

Abstract
The G6PC3 gene encodes the ubiquitously expressed glucose-6-phosphatase enzyme (G-6-Pase β or G-6-Pase 3 or G6PC3). Bi-allelic G6PC3 mutations cause a multi-system autosomal recessive disorder of G6PC3 deficiency (also called severe congenital neutropenia type 4, MIM 612541). To date, at least 57 patients with G6PC3 deficiency have been described in the literature.
G6PC3 deficiency is characterized by severe congenital neutropenia, recurrent bacterial infections, intermittent thrombocytopenia in many patients, a prominent superficial venous pattern and a high incidence of congenital cardiac defects and uro-genital anomalies. The phenotypic spectrum of the condition is wide and includes rare manifestations such as maturation arrest of the myeloid lineage, a normocellular bone marrow, myelokathexis, lymphopaenia, thymic hypoplasia, inflammatory bowel disease, primary pulmonary hypertension, endocrine abnormalities, growth retardation, minor facial dysmorphism, skeletal and integument anomalies amongst others. Dursun syndrome is part of this extended spectrum. G6PC3 deficiency can also result in isolated non-syndromic severe neutropenia. G6PC3 mutations in result in reduced enzyme activity, endoplasmic reticulum stress response, increased rates of apoptosis of affected cells and dysfunction of neutrophil activity.
In this review we demonstrate that loss of function in missense G6PC3 mutations likely results from decreased enzyme stability. The condition can be diagnosed by sequencing the G6PC3 gene. A number of G6PC3 founder mutations are known in various populations and a possible genotype-phenotype relationship also exists. G6PC3 deficiency should be considered as part of the differential diagnoses in any patient with unexplained congenital neutropenia.
Treatment with G-CSF leads to improvement in neutrophil numbers, prevents infections and improves quality of life. Mildly affected patients can be managed with prophylactic antibiotics. Untreated G6PC3 deficiency can be fatal. Echocardiogram, renal and pelvic ultrasound scans should be performed in all cases of suspected or confirmed G6PC3 deficiency. Routine assessment should include biochemical profile, growth profile and monitoring for development of varicose veins or venous ulcers.
Keywords: Ubiquitously expressed glucose-6-phosphatase enzyme, G-6-Pase, G-6-Pase 3, G6PC3, Severe congenital neutropenia type 4, Neutropenia, Dursun syndrome, Prominent superficial venous pattern, Congenital cardiac defects, Uro-genital anomalies
Review
Introduction
The glucose-6-phosphatase enzyme catalyzes the final step of glycogenolysis, hydrolysis of glucose-6-phosphate in the endoplasmic reticulum (ER). In humans, there are three glucose-6-phosphatase enzymes encoded by a gene-family consisting of G6PC1, G6PC2 and G6PC3[1] (Figure 1). The enzymes encoded by the three genes, differ in their expression patterns and kinetic properties. G6PC1 is expressed in the liver, kidney and small intestine. G6PC2 is expressed mainly in the pancreas and G6PC3 is ubiquitously expressed. The transport of glucose-6-phosphate from cytosol to ER is facilitated by glucose-6-phosphate translocase encoded by ubiquitously expressed SLC37A4.
Figure 1.

The glucose-6-phosphatase system. A schematic representation of the glucose-6-phosphatase system. Enzymes and genes are displayed in red font, expression sites of the genes are provided within brackets and associated disorders are highlighted in bold.
Bi-allelic G6PC1 mutations cause type 1a glycogen storage disease (GSD1a, MIM 232200) [2]. GSD1a is characterized by fasting hypoglycaemia, hepatomegaly, growth retardation, fasting lactic acidosis, hyperlipidaemia and hyperuricaemia. Long-term complications of GSD1a include hepatic adenomas, pancreatitis, gout, renal failure, pulmonary hypertension, polycystic ovaries, osteoporosis, platelet dysfunction, and mild to moderate learning difficulties [3]. Bi-allelic SLC37A4 mutations result in glycogen storage disease 1b (GSD1b, MIM 232220) [4]. The phenotype of GSD1b is similar to that of GSD1a, but patients additionally develop chronic neutropenia.
