform pressure measurements in right-heart chambers and pulmonary artery (to be also used to monitor the effect of therapy) and the possibility to continue, immediately after the diagnostic assessment, with catheter-based management by thrombus fragmentation or, possibly, selective infusion of a thrombolytic agent to the site of thrombosis. Conventional PA can also be used in urgent cases of diagnostic uncertainty with advantage in emergencies posing a diagnostic quandary such as a patient presenting as an acute coronary syndrome when there is a need to exclude PE in the differential diagnosis (Figure 4).
Figure 4).
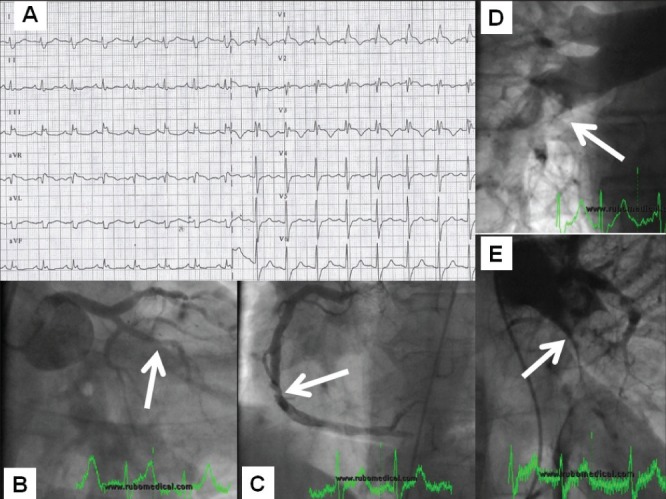
Patient referred as a case of acute coronary syndrome with ST-segment elevation and cardiogenic shock (electrocardiography [panel A – typical picture of right bundle branch block with ST-segment elevation]). Coronary angiography failed to explain the severity of status (panel B) with a normal finding on the left main coronary artery and an (at most) borderline stenosis (arrow) on the right coronary artery (panel C). Pulmonary artery angiography documented an extensive pulmonary embolism (filling defects indicated with arrows – right and left pulmonary arteries in panels D and E, respectively). The procedure continued with local thrombolytic delivery into the pulmonary artery and catheter-based fragmentation with prompt resolution of complaints and hemodynamic stabilization
Echocardiography:
Echocardiography, which should be available at all hours in any intensive care unit, is currently considered – in addition to physical examination – the main adjunct method of examination in acute PE. Echocardiography offers a potential for emergency risk stratification based on evaluation of the hemodynamic impact of the disease on the right-heart chambers while also allowing for comprehensive non-invasive assessment of the patient’s hemodynamic status.
Echocardiography in massive acute PE:
Although physical examination plays the pivotal role in massive/high-risk PE, echocardiography is an irreplaceable tool for assessing PE patients in critical condition (35). Given the hemodynamic instability, these patients are usually transferred to the intensive care unit and are thus unable to undergo morphological examination and/or transportation for assessment (ie, CT angiography, and V-P scan are absolutely inappropriate in this situation). In addition, the diagnostic algorithm in the current guidelines for the diagnosis and management of PE (2,3) – particularly if acute PE is highly suspected (signs of DVT, presence of a risk factor) – it is appropriate to urgently opt, in a hemodynamically unstable patient, for a more aggressive therapeutic intervention (ie, thrombolysis) even without direct morphological confirmation of the diagnosis of PE. However, the high suspicion should be verified by echocardiography-documented signs of the hemodynamic consequences (ie, evidence of acute RV overload) (Table 9). Therefore, bedside echocardiography in the critically unstable patient, or in a patient after cardiopulmonary resuscitation, should be performed immediately on admission to the intensive care unit.
TABLE 9.
Echocardiographic signs of right-heart overload/failure
| Increase in RV/LV dimension ratio |
| Dilation of nonhypertrophic RV (up to 5 mm) |
| Contractile asynergy, hypokinesia or akinesia of the RV free wall |
| Paradoxical ventricular septal motion, ‘D-shape’ of the interventricular septum |
| RV dilation |
| Pulmonary artery dilation |
| Regurgitation flow on tricuspid valve >2.8 m/s = gradient 31 mmHg |
| Reduced inferior vena cava fluctuations with respiration (<40%) |
Echocardiography in submassive acute PE:
Echocardiography is the ultimate technique to discriminate submassive (intermediate-risk) PE from small PE; two conditions with completely different prognoses (36–37). The body of evidence as to whether the presence of individual echocardiographic signs of impact of the disease on right-heart chambers can be helpful in guiding further therapy remains rather small. The reason for this is that most published studies do not define and distinguish overload and right-heart dysfunction; neither do they assess the overall hemodynamic impact such as LV diastolic failure due to ventricular interdependence in the presence of RV pressure overload and noninvasive measurement of other parameters such as cardiac output. According to the European Society of Cardiology guidelines, intermediate-risk PE typically presents as a combination of RV dilation, RV free wall hypokinesia, increase in the right-to-left ventricular dimension ratio in diastole, shape of the left ventricle in parasternal projection to the short axis (D-shape), vena cava inferior dilatation, restricting its collapse depending on breathing and the presence of signs of pulmonary hypertension (Figure 5, Table 9). Although these parameters no doubt suggest the impact of PE on the right-heart chambers, they have a number of limitations. These include, in particular, correlation of RV dilation with its systolic dysfunction and assessment of RV free wall hypokinesis, which can be confounded by considerable interindividual variability. It would, therefore, be most appropriate to more accurately specify the echocardiographic picture of the impact on RV function using additional parameters defining RV systolic function and LV diastolic function in more detail, with respect to ventricular interdependence and noninvasive assessment of cardiac output. These echocardiographic parameters could include (tricuspid/mitral annular plane systolic excursions) (38) assessed using M-mode, fractional change in RV area and tissue Doppler echocardiography-documented tricuspid annular plane excursions. While these parameters can be readily assessed using the more recent echocardiography devices, only few studies verifying the clinical utility have been published to date (39).
Figure 5).
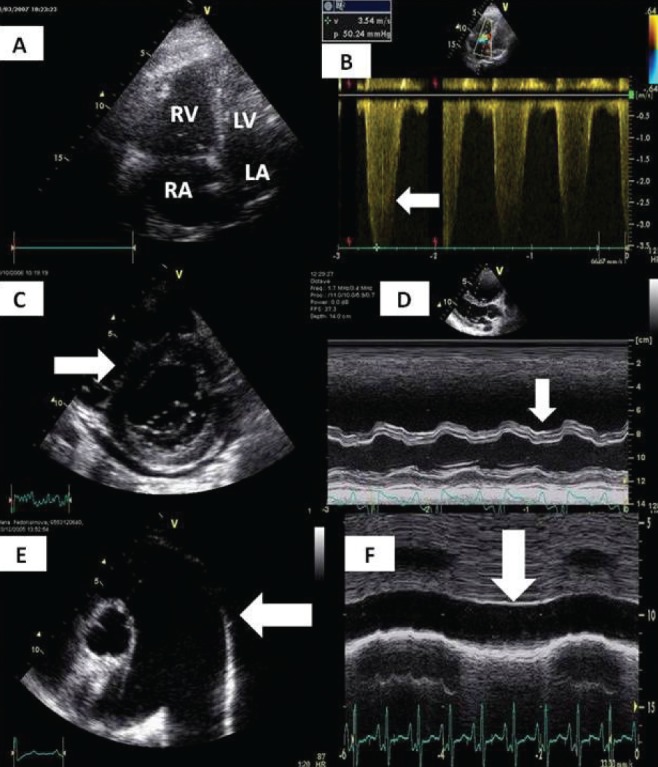
Classical echocardiographic signs of massive and submassive pulmonary embolisms. A Dilation of the right atrium (RA) and right ventricle (RV). B Jet of tricuspid regurgitation corresponding with severe pulmonary hypertension, gradient >50 mmHg. C ‘D-Shape’ of left ventricle (LV). D Abnormal motion of interventricular septum. E Dilation of the pulmonary artery. F Dilation and absence of respiratory variability of inferior vena cava corresponding with increased RA pressure. LA Left atrium
Echocardiography in a small PE:
The echocardiographic picture of the hemodynamic impact of small PE is, by definition, normal. However, echocardiography can provide invaluable information even in hemodynamically and ‘echocardiography-hemodynamically’ normal PE. This useful information can be derived from visualization of a mobile thrombus in right-heart chambers, PFO or other conditions that could affect the prognosis and risk stratification of the PE per se, and will thus facilitate the differential diagnosis of the condition. To explain, a subanalysis of the International Cooperative Pulmonary Embolism Registry (ICOPER) data showed the presence of mobile thrombi in right-heart chambers to be associated with more severe forms of PE and higher in-hospital mortality (up to 45%) (40). Echocardiographically documented presence of mobile thrombi is reported in 4% to 18% depending on the interval between the examination and onset of symptoms, and each centre’s routine policy to perform echocardiography in all PE patients. Detection of PFO, also associated with the risk of systemic embolization and almost double in-hospital mortality, is doubtedly an additional benefit (40–43).
DIFFERENTIAL DIAGNOSIS
As the most typical feature of PE, dyspnea can be most often erroneously considered to be related to acute left-heart insufficiency; if accompanied by chest pain, acute coronary syndrome with heart failure may be difficult to differentiate. Dyspnea with chest pain can also be present in other conditions such as pneumonia, pneumothorax or acute exacerbation of COPD (see above). Initial discrimination of individual conditions is critical because misdiagnosis can substantially complicate future therapeutic procedures such as arterial puncture (Figures 4 and 5).
NONTHROMBOTIC PE
Nonthrombotic PEs generally include foreign body embolization, septic emboli, fat or air embolism, amniotic fluid embolism and tumorous mass embolization.
Embolization of foreign bodies is mostly iatrogenic and caused by carelessness or careless handling of the instrumentarium. Individual cases may include pieces of catheters breaking off, dislodged guide-wires, caval filters and/or parts of implanted stents or other invasive devices subsequently travelling along blood vessels. Most of these objects become entrapped in the right heart where they can exert arrhythmogenic effects or in the pulmonary artery. Their removal is again accomplished using catheter-based techniques, which are frequently successful.
Septic embolization is most often associated with tricuspid endocarditis (typically in drug addicts), or endocarditis on foreign bodies such as catheters or pacing electrodes. Patients presenting with fever, cough, hemoptysis, pulmonary infiltrates and sepsis are at a particularly high risk. This possibility should always be considered in patients with evidence of Gram-positive infection.
Fat embolism is an often reported, although rare, real-life clinical finding typically associated with long bone fractures. It can be also associated with liposuction, lipid or propofol infusion or extensive necrosis of a steatotic liver. Manifestations may be insidious, but also fulminant with RV failure and hemodynamic compromise. The pathogenesis of fat embolism remains unclear; however, management is nonspecific and supportive.
Air embolism occurs on opening the communication between the vascular system and air (surgical wound, poorly sealed or open vascular access sites, inadequate compression of the site following removal of a venous catheter, etc). Air embolism may occur both in veins and arteries, with symptoms being systemic and frequently dramatic. Presentations are dependent on the amount of air entering the vascular system, with lethal amounts reported to be approximately 200 mL (approximately 3 mL/kg to 5 mL/kg of body weight). Once in the venous system, the air causes obstruction in the RV outflow tract or in the pulmonary arteries; both may result in circulatory failure. Every effort should be made to prevent additional amounts of air entering the vascular system and to maximize hemodynamic support. Patients are positioned on their left side with their head tilted down; a beneficial effect of hyperbaroxia has been reported in cases with air reaching the brain.
Amniotic fluid may pose a fatal complication in pregnancy. Although a rare occurrence (one in 8000 to 80,000 pregnancies), it is associated with high maternal and fetal mortality. Manifestations can again be mild to fatal in the form of coagulopathy and organ dysfunction. Therapy is, again, supportive.
Tumourous mass embolization can occur in breast malignancies, prostate, renal, hepatic, gastric or pancreatic cancer. Tumourous mass embolization may present as a typical PE or lung infarction, occasionally as an acute pulmonary hypertension secondary to obstruction of small vessels or the lymphatic system (typically generalized gastric cancer). By nature, in most cases, treatment is only supportive.
Footnotes
FUNDING: The manuscript preparation was supported by PRVOUK-P35/LF1/ and the operational programme Prague Competetiveness (project reg. no. CZ.2.16/3.1.00/24012).
DISCLOSURES: The authors have no financial disclosures or conflicts of interest to declare.
REFERENCES
- 1.Oger E. Incidence of venous thromboembolism in a community-based study in western France. Thromb Haemost. 2000;83:657–60. [PubMed] [Google Scholar]
- 2.Widimský J, Malý J, Eliáš P, et al. Doporučení pro diagnostiku a léčbu akutní plicní embolie. Vnitř. Lék. 2008;54:1S25–1S72. [Google Scholar]
- 3.Torbicki A, Perrier A, Konstantidines S, et al. Guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J. 2008;29:2276–315. doi: 10.1093/eurheartj/ehn310. [DOI] [PubMed] [Google Scholar]
- 4.Meignan M, Rosso J, Gauthier H, et al. Systematic lung scans reveal a high frequency of silent pulmonary embolism in patients with proximal deep venous thrombosis. Arch Intern Med. 2000;160:159–64. doi: 10.1001/archinte.160.2.159. [DOI] [PubMed] [Google Scholar]
- 5.Pineda LA, Hathwar VS, Grant BJ. Clinical suspicion of fatal pulmonary embolism. Chest. 2001;120:791–5. doi: 10.1378/chest.120.3.791. [DOI] [PubMed] [Google Scholar]
- 6.Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: Clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER) Lancet. 1999;353:1386–9. doi: 10.1016/s0140-6736(98)07534-5. [DOI] [PubMed] [Google Scholar]
- 7.Joffe HV, Kucher N, Tapson VF, et al. Upper-extremity deep vein thrombosis: A prospective registry of 592 patients. Circulation. 2004;110:1605–11. doi: 10.1161/01.CIR.0000142289.94369.D7. [DOI] [PubMed] [Google Scholar]
- 8.British Thoracic Society guidelines for the management of suspected acute pulmonary embolism. Thorax. 2003;58:470–83. doi: 10.1136/thorax.58.6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McIntyre KM, Sasahara AA. The hemodynamic response to pulmonary embolism in patients without prior cardiopulmonary disease. Am J Cardiol. 1971;28:288–94. doi: 10.1016/0002-9149(71)90116-0. [DOI] [PubMed] [Google Scholar]
- 10.Konstantinides S, Geibel A, Heusel G, et al. Heparin plus alteplase compared with heparin alone in patients with submassive pumonary embolism. N Engl J Med. 2002;347:1143. doi: 10.1056/NEJMoa021274. [DOI] [PubMed] [Google Scholar]
- 11.Bĕlohlávek J, Dytrych V, Linhart A. Pulmonary embolism, part II: Management. Exp Clin Cardiol. 2013;2:139–47. [PMC free article] [PubMed] [Google Scholar]
- 12.Grifoni S, Olivotto I, Cecchini P, et al. Short-term clinical outcome of patients with acute pulmonary embolism, normal blood pressure, and echocardiographic right ventricular dysfunction. Circulation. 2000;101:2817–22. doi: 10.1161/01.cir.101.24.2817. [DOI] [PubMed] [Google Scholar]
- 13.Král A, Bělohlávek J, Dytrych V, et al. Současná léčba pacientů s akutní a subakutní plicní embolií s ohledem na nově publikovaná doporučení diagnostiky a léčby tohoto onemocnění. Cor Vasa. 2009;51:11–12. [Google Scholar]
- 14.Widimský J. Několik poznámek k plicní embolii. Cor Vasa. 2009;51:11–12. [Google Scholar]
- 15.Jimenez D, Diaz G, Valle M. Prognostic value of syncope in the presentation of pulmonary embolism. Arch Bronconeumol. 2005;41:385–8. doi: 10.1016/s1579-2129(06)60246-2. [DOI] [PubMed] [Google Scholar]
- 16.Piazza G, Goldhaber SZ. Chronic thrombembolic pulmonary hypertension. N Engl J Med. 2011;364:351–60. doi: 10.1056/NEJMra0910203. [DOI] [PubMed] [Google Scholar]
- 17.Lucassen W, Geersing GJ, Erkens PM, et al. Clinical decision rules for excluding pulmonary embolism: A meta-analysis. Ann Intern Med. 2011;155:448. doi: 10.7326/0003-4819-155-7-201110040-00007. [DOI] [PubMed] [Google Scholar]
- 18.Hugli O, Righini M, Le Gal G, et al. The pulmonary embolism rule-out criteria (PERC) rule does not safely exclude pulmonary embolism. J Thromb Haemost. 2011;9:300. doi: 10.1111/j.1538-7836.2010.04147.x. [DOI] [PubMed] [Google Scholar]
- 19.Haap MM, Gatidis S, Horger M, et al. Computed tomography angiography in patients with suspected pulmonary embolism – too often considered? Am J Emerg Med. 2012;30:325–30. doi: 10.1016/j.ajem.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 20.Burge AJ, Freeman KD, Klapper PJ, et al. Increased diagnosis of pulmonary embolism without a corresponding decline in mortality during the CT era. Clin Radiol. 2008;63:381–6. doi: 10.1016/j.crad.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Bělohlávek J, Beran S, Linhart A. Critical ostial celiac trunk stenosis presenting as abdominal angina during massive pulmonary embolism with cardiogenic shock. J Invasive Cardiol. 2009;21:139–40. [PubMed] [Google Scholar]
- 22.Bělohlávek J, Aschermann M. Doporučený postup pro diagnostiku a léčbu akutních koronárních syndromů bez elevací ST úseků na EKG. Vnitř Lék. 2008;54(Supl 1):1S7–1S23. Cor Vasa 2008:50(Suppl):1S7–1S23. Česká kardiologická společnost. [Google Scholar]
- 23.Douketis J, Tosetto A, Marcucci M, et al. Patient-level meta-analysis: Effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med. 2010;153:523–31. doi: 10.7326/0003-4819-153-8-201010190-00009. [DOI] [PubMed] [Google Scholar]
- 24.Becattini C, Vedovati MC, Agnelli G. Prognostic value of troponins in acute pulmonary embolism. Circulation. 2007;116:427–33. doi: 10.1161/CIRCULATIONAHA.106.680421. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez O, Trinquart L, Colombet I, et al. Prognostic value of right ventricular dysfunction in patients with haemodynamically stable pulmonary embolism: A systematic review. Eur Heart J. 2008;29:1569–77. doi: 10.1093/eurheartj/ehn208. [DOI] [PubMed] [Google Scholar]
- 26.Jiménez D, Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism. Chest. 2009;136:974–82. doi: 10.1378/chest.09-0608. [DOI] [PubMed] [Google Scholar]
- 27.Kostrubiec M, Pruszczyk P, Kaczynska A, Kucher N. Persistent NT-proBNP elevation in acute pulmonary embolism predicts early death. Clin Chim Acta. 2007;382:124–8. doi: 10.1016/j.cca.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Widimský J. Diagnostika a léčba akutní plicní embolie v roce 2010. Vnitřní lékařství. 2011;57:5–21. [PubMed] [Google Scholar]
- 29.Schoepf UJ, Kucher N, Kipfmueller F, et al. Right ventricular enlargement on chest computed tomography: a predictor of early death in acute pulmonary embolism. Circulation. 2004;110:3276–80. doi: 10.1161/01.CIR.0000147612.59751.4C. 16; [DOI] [PubMed] [Google Scholar]
- 30.Kang KD, Thilo C, Schoepf J, et al. CT Signs of right ventricular dysfunction: Prognostic role in acute pulmonary embolism. JACC Cardiovasc Imaging. 2011;4:841–9. doi: 10.1016/j.jcmg.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Mastora I, Remy-Jardin M, Masson P, et al. Severity of acute pulmonary embolism: Evaluation of a new spiral CT angiographic score in correlation with echocardiographic data. Eur Radiol. 2003;13:29–35. doi: 10.1007/s00330-002-1515-y. [DOI] [PubMed] [Google Scholar]
- 32.Qanadli SD, El Hajjam M, Vieillard-Baron A, et al. New CT index to quantify arterial obstruction in pulmonary embolism: comparison with angiographic index and echocardiography. AJR Am J Roentgenol. 2001;176:1415–20. doi: 10.2214/ajr.176.6.1761415. [DOI] [PubMed] [Google Scholar]
- 33.Thieme SF, Ashoori N, Bamberg F, et al. Severity assessment of pulmonary embolism using dual energy CT – correlation of a pulmonary perfusion defect score with clinical and morphological parameters of blood oxygenation and right ventricular failure. Eur Radiol. 2011;22:269–78. doi: 10.1007/s00330-011-2267-3. [DOI] [PubMed] [Google Scholar]
- 34.Cosmi B, Nijkeuter M, Valentino M, et al. Residual emboli on lung perfusion scan or multidetector computed tomography after a first episode of acute pulmonary embolism. Int Emerg Med. 2011;6:521–8. doi: 10.1007/s11739-011-0577-8. [DOI] [PubMed] [Google Scholar]
- 35.Vieillard-Baron A, Page B, Augarde R, et al. Acute cor pulmonale in massive pulmonary embolism: incidence, echocardiographic pattern, clinical implications and recovery rate. Intensive Care Med. 2001;27:1481–6. doi: 10.1007/s001340101032. [DOI] [PubMed] [Google Scholar]
- 36.Goldhaber SZ. Echocardiography in the management of pulmonary embolism. Ann Intern Med. 2002;136:691–700. doi: 10.7326/0003-4819-136-9-200205070-00012. [DOI] [PubMed] [Google Scholar]
- 37.Kreit JW. The impact of right ventricular dysfunction on the prognosis and therapy of normotensive patients with pulmonary embolism. Chest. 2004;125:1539–45. doi: 10.1378/chest.125.4.1539. [DOI] [PubMed] [Google Scholar]
- 38.Gromadziński L, Ciurzyński M, Januszko-Giergielewicz B, et al. Diagnostic value of mitral and tricuspid annular excursion in the diagnostics of acute pulmonary embolism patients with chronic heart failure. Int J Cardiol. 2011;149:118–9. doi: 10.1016/j.ijcard.2011.01.070. [DOI] [PubMed] [Google Scholar]
- 39.Dytrych V, Bělohlávek J, Král A, Linhart A. Zásadní role echokardiografie u akutní plicní embolie. Interv Akut Kardiol. 2011;10(Suppl A):17–19. [Google Scholar]
- 40.Torbicki A, Galie N, Covezzoli A, et al. Right heart thrombi in pulmonary embolism: Results from the International Cooperative Pulmonary Embolism Registry. J Am Coll Cardiol. 2003;41:2245–52. doi: 10.1016/s0735-1097(03)00479-0. [DOI] [PubMed] [Google Scholar]
- 41.Chapoutot L, Nazeyrollas P, Metz D, et al. Floating right heart thrombi and pulmonary embolism: Diagnosis, outcome and therapeutic management. Cardiology. 1996;87:169–74. doi: 10.1159/000177081. [DOI] [PubMed] [Google Scholar]
- 42.Schuchlenz H, Weihs W, Horner S, et al. The association between the diameter of a patent foramen ovale and the risk of embolic cerebrovascular events. Am J Med. 2000;109:456–62. doi: 10.1016/s0002-9343(00)00530-1. [DOI] [PubMed] [Google Scholar]
- 43.Konstantinides S, Geibel A, Kasper W, et al. Patent foramen ovale is an important predictor of adverse outcome in patients with major pulmonary embolism. Circulation. 1998;97:1946. doi: 10.1161/01.cir.97.19.1946. [DOI] [PubMed] [Google Scholar]