

Abstract
BACKGROUND:
Low-dose hydroxychloroquine (HCQ) and ataxia-telangiectasia-mutated (ATM) protein kinase have recently been postulated to be beneficial for the prevention of the age-associated metabolic syndrome including hypertension, hypercholesterolemia and glucose intolerance; however, the effects of low-dose HCQ on the expression of ATM downstream phosphorylated Akt (protein kinase B) and p53 proteins and cardiomyocyte apoptosis in the peri-infarct myocardium remain unclear.
OBJECTIVE:
To explore the effects of low-dose HCQ on the expression of phosphorylated Akt and p53 proteins and cardiomyocyte apoptosis in the peri-infarct myocardium in a rat model.
METHODS:
Myocardial infarction (MI) was induced experimentally in a subset of rats, while others underwent sham operation (sham). Three days after operation, surviving Sprague-Dawley male rats were divided into MI+HCQ, MI, sham+HCQ and sham groups. MI+HCQ and sham + HCQ groups were treated with HCQ (3.4 mg/kg); and MI and sham groups were treated with phosphate buffered (ie, physiological) saline (10 mL/kg) by gavage every day for 12 weeks. The expression of phosphorylated Akt and p53 proteins and cardiomyocyte apoptosis in the peri-infarct myocardium was detected by Western blot and terminal deoxynucleotidyl transferase dUTP nick end labelling, respectively.
RESULTS:
Twelve weeks after treatment, the expression of phosphorylated Akt protein was significantly increased (P<0.05). Expression of phosphorylated p53 protein was not significantly different (P>0.05) in the peri-infarct myocardium of the MI+HCQ group from that in the MI group. The cardiomyocyte apoptosis rate in the peri-infarct myocardium was significantly decreased in the MI+HCQ group compared with the MI group (P<0.05).
CONCLUSION:
Low-dose HCQ can significantly increase the expression of phosphorylated Akt protein without significantly impacting expression of phosphorylated p53 protein in the peri-infarct myocardium. Accordingly, it can inhibit cardiomyocyte apoptosis in the peri-infarct myocardium.
Keywords: Akt (protein kinase B), Cardiomyocyte apoptosis, Hydroxychloroquine (HCQ), Myocardial infarction (MI), P53 (protein 53)
Chloroquine is an activator of ataxia-telangiectasia-mutated (ATM) protein kinase; the ATM protein is mutated in individuals with ataxia-telangiectasia (1). Loss-of-function mutated ATM has been found to be associated with accelerated atherosclerosis and multiple features of the age-associated metabolic syndrome including hypertension, hypercholesterolemia and glucose intolerance (1,2). ATM is a phosphatidylinositol-3-phosphate kinase family member (3), and it is also an upstream activator of Akt (protein kinase B) and p53 proteins (3–5). Phosphorylated Akt (p-Akt) protein has been shown to be a prosurvival molecule in cardiomyocytes and its overexpression in cardiomyocytes has been demonstrated to inhibit the progression of postinfarction cardiac remodelling (6). In contrast, phosphorylated p53 (p-p53) protein acts as a proapoptosis molecule in cardiomyocytes and its expression in cardiomyocytes was found to accelerate the progression of cardiac remodelling (5,7). However, low-dose chloroquine and hydroxychloroquine (HCQ) were reported to relieve the metabolic syndrome and the extent of atherosclerosis by activating ATM (4,8,9), implying protective roles for low-dose chloroquine and HCQ, most likely in patients with coronary artery disease with ischemic cardiomyopathy. To address this issue, we performed the present study to confirm whether low-dose HCQ could primarily elevate ATM downstream p-Akt protein expression with minimal impact on p-p53 protein expression in the peri-infarct myocardium. Accordingly, low-dose HCQ can inhibit cardiomyocyte apoptosis in the peri-infarct myocardium via activating the ATM-p-Akt pathway and, thus, attenuate postinfarction cardiac remodelling, ischemic cardiomyopathy and heart failure.