The ubiquitously expressed human glucose-6-phosphatase enzyme (G-6-Pase β or G-6-Pase 3 or G6PC3, EC = 3.1.3.9), encoded by the G6PC3 gene, was independently discovered by two groups in 2003 [5,6]. g6pc3−/− mice demonstrate neutropenia, defects in neutrophil respiratory burst, chemotaxis, calcium flux, increased susceptibility to bacterial infection, elevated plasma glucagon and reduced plasma cholesterol [7,8]. In humans, homozygous or compound heterozygous G6PC3 mutations cause an autosomal recessive multi-system syndrome of severe congenital neutropenia type 4 (SCN4 or G6PC3 deficiency, MIM 612541) [9]. We described the extended phenotype associated with the condition [10,11], and demonstrated that Dursun syndrome (MIM 613034) is part of this spectrum [12]. G6PC3 mutations can also result in non-syndromic severe neutropenia [13,14].
In the last decade, significant progress has been made from the identification of the human ubiquitous glucose-6-phosphatase enzyme to understanding its role in physiology and disease. Here we provide an up-to-date review of the clinical, molecular and genetic aspects of G6PC3 deficiency.
Epidemiology
Congenital neutropenia has an estimated minimum prevalence of 6 per million [15]. In addition to G6PC3 deficiency, mutations in ELANE, GFI1, HAX1, CSF3R and WAS are known to cause congenital neutropenia [16]. Neutropenia constitutes the phenotype of a number of multi-system conditions (listed under differential diagnoses). Xia et al. studied 162 patients from the North American severe congenital neutropenia tissue repository [17]. In this cohort, bi-allelic pathogenic G6PC3 mutations were detected in two samples (1.2%), ELANE mutations in 55.6% of patients and no known genetic cause could be identified in about 40% of patients. In a recent British study of 108 kindreds that were negative for ELANE, HAX1 and WAS mutations, four patients from three families were diagnosed with G6PC3 deficiency (2.8%) [14]. These two studies provide an estimate of the frequency of G6PC3 deficiency, but the true prevalence of the condition remains unknown. To date, at least 57 patients with G6PC3 deficiency have been described in the literature (Tables 1, 2 and 3).
Table 1.
Patients with homozygous missense G6PC3 mutation
| Fam | Pt. no. | Eth | Sex | Age | Mutation | Protein | Atypical hematological features | Bone marrow | Vascular features | Cardiac features | Renal and genital defects | Other features | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
1 |
Pak |
F |
10 |
c.[130C > T]; [130C > T] |
p.[P44S]; [P44S] |
None |
Normal cellularity and neutrophil maturation. |
None |
None |
None |
None |
[13,21] |
| 2 |
2 |
Pak |
M |
20 |
c.[130C > T]; [130C > T] |
p.[P44S]; [P44S] |
None |
Normocellular with mild left-sided shift. |
None |
None |
None |
A single episode of myositis |
[14,30] |
| 3 |
3 |
Pak |
M |
10 |
c.[130C > T]; [130C > T] |
p.[P44S]; [P44S] |
None |
Normocellular |
None |
None |
None |
None |
[14] |
| 4 |
4 |
Turk |
M |
18 m |
c.[346A > G]; [346A > G] |
p.[M116V]; [M116V] |
T-cell lymphopenia, monocytosis and anemia. |
Dysplastic |
Primary PHT since infancy. |
ASD |
Bilateral inguinal herniae and undescended testes |
Diagnosed with Dursun syndrome. Thymus hypoplasia, proximally placed thumbs, broad nasal bridge, pectus carinatum and high arched palate. Died at 18 m due to severe respiratory distress. |
[12,18] |
| 5 |
Turk |
F |
18 m |
Lymphopenia, monocytosis and anemia. |
Dysplastic |
Primary PHT since infancy. |
ASD |
None |
Diagnosed with Dursun syndrome. Thymus hypoplasia, proximally placed thumbs, broad nasal bridge, pectus carinatum, single palmar creases and high arched palate. Died at 18 m due to severe respiratory distress. |
[12,18] |
|||
| 5 |
6 |
NA |
NA |
NA |
c.[347 T > A]; [347 T > A] |
p.[M116K]; [M116K] |
None |
Not described |
Prominent veins |
ASD, mild mitral and tricuspid insufficiency |
None |
Learning difficulties and Hypogonadotrophic hypogonadism. |
[26] |
| 6 |
7 |
Pak |
F |
13 |
c.[347 T > C]; [347 T > C] |
p.[M116T]; [M116T] |
None |
Normal cellularity and neutrophil maturation. |
None |
None |
None |
None |
[13,21] |
| 7 |
8 |
NA |
F |
NA |
c.[353C > G]; [c.353C > G]* |
p.[T118R]; [T118R] |
None |
Not described |
Not described |
ASD |
Not described |
Not described |
[17] |
| 8 |
9 |
Turk* |
F |
13 |
c.[461 T > C]; [461 T > C]* |
p.[L154P]; [L154P] |
None |
Hypercellular marrow, myeloid hyperplasia, no maturation arrest. |
Prominent veins on neck, chest, and abdomen. |
Mild mitral regurgitation |
None |
Frontal bossing, depressed nasal bridge, upturned nose and retrognathia. Learning difficulties. Poorly developed secondary sexual characteristics. Elevated serum total cholesterol and LDL cholesterol. |
[27] |
| 9 |
10 |
Turk |
F |
12 |
c.[554 T > C]; [554 T > C] |
p.[L185P]; [L185P] |
None |
Not described |
Prominent veins |
ASD and pulmonary valve stenosis. |
None |
None |
[9] |
| 10 |
11 |
Turk |
F |
|
c.[623 T > G]; [623 T > G] |
p.[L208R]; [L208R] |
None |
Not described |
None |
PFO and tricuspid insufficiency. |
None |
None |
[14] |
| 11 |
12 |
Arab |
F |
12 |
c.[758G > A]; [758G > A] |
p.[R253H]; [p.R253H] |
None |
Reduced mature neutrophils. |
Prominent veins |
ASD and small PDA. |
Right grade III VUR. Discontinuous labia majora and minora. |
Close set, down sloping eyes, low set ears |
[11] |
| 12 |
13 |
Arm, Turk |
M |
6 |
c.[758G > A]; [758G > A] |
p.[R253H]; [p.R253H] |
None |
Reduced mature neutrophils. |
Prominent veins |
ASD |
Bilateral cryptorchidism |
None |
[9] |
| 14 |
Arm, Turk |
F |
3 |
None |
Reduced mature neutrophils. |
Prominent veins and pulmonary venous anomaly. |
Cor triatriatum |
None |
None |
[9] |
|||
| 15 |
Arm, Turk |
F |
11 |
None |
Not described |
Prominent veins |
ASD and mitral insufficiency. |
None |
None |
[9] |
|||
| 16 |
Arm, Turk |
M |
6 |
None |
Reduced mature neutrophils. |
Prominent veins |
ASD |
Bilateral cryptorchidism |
None |
[9,30] |
|||
| 13 |
17 |
Arm, Turk |
M |
4 |
c.[758G > A]; [758G > A] |
p.[R253H]; [p.R253H] |
None |
Reduced mature neutrophils. |
Prominent veins |
Not reported |
Not reported |
Poor growth |
[9] |
| 14 |
18 |
Arab, Israel |
F |
29 |
c.[758G > A]; [758G > A] |
p.[R253H]; [p.R253H] |
None |
Hypercellular marrow with myeloid hyperplasia. Increased blast-like forms and megakaryocytes including atypical forms. |
Prominent veins and varicose veins. |
None |
None |
SGA, mild learning difficulties. Hypothyroidism. Dyslipidemia and elevated pancreatic amylase and uric acid. |
[10] |
| 19 |
Arab, Israel |
M |
26 |
None |
Mildly decreased myeloid cells. Increased megakaryocytes. |
Prominent veins and varicosities. |
None |
None |
SGA, FTT and mild learning difficulties. Hypothyroidism and secondary sexual characteristics. |
[10] |
|||
| 20 |
Arab, Israel |
F |
25 |
None |
Hypercellular marrow. Mild dysmyelopoetic changes. Decreased erythropoiesis. |
Prominent veins and varicosities. |
ASD and PDA |
None |
Poor growth, mild learning difficulties and delayed menarche. |
[10] |
|||
| 21 |
Arab, Israel |
M |
2 |
Monocytosis, lymphopenia. |
Normocellular |
Prominent veins on face. PHT. |
ASD, PDA and critical PS. |
Bilateral crytorchidism |
Liver calcifications. Mild learning difficulties. Pectus excavatum. Raised gamma glutamyltranspeptidases. |
[10] |
|||
| 15 |
22 |
Cauc |
M |
9 |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
None |
Not described |
Prominent veins |
ASD |
Micropenis |
Mild developmental delay. Hypoplastic nipples, malar flattening, bilateral metatarsus adductus and thick erythematous skin on palms and soles. |
[11] |
| 16 |
23 |
Cauc |
M |
11 |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
None |
Reduced mature neutrophils. |
Prominent veins |
PDA, ASD, bicuspid aortic valve. |
Cryptorchidism andmicropenis |
Mild developmental delay. Clubbing. Palmar erythema, inverted nipples and malar flattening. |
[11] |
| 17 |
24 |
White Ger |
F |
7 |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
None |
Not described |
Prominent veins |
ASD |
Urachal fistula |
Microcephaly |
[9] |
| 18 |
25 |
White Ger |
M |
17 |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
None |
Not described |
Prominent veins |
ASD |
Bilateral cryptorchidism and genital dysplasia |
Growth retardation |
[9] |
| 19 |
26 |
NA |
M |
NA |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
None |
Not described |
Prominent veins |
ASD |
Bilateral cryptorchidism and genital dysplasia |
Hypogammaglobunemia, microcephaly |
[26] |
| 20 |
27 |
Cauc |
M |
13 |
c.[778G > C]; [778G > C] |
p.[G260R]; [G260R] |
Iron deficiency anaemia |
Pyknotic neutrophils, no maturation arrest. Myelokathexis. Emperipolesis of neutrophils in 12% of megakaryocytes. Atypical mononuclear megakaryocytes |
Prominent veins. Mild PHT. |
PFO, thickened mitral valve and dilated right ventricle. |
Bilateral cryptorchidism, unilateral testicular agenesis, testicular microlithiasis and inguinal hernia. |
Poor growth, microcephaly. Buffalo hump, ligamentous laxity, mild proximal muscle weakness and mild sensorineural hearing loss. |
[20] |
| 28 |
Cauc |
F |
9 |
Iron deficiency anaemia |
Pyknotic Neutrophils, no maturation arrest. Myelokathexis. Emperipolesis of neutrophils in 12% of megakaryocytes. Atypical mononuclear megakaryocytes |
Prominent veins |
Large ASD |
None |
Poor growth, microcephaly, ligamentous laxity, mild proximal muscle weakness and mild sensorineural hearing loss. |
[20] |
|||
| 21 | 29 | Turk | M | ? | c.[779G > A]; [779G > A] | p.[G260D]; [G260D] | None | Maturation arrest at myelocyte/promyelocyte stage | Prominent veins | ASD | Left grade II hydronephrosis | Triangular face, frontal bossing, micrognathia and depressed nasal bridge. Cutis laxa. Bilateral hearing loss. Growth retardation. | [11] |
Table 2.
Patients with homozygous G6PC3 truncating, frameshift and splice-site mutations
| Fam | Pt. no. | Eth | Sex | Age | Mutation | Protein | Atypical hemat features | Bone marrow | Vascular features | Cardiac features | Renal and genital defects | Other features | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 22 |
30 |
White Greek |
F |
13 |
c.[141C > G]; [141C > G] |
p.[Y47X]; [Y47X] |
None |
Not described |
Prominent veins |
None |
None |
None |
[9] |
| 23 |
31 |
Arab, Leb |
M |
5 |
c.[144C > A]; [144C > A] |
p.[Y48X]; [Y48X] |
None |
Not described |
No |
None |
Cryptorchidism and bilateral inguinal hernia. |
Cleft palate |
[9] |
| 24 |
32 |
Pak |
M |
28 |
c.[190_210del]; [190_210del] |
p.[T64_I70del]; [T64_I70del] |
None |
Not described |
None |
ASD |
None |
Granulomatous inflammatory bowel disease, splenomegaly, digital clubbing and short stature. |
[14,30] |
| 24 |
33 |
Pak |
F |
16 |
None |
Not described |
None |
None |
None |
Granulomatous inflammatory bowel disease, splenomegaly, digital clubbing and short stature. |
[14,30] |
||
| 25 |
34 |
Hisp |
F |
12 |
c.[210delC]; [210delC] |
p.[I70fsX46]; [I70fsX46] |
None |
Maturation arrest at myelocyte/promyelocyte stage |
Prominent veins |
Small ASD |
None |
Triangular face, depressed nasal bridge, growth retardation, growth hormone deficiency, enlarged anterior pituitary lobe. |
[11] |
| 26 |
35 |
Hisp |
M |
14 |
c.[210delC]; [210delC] |
p.[I70fsX46]; [I70fsX46] |
None |
Maturation arrest at myelocyte/promyelocyte stage |
Prominent veins |
ASD |
None |
Triangular face, depressed nasal bridge, osteoporosis, Kawasaki’s disease, growth retardation and delayed puberty. |
[11] |
| 27 |
36 |
NA |
M |
NA |
c.[210delC]; [210delC] |
p.[I70fsX46]; [I70fsX46]*† |
None |
Not described |
Not described |
ASD and coronary aneurysm |
Not described |
Not described |
[17] |
| 28 |
37 |
Hisp |
M |
9 |
c.[218 + 1G > A]; [218 + 1G > A] |
p.[?];[?] |
None |
Reduced mature neutrophils and increased reticular staining. |
Prominent veins |
ASD |
Right inguinal hernia. Bilateral cryptorchidism. |
Frontal bossing upturned nose, recessed chin and triangular face. |
[11] |
| 29 |
38 |
Mor |
M |
22 |
c.[257delA]; [257delA]* |
p.[E86fs]; [E86fs] |
Iron deficiency anaemia |
Maturation arrest at promyelocyte stage |
Prominent veins |
ASD |
Bilateral cryptorchidism |
Poor growth |
[38] |
| 30 |
39 |
Persian |
M |
1 |
c.[416G > T]; [416G > T] |
p.[?];[?]* ‡ |
None |
Maturation arrest at myelocyte/promyelocyte stage |
Prominent veins |
ASD |
None |
None |
[11] |
| 31 |
40 |
Persian |
M |
9 m |
c.[416G > T]; [416G > T]* |
p.[?];[?]* ‡ |
None |
Maturation arrest inmyelocyte stage |
Not reported |
ASD |
None |
Failure to thrive. Died at 9 m due to severe lung infection. |
[37] |
| 32 |
41 |
Hisp |
M |
11 |
c.[766_777del]; [766_777del] |
p.[S255fs]; [S255fs] |
Mild normocytic anaemia |
Maturation arrest at myelocyte/promyelocyte stage |
Prominent veins |
ASD, MR and TR. |
Left inguinal hernia. Right cryptorchidism. |
Broad face, prominent ears, small nose, big mouth, narrow forehead and short philtrum. Mild hepatomegaly. Bilateral inner ear hearing loss. |
[11] |
| 33 |
42 |
Pak |
F |
1 |
c.[766_777del]; [766_777del] |
p.[S255fs]; [S255fs] |
None |
Reduced mature neutrophils |
None |
Hypoplastic left ventricle (mild) |
None |
Congenital ptosis and growth retardation. |
[11] |
| 34 |
43 |
Hisp |
M |
19 |
c.[766_777del]; [766_777del] |
p.[S255fs]; [S255fs] |
Anaemia |
Reduced mature neutrophils and magakaryocyte hyperplasia. |
Prominent veins |
ASD, MR and TR. |
Right cryptorchidism. |
Sensorineural hearing loss and prominent ears. |
[28] |
| 35 |
44 |
Irish |
F |
38 |
c.[829C > T]; [829C > T] |
p.[Q277X]; [Q277X] |
None |
Hyperplasia of granulocyte precursors with maturation arrest |
Varicose veins and PHT at 35y |
ASD |
Ureteric re-implantation at 12 months |
Crohn's disease diagnosed at 7y, midface hypoplasia, full lips and prognathism. |
[22,23] |
| 45 |
Irish |
M |
37 |
None |
Not described |
None |
Mitral valve prolapse |
Ureteric re-implantation at 2y |
Crohn's disease diagnosed at 15y, mid-face hypoplasia, full lips and prognathism. Died at 37y due to multi-organ failure following infective endocarditis. |
[22] |
|||
| 36 |
46 |
Persian |
F |
11 |
c.[935dupT]; [935dupT] |
p.[N313fs]; [N313fs] |
None |
Maturation arrest atmyelocyte/promyelocyte stage |
None |
ASD |
Mild VUR at 7 m (resolved at 2y). |
Triangular face, depressed nasal bridge, growth retardation and cutis laxa. |
[11] |
| 37 |
47 |
Persian |
M |
2y |
c.[935dupT]; [935dupT] |
p.[N313fs]; [N313fs] |
None |
Maturation arrest inmyelocyte stage |
Prominent veins |
ASD |
Unilateralhydronephrosis |
None |
[37] |
| 38 | 48 | Persian | M | 10 | c.[935dupT]; [935dupT] | p.[N313fs]; [N313fs] | None | Not described | