

Abstract
BACKGROUND/OBJECTIVES:
Previous studies using isolated mitochondria have provided new insight into the mechanisms and interventions for ischemia and reperfusion (I/R) injury. In in vitro experiments involving isolated mitochondria, hypoxia and reoxygenation (H/R) has been widely used to mimic I/R injury. However, in in vitro H/R mitochondrial experiments, the effects of various substrates on mitochondrial oxidative phosphorylation are unclear. In the present study, the effects of in vitro I/R injury on mitochondrial oxidative phosphorylation under different substrate conditions were investigated.
METHODS:
Hypoxia was achieved following complete consumption of oxygen by mitochondria isolated from rat heart tissue in an experimental chamber. The H/R protocol involved 30 min hypoxia followed by 15 min reoxygenation in a chamber opened to the atmosphere. Mitochondrial respiration and respiratory control ratio (RCR) were measured.
RESULTS:
When pyruvate/malate were used as substrates, H/R significantly decreased state 3 respiration (28.2±12 nmol O2/min/mg protein) and RCR (2.7±0.8) compared with the control (121.4±32.5 nmol O2/mg protein/min and 7.8±1.2, respectively). In contrast, when succinate was used without rotenone, H/R significantly increased state 3 respiration (57.0±11.2 nmol O2/mg protein/min) and RCR (2.0±0.3) compared with the control (48.2±12.3 nmol O2/mg protein/min and 1.3±0.2, respectively).
CONCLUSIONS:
The present study demonstrated that mitochondrial oxidative phosphorylation can be modulated by H/R in vitro depending on substrate conditions.
Keywords: Ischemia/reperfusion injury, Isolated mitochondria, Oxidative phosphorylation
Isolated mitochondria have been widely used to investigate mitochondrial bioenergetics. Oxygen consumption recordings using isolated mitochondria revealed mitochondrial coupling between oxidation and phosphorylation under physiological and pathological conditions such as ischemia and reperfusion (I/R). It is well accepted that mitochondria have a crucial role in I/R injury and that I/R-induced mitochondrial dysfunction leads to cell death (1,2). Indeed, isolated mitochondria from myocardium damaged by I/R, cardiac arrest (3) or septic shock (4) exhibit impaired oxidative phosphorylation activity. Intact isolated mitochondria from normal myocardium have also been used to elucidate the mechanisms of I/R injury. Several studies (5–7) have demonstrated that intact mitochondria exposed to hypoxia followed by normoxia may be impaired and mimic pathological mitochondria obtained from I/R-injured myocardium. Thus, an in vitro model of hypoxia and reoxygenation (H/R)-injured mitochondria may be expected to provide new insight into the mechanisms and interventions for I/R injury. On the other hand, the effects of different substrates on mitochondrial oxidative phosphorylation and the appropriate substrate conditions for in vitro H/R mitochondrial experiments remain controversial because ischemia and/or reperfusion may dramatically modulate substrate conditions (8,9). In the present study, we focused on the effects of in vitro H/R injury on oxidative phosphorylation in isolated mitochondrial under different substrate conditions.
METHODS
All experimental procedures and protocols used in the present study were reviewed and approved by the Animal Care and Use Committee of the Sapporo Medical University (Sapporo, Hokkaido, Japan).
Isolation of rat heart mitochondria
Male Wistar rats (200 g to 250 g) were anesthetized using an intraperitoneal injection of pentobarbital (50 mg/kg), after which mitochondria were isolated from the heart using differential centrifugation as previously described (10). Hearts were excised and washed in isolation buffer (200 mM mannitol, 50 mM sucrose, 5 mM KH2PO4, 5 mM 3-(N-morpholino) propanesulfonic acid, 1 mM EGTA, 0.1% bovine serum albumin, pH 7.3 adjusted with 5 M KOH) and minced into small pieces at 4°C. The suspension was homogenized in the presence of 5 U/mL Bacillus licheniformis protease with a T 25 dispenser (IKA-Werke, Germany). The mitochondrial pellets were resuspended in isolation buffer at a concentration of 10 mg/mL to 20 mg/mL, stored on ice and used for experiments within 4 h. Protein concentrations were determined using a modified Lowry assay kit (Bio-Rad, USA).
Measurement of mitochondrial oxygen consumption
Mitochondrial oxygen consumption was measured using an oxygen electrode (Hansatech Instruments, United Kingdom) maintained at 30 °C with experiments conducted in respiration buffer (130 mM KCl, 5 mM K2HPO4, 20 mM 3-(N-morpholino) propanesulfonic acid, 2.5 mM EGTA, 1 μM Na4P2O7 and 0.1% bovine serum albumin, pH 7.4) containing 0.5 mg/mL mitochondrial protein. State 2 respiration was initiated using the complex I substrates pyruvate (5 mM) and malate (5 mM) or the complex II substrate succinate (5 mM) in the presence and absence of the complex I inhibitor rotenone (1 μM). State 3 was measured in the presence of 250 μM ADP, and state 4 was monitored after complete ADP consumption.
Ischemia reperfusion experiments with isolated mitochondria
Hypoxia was achieved following complete oxygen consumption by the mitochondria in the experimental chamber. The H/R protocol involved 30 min hypoxia and 15 min reoxygenation following opening of the chamber to the atmosphere.
State 3, state 4 and respiratory control ratios (RCR) were measured before and after H/R. All data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA followed by Tukey’s test; P<0.05 was considered to be statistically significant.
RESULTS
Figure 1 shows a representative trace of oxygen consumption in in vitro H/R mitochondrial experiments.
Figure 1).
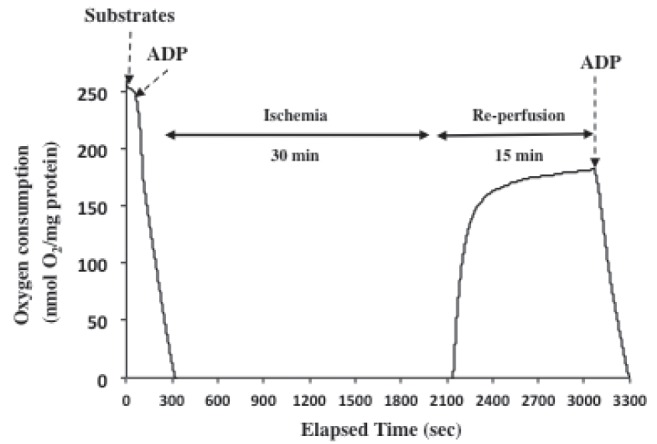
Representative trace demonstrating oxygen consumption and in vitro ischemia/reperfusion mitochondrial experiments. Mitochondrial respiration was initiated by substrates followed by adenosine diphosphate (ADP)
Figure 2 shows the effects of H/R on mitochondrial respiration initiated by pyruvate/malate or succinate in the absence and presence of rotenone. When pyruvate/malate were used as substrates, H/R significantly decreased state 3 respiration (28.2±12.7 nmol O2/min/mg protein) and RCR (2.7±0.8) compared with the control (121.4±32.5 nmol O2/mg protein/min and 7.8±1.2, respectively). In contrast, when succinate was used in the absence of rotenone, H/R significantly increased state 3 respiration (48.2 nmol O2/mg protein/min) compared with the control (57.0 nmol O2/mg protein/min) while state 4 respiration was unchanged. The RCR (2.0±0.3) with only succinate after H/R was significantly higher than the control (1.3±0.2). On the other hand, H/R significantly decreased state 3 respiration and RCR with succinate in the presence of rotenone in the same manner as pyruvate/malate.
Figure 2).
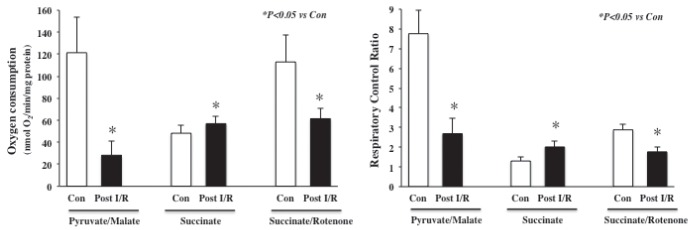
Effects of in vitro ischemia/reperfusion (I/R) on mitochondrial respiration (left panel) and respiratory control ratio (RCR) (right panel) using the substrates pyruvate/malate, succinate only, succinate/rotenone and adenosine diphosphate. In vitro I/R significantly decreased oxygen consumption rate and RCR when pyruvate/malate or succinate/rotenone were used as substrates. When succinate was used in the absence of rotenone, in vitro I/R significantly increased mitochondrial respiration and RCR. Data are presented as mean ± SD; n=8/group. Con Control
DISCUSSION
The present study showed that in vitro H/R injury could modulate oxidative phosphorylation in isolated mitochondria, depending on substrate conditions. Interestingly, in vitro H/R injury improved the RCR with succinate only in the absence of rotenone. In experiments using pyruvate/malate, in vitro H/R decreased the RCR. Previous studies have revealed that complex I is a major site of damage to the respiratory chain in ischemia and activity of complex I is markedly decreased after 20 min to 30 min of ischemia due to the effects of low pH on the enzyme, while downstream electron transport chains are relatively resistant to I/R injury (11). Succinate is converted to fumarate and malate and then oxaloacetate in the Krebs cycle when rotenone is not present (12). Oxaloacetate participates in a direct feedback inhibition of complex II that results in decreased oxidation of succinate. When complex I activity is inhibited, NADH oxidation and, thereby, the level of NADH+ are decreased, which, in turn, impairs the oxidation of malate to oxaloacetate (13). Collectively, our results suggest that in vitro H/R should impair complex I during hypoxia and, more importantly, succinate-initiated oxidative phosphorylation may be preserved after H/R via decreased accumulation of oxaloacetate from succinate oxidation.
In experiments involving isolated mitochondria, complex I-linked substrates (eg, pyruvate, malate and glutamate) and the complex II-linked substrate succinate have been widely used as physiological substrates to initiate mitochondrial oxidative phosphorylation. On the other hand, pathological conditions, such as ischemia, have been shown to result in eight- to 10-fold decreases in the concentrations of glycolytic intermediates and mitochondrial complex I-linked oxidative substrates, while the succinate concentration increased threefold in rat cerebral mitochondria (14,15). Nevertheless, in vitro studies using H/R-injured mitochondria have not focused on these facts and there are no reports that refer to these issues. In contrast to our results, Shiva et al (7) demonstrated that in vitro H/R-injured mitochondria exhibit a decreased RCR with succinate in the absence of rotenone, while nitrite administration during ischemia preserved the RCR. In their experiments, 30 min hypoxia-injured mitochondria were recentrifuged and the buffer, which should include Ca2+ and/or reactive oxygen species generated during ischemia, was replaced with fresh buffer before reperfusion. These experimental differences may be responsible for the contrasting results obtained in that study. Pravdic et al (5) showed that in vitro H/R injury decreased the RCR with pyruvate/malate as substrates and that volatile anesthetic postconditioning preserved the RCR. Ozcan et al (16) also reported that in vitro H/R elicited an important decrease of ADP-induced respiration with pyruvate/malate as substrates and the prototype K+ channel opener diazoxide preserved oxidative phosphorylation. These results of oxidative phosphorylation are consistent with the present study using pyruvate/malate, but other substrate conditions were not considered in those experiments. In in vitro and in vivo experiments with H/R- or I/R-injured mitochondria using succinate as a substrate, rotenone is usually added to inhibit reverse electron flow to complex I and prevent accumulation of oxaloacetate, which inhibits succinate dehydrogenase. Rotenone is a useful agent to assess the electron flow and oxidative phosphorylation from complex II to complex V. However, important aspects to consider are that substrate conditions in the presence of rotenone never occur in vivo under physiological or pathological conditions, and the agent is a neurotoxic agent being used to induce a Parkinson-like syndrome as an experimental model in rats (17).
The current results should be interpreted within the constraints of several limitations. Experiments were conducted in an established model of isolated mitochondria; thus, these results may not be directly comparable with cardiac cells and hearts, because the endogenous mechanisms should be involved in mitochondrial bioenergetics (11). Although Lim et al (18) demonstrated that succinate-linked respiration increased in isolated mitochondria from Langendorff-perfused hearts after 30 min global ischemia followed by 30 min reperfusion, additional in vivo experiments focusing on the differences of cardiac mitochondrial substrates condition between normoxia and hypoxia should be performed.
CONCLUSION
The present study indicates that mitochondrial oxidative phosphorylation can be modulated depending on the substrate and experimental conditions, and careful consideration of the substrates used for experiments involving H/R-injured mitochondria is needed.
Footnotes
DISCLOSURES: This research was supported solely by institutional and/or departmental sources. None of the authors have any financial interests in products related to this study.
REFERENCES
- 1.Camara A, Bienengraeber M, Stowe D. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol. 2011;2:13. doi: 10.3389/fphys.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–58. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- 3.Yeh ST, Lee HL, Aune SE, Chen CL, Chen YR, Angelos MG. Preservation of mitochondrial function with cardiopulmonary resuscitation in prolonged cardiac arrest in rats. J Mol Cell Cardiol. 2009;47:788–97. doi: 10.1016/j.yjmcc.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larche J, Lancel S, Hassoun SM, et al. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;48:377–85. doi: 10.1016/j.jacc.2006.02.069. [DOI] [PubMed] [Google Scholar]
- 5.Pravdic D, Mio Y, Sedlic F, et al. Isoflurane protects cardiomyocytes and mitochondria by immediate and cytosol-independent action at reperfusion. Br J Pharmacol. 2009;160:220–32. doi: 10.1111/j.1476-5381.2010.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zenebe WJ, Nazarewicz RR, Parihar MS, Ghafourifar P. Hypoxia/reoxygenation of isolated rat heart mitochondria causes cytochrome c release and oxidative stress; evidence for involvement of mitochondrial nitric oxide synthase. J Mol Cell Cardiol. 2007;43:411–9. doi: 10.1016/j.yjmcc.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiva S, Sack MN, Greer JJ, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bittl JA, Shine KI. Protection of ischemic rabbit myocardium by glutamic acid. Am J Physiol. 1983;245:H406–12. doi: 10.1152/ajpheart.1983.245.3.H406. [DOI] [PubMed] [Google Scholar]
- 9.Hoyer S, Krier C. Ischemia and aging brain. Studies on glucose and energy metabolism in rat cerebral cortex. Neurobiol Aging. 1986;7:23–9. doi: 10.1016/0197-4580(86)90022-9. [DOI] [PubMed] [Google Scholar]
- 10.Hirata N, Shim YH, Pravdic D, et al. Isoflurane differentially modulates mitochondrial reactive oxygen species production via forward versus reverse electron transport flow: Implications for preconditioning. Anesthesiology. 2011;115:531–40. doi: 10.1097/ALN.0b013e31822a2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solani G, Harris DA. Biochemical dysfunction in heart mitochondria exposed to ischemia and reperfusion. Biochem J. 2005;390:377–94. doi: 10.1042/BJ20042006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackrell BA, Kearney EB, Mayr M. Role 3f oxaloacetate in the regulation of mammalian succinate dehydrogenase. J Biol Chem. 1974;294:2021–7. [PubMed] [Google Scholar]
- 13.Esteitie N, Hinttala R, Wibom R, et al. Secondary metabolic effects in complex I deficiency. Ann Neurol. 2005;58:544–52. doi: 10.1002/ana.20570. [DOI] [PubMed] [Google Scholar]
- 14.Folbergrová J, Ljunggren B, Norberg K, Siesjö BK. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 1974;80:265–79. doi: 10.1016/0006-8993(74)90690-8. [DOI] [PubMed] [Google Scholar]
- 15.Benzi G, Arrigoni E, Marzatico F, Villa RF. Influence of some biological pyrimidines on the succinate cycle during and after cerebral ischemia. Biochem Pharmacol. 1979;28:2545–50. doi: 10.1016/0006-2952(79)90024-8. [DOI] [PubMed] [Google Scholar]
- 16.Ozcan C, Bienengraeber M, Djeza PP, Terzic A. Potassium channel openers protect cardiac mitochondria by attenuating oxidative stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H531–9. doi: 10.1152/ajpheart.00552.2001. [DOI] [PubMed] [Google Scholar]
- 17.Levere XM, Fontaine E. Role of substrates in the regulation of mitochondrial function in situ. IUBMB Life. 2001;52:221–9. doi: 10.1080/15216540152846037. [DOI] [PubMed] [Google Scholar]
- 18.Lim KH, Javadov SA, Das M, Clarke SJ, Suleman MS, Halestrap AP. The effects of ischemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol. 2002;545:961–74. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]