METHODS
Animals
Fifty male 12-week-old Sprague Dawley rats (mean [± SD] body weight [BW] 250±30 g) were housed in cages in a climate-controlled environment with 12 h light/12 h dark schedules, and adlibitum water and chow. The experiments were performed according to the guidelines (EC Directive 86/609/ECC) for the care and use of laboratory animals of Chongqing Medical University, Chongqing, China. The following is a list of reagents used in the present study: HCQ sulfate tablet (No. H19990263, Shanghai Zhongxi Pharmaceutical Co, Ltd, China); Doppler echocardiography Vivid7 with a 7.5-MHz sector scan probe (General Electric Co, USA); anti-p-Akt (ser473) rabbit monoclone antibody (No. 4060, Cell Signaling Technology Inc, USA); Anti-p-p53 (ser 15) rabbit polyclone antibody (No. sc-11764-R, Santa Cruz Biotechnology Inc, USA); Anti-β-actin rabbit polyclonal antibody (No. bs-0061R, Beijing Biosynthesis Biotechnology Co, Ltd, China); In situ cell apoptosis detection kit (No. MK1020, Wuhan Boster Biological Technology Co, Ltd, China).
Experimental myocardial infarction
Myocardial infarction (MI) was induced in the rats (10). Briefly, the animals were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally). After endotracheal intubation and initiation of mechanical ventilation, the heart was exposed through a left thoracotomy and the proximal left anterior descending coronary artery was permanently ligated with a 6-0 silk suture. The successful coronary occlusion was confirmed by visual cyanosis of the anterior wall of the left ventricle (LV) and ST-segment elevation on electrocardiogram; sham operation was performed in the same procedure but without ligation of the coronary artery; the chest was subsequently closed. The rats were allowed to recover under care.
Treatment with HCQ
Three days after operation, MI and sham operation was confirmed by echocardiography. The surviving rats were divided into MI+HCQ (n=11); MI (n=15); sham+HCQ (n=7); and sham (n=7) groups. The MI+HCQ and sham+HCQ groups were treated with HCQ (3.4 mg/kg) (9); the MI and sham groups were treated with phosphate buffered (ie, physiological) saline (PBS, 10 mL/kg) by gavage every day for 12 weeks. Twelve weeks after treatment, there were 10 surviving rats in the MI+HCQ group, nine in the MI group, seven in the sham+HCQ group and seven in the sham group.
Echocardiography
Under slight anesthesia with sodium pentobarbital (25 mg/kg, intraperitoneally), the transthoracic M-mode echocardiograms guided by two-dimensional long-axis images were obtained through the anterior and posterior LV walls at the level of the papillary muscles. The LV end-diastolic diameter (LVEDD), LV posterior wall end-diastolic thickness (LVPWEDT), LV ejection fraction (LVEF) and heart rate (HR) were measured from the M-mode tracings according to the American Society for Echocardiology leading-edge method (10). For each measurement, data from at least three consecutive cardiac cycles were averaged.
Histology
Hearts were rapidly excised from fully anesthetized rats 12 weeks after treatment, and washed in PBS. With the atrium removed, the heart weight (HW) was measured, and the ratio of the HW to BW (HW/BW) was obtained. The LV was separated from the right ventricle. The infarcted and noninfarcted zones in the LV were outlined by visual inspection. The infarcted myocardium in the LV was immersed in fixative solution, dehydrated, subsequently embedded in paraffin and used for detection of infarction size (IS). Briefly, histological slices 5 mm thick were obtained and stained with hematoxylin and eosin. Endocardial and epicardial circumferences of the infarcted tissue and the LV were determined using image analysis software (Image-pro plus 6.0, Media Cybernetics, USA). IS was calculated as (endocardial + epicardial circumference of the infarcted tissue)/(endocardial + epicardial circumference of the LV). The peri-infarct noninfarcted myocardium in the LV was cut into two transverse slices. The first slice was stored at −80°C and used for detection by Western blot. The other slice was fixed by overnight immersion in 4% (w/v) paraformaldehyde in 100 mM sodium phosphate buffer (pH 7.4), and embedded in paraffin, and cut into 4 μm sections. The sections were used for detecting cardiomyocyte apoptosis.
Western blot
Myocardium tissue homogenates and lysates were prepared. An equal amount of protein was separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis and immunoblotted onto polyvinylidene fluoride filters. The filter was blocked with 3% bovine serum albumin and subsequently incubated with the appropriate primary antibodies (anti-p-Akt, anti-p-p53 and anti-β-actin antibody) followed by peroxidase-conjugated secondary antibodies. Positive signal bands were detected and quantified using the ChemiDoc XRS detection system (Bio-RAD, USA); the ratios of absorbance of p-Akt to β-actin and p-p53 to β-actin were obtained, which represent the relative expression levels of p-Akt and p-p53.
Detection of cardiomyocyte apoptosis using terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling
The section was dewaxed and hydrated, then according to the instruction of terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) in situ cell apoptosis detection kit, incubated in proteinase K working solution at 37 °C in a humidified atmosphere for 15 min. Fifty microlitres of TUNEL reaction mixture was added and incubated for 60 min at 37°C. After three rinses in PBS, 50 μL of converter-peroxidase was added to the section and incubated for an additional 30 min at 37°C, rinsed again three times with PBS, followed by addition of 100 μL diaminobenzidine substrate. Finally, the section was counterstained with hematoxylin and analyzed by light microscopy. The images were analyzed using a computerized image analysis system (Image-pro plus 6.0). Cells with a dark brown nucleus were defined as apoptotic cells, while the cells with a light brown nucleus were defined as normal. Twenty fields per section were randomly selected and observed. The apoptotic cells found in each field were counted. The apoptotic rate was calculated as the number of apoptotic cardiomyocytes divided by the total number of cardiomyocytes in each field and averaged.
Statistical analysis
Each of the experiments were repeated at least three times. Data are presented as mean ± SD. All analyses were performed using SPSS version 17.0 (IBM Corporation, USA). Differences among groups were evaluated using one-way ANOVA. A two-sided Tukey post hoc test was performed to compare mean values between groups; P<0.05 was considered to be statistically significant.
RESULTS
Effects of HCQ on cardiac structure and function
Twelve weeks after treatment, parameters in the surviving rats were measured and evaluated: there were significantly smaller LVEDD and HW/BW, greater LVPWEDT and increased LVEF in the MI+HCQ group compared with the MI group (all P<0.05, Table 1), although the HW/BW ratio and IS remained larger, and the LVEF was still decreased significantly in the MI+HCQ group compared with the sham+HCQ group; and the LVPWEDT remained greater, the HW/BW and IS remained greater, and the LVEF remained significantly decreased in the MI+HCQ group compared with the sham group (all P<0.05, Table 1). Furthermore, there were significantly greater LVEDD, HW/BW and IS, smaller LVPWEDT, higher HR and decreased LVEF in the MI group compared with the sham+HCQ and sham groups (All P<0.05, Table 1), while the LVPWEDT was significantly greater in the sham+HCQ group compared with the sham group (P<0.05, Table 1).
TABLE 1.
Cardiac structure and function
| Group | LVEDD, mm | LVPWEDT, mm | HW/BW, mg/g | IS | Heart rate, beats/min | LVEF |
|---|---|---|---|---|---|---|
| 0 weeks | ||||||
| MI+HCQ (n=10) | 5.61±0.61 | 1.24±0.06 | – | – | 447.34±35.61 | 0.67±0.06 |
| MI (n=9) | 5.60±0.62 | 1.24±0.06 | – | – | 448.14±34.75 | 0.68±0.06 |
| Sham+HCQ (n=7) | 5.59±0.60 | 1.22±0.07 | – | – | 439.25±36.35 | 0.69±0.07 |
| Sham (n=7) | 5.58±0.59 | 1.22±0.06 | – | – | 438.43±35.26 | 0.70±0.08 |
| 12 weeks | ||||||
| MI+HCQ (n=10) | 6.36±0.62* | 1.50±0.09*‡ | 2.62±0.18*†‡ | 0.33±0.04†‡ | 435.37±35.24 | 0.62±0.06*†‡ |
| MI (n=9) | 7.20±0.63†‡ | 1.25±0.08†‡ | 2.88±0.19†‡ | 0.32±0.03†‡ | 452.87±36.32†‡ | 0.48±0.05†‡ |
| Sham+HCQ (n=7) | 5.96±0.61 | 1.52±0.10‡ | 2.31±0.17 | 0 | 411.23±35.61 | 0.70±0.07 |
| Sham (n=7) | 5.74±0.62 | 1.37±0.09 | 2.23±0.16 | 0 | 410.27±36.27 | 0.71±0.06 |
Data presented as mean ± SD.
P<0.05 compared with myocardial infarction (MI) group;
P<0.05 compared with sham + hydroxychloroquine (HCQ) group;
P<0.05 compared with sham operation (Sham) group. BW Body weight; HR Heart rate; HW Heart weight; IS Infarction size; LVEDD Left ventricular end-diastolic diameter; LVEF LV ejection fraction; LVPWEDT LV posterior wall end-diastolic thickness
Effects of HCQ on expression of p-Akt and p-p53 proteins in the myocardium
Twelve weeks after treatment, the myocardium exhibited significant increase in p-Akt protein expression in the MI+HCQ group compared with the MI, sham+HCQ and sham groups (all P<0.05, Figure 1), while p-p53 protein expression was significantly increased in the myocardium in the MI+HCQ group compared with the sham+HCQ and sham groups (all P<0.05, Figure 1). The MI group demonstrated higher p-p53 protein expression compared with the sham+HCQ and sham groups (all P<0.05, Figure 1).
Figure 1).
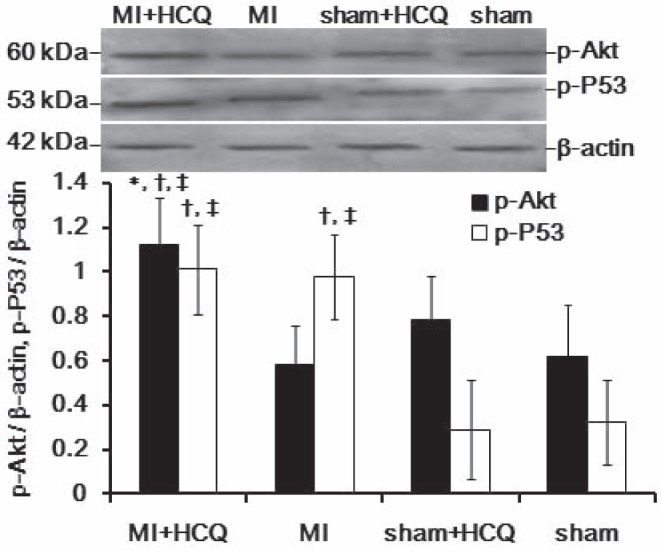
Representative Western blots of phosphorylated Akt (p-Akt) and phosphorylated p53 (p-P53) protein expression in the myocardium. *P<0.05 compared with myocardial infarction (MI) group; †P<0.05 compared with sham operation (sham) + hydroxychloroquine (HCQ) group; ‡P<0.05 compared with sham group. Data presented as mean ± SD
Effect of HCQ on cardiomyocyte apoptosis
Twelve weeks after treatment, the peri-infarct cardiomyocyte apoptosis rate was significantly decreased in the MI+HCQ group compared with the MI group, although the MI+HCQ and MI groups still demonstrated higher rates of cardiomyocyte apoptosis compared with the sham+HCQ and sham groups (all P<0.05, Figure 2).
Figure 2).
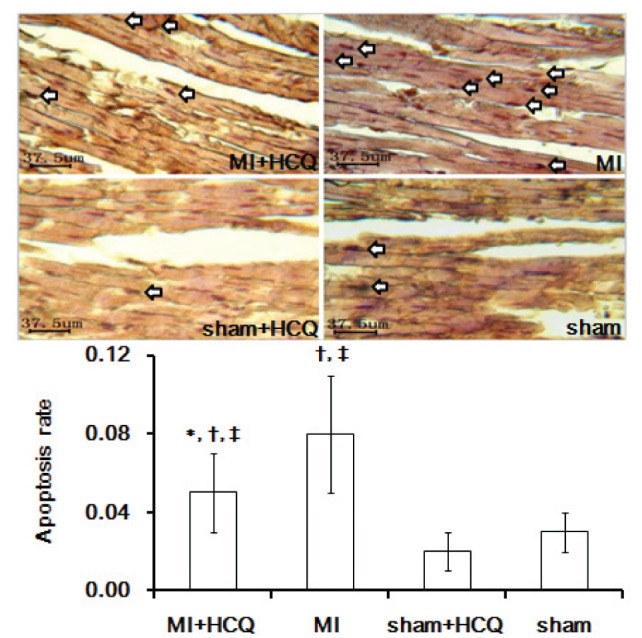
The representative terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) images of cardiomyocyte apoptosis in the myocardium (original magnification ×400) The arrows indicate apoptotic cardiomyocytes. *P<0.05 compared with myocardial infarction (MI) group; †P<0.05 compared with sham operation (sham) + hydroxychloroquine (HCQ) group; ‡P<0.05 compared with sham group. Data presented as mean ± SD
DISCUSSION
In the present study, we demonstrated that low doses of HCQ upregulated the expression of p-Akt protein with minimal impact on p-p53 protein expression in the peri-infarct myocardium. The inhibitory effect of low-dose HCQ on peri-infarct cardiomyocyte apoptosis, postinfarction ventricular dilation remodelling, ischemic cardiomyopathy and cardiac dysfunction was also observed.
ATM has been reported to play a crucial role in the maintenance of genomic integrity and the prevention of a wide variety of adverse cellular events including apoptosis and cellular senescence (11). In 2008, a study suggested that ATM is present in the heart (12) and can be activated by DNA damage induced by hypoxia, reoxygenation and oxidative stress (13,14). A more recent study has supported its role in preventing cardiomyocyte apoptosis and myocardial remodelling (15). However, the underlying mechanism remains unclear. We hypothesized whether HCQ – an activator of ATM – could function similarly in preventing postinfarction cardiac remodelling, ischemic cardiomyopathy and heart failure.
In the present study, low-dose HCQ was found to upregulate the expression of p-Akt protein with minimal impact on p-p53 protein expression in the peri-infarct myocardium. Furthermore, its inhibitory effect on peri-infarct cardiomyocyte apoptosis, postinfarction ventricular dilation remodelling, ischemic cardiomyopathy and cardiac dysfunction was also observed as expected. These findings indicate that the activated ATM-p-Akt pathway, induced by low-dose HCQ, contributes to its protective role in peri-infarct myocardium through inhibiting cardiomyocyte apoptosis, postinfarction ventricular dilation remodelling, ischemic cardiomyopathy and cardiac dysfunction. Our results are consistent with studies by Foster et al (5,15) and others (16). Postinfarction cardiac remodelling, ischemic cardiomyopathy and heart failure have become increasingly common in China (17). The pharmacological manipulation of ATM-mediated axis activity via low-dose HCQ could be useful in slowing the development of the age-associated cardiometabolic diseases (18,19).
In conclusion, low-dose HCQ can reduce peri-infarct cardiomyocyte apoptosis through upregulating the expression of p-Akt protein while minimally affecting p-p53 protein expression in the peri-infarct myocardium.
Acknowledgments
This study was supported by the Natural Science Research Fund of the Chongqing Science & Technology Commission in Chongqing City, China (CSTC, No. 2007BB5276), and the Medical Science & Technology Research Fund of Health Bureau of Chongqing City, China (No. 04-2-154 and No. 2009-2-290).
REFERENCES
- 1.Mercer JR, Cheng KK, Figg N, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–31. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu D, Yang H, Xiang W, et al. Heterozygous mutation of ataxia-telangiectasia mutated gene aggravates hypercholesterolemia in apoE-deficient mice. J Lipid Res. 2005;46:1380–7. doi: 10.1194/jlr.M400430-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Halaby MJ, Hibma JC, He J, Yang DQ. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal. 2008;20:1555–63. doi: 10.1016/j.cellsig.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Razani B, Feng C, Semenkovich CF. p53 is required for chloroquine-induced atheroprotection but not insulin sensitization. J Lipid Res. 2010;51:1738–46. doi: 10.1194/jlr.M003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster CR, Zha Q, Daniel LL, Singh M, Singh K. Lack of ataxia telangiectasia mutated kinase induces structural and functional changes in the heart: Role in beta-adrenergic receptor-stimulated apoptosis. Exp Physiol. 2012;97:506–15. doi: 10.1113/expphysiol.2011.061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006;281:32841–51. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009;47:698–705. doi: 10.1016/j.yjmcc.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Schneider JG, Finck BN, Ren J, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–89. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Morris SJ, Wasko MC, Antohe JL, et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res (Hoboken) 2011;63:530–4. doi: 10.1002/acr.20393. [DOI] [PubMed] [Google Scholar]
- 10.Litwin SE, Katz SE, Morgan JP, Douglas PS. Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation. 1994;89:345–54. doi: 10.1161/01.cir.89.1.345. [DOI] [PubMed] [Google Scholar]
- 11.Golubnitschaja O. Cell cycle checkpoints: The role and evaluation for early diagnosis of senescence, cardiovascular, cancer, and neurodegenerative diseases. Amino Acids. 2007;32:359–71. doi: 10.1007/s00726-006-0473-0. [DOI] [PubMed] [Google Scholar]
- 12.Kato H, Takashima S, Asano Y, et al. Identification of p32 as a novel substrate for ATM in heart. Biochem Biophys Res Commun. 2008;366:885–91. doi: 10.1016/j.bbrc.2007.11.175. [DOI] [PubMed] [Google Scholar]
- 13.Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem. 2003;278:12207–13. doi: 10.1074/jbc.M212360200. [DOI] [PubMed] [Google Scholar]
- 14.Watters DJ. Oxidative stress in ataxia telangiectasia. Redox Rep. 2003;8:23–9. doi: 10.1179/135100003125001206. [DOI] [PubMed] [Google Scholar]
- 15.Foster CR, Singh M, Subramanian V, Singh K. Ataxia telangiectasia mutated kinase plays a protective role in beta-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Mol Cell Biochem. 2011;353:13–22. doi: 10.1007/s11010-011-0769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorr MW, Stevens SC, Wold LE. Ataxia telangiectasia mutated kinase in the heart: Currency for myocyte apoptosis. Exp Physiol. 2012;97:476. doi: 10.1113/expphysiol.2011.063875. [DOI] [PubMed] [Google Scholar]
- 17.Li G, Wang ZH, Zhu BB, Zhang CJ. Plasma uric acid is associated with postinfarction cardiac remodeling in elderly with old myocardial infarction. Int J Gerontol. 2012;6:174–6. [Google Scholar]
- 18.Shackelford RE. Pharmacologic manipulation of the ataxia-telangiectasia mutated gene product as an intervention in age-related disease. Med Hypotheses. 2005;65:363–9. doi: 10.1016/j.mehy.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Shoelson SE. Banking on ATM as a new target in metabolic syndrome. Cell Metab. 2006;4:337–8. doi: 10.1016/j.cmet.2006.10.009. [DOI] [PubMed] [Google Scholar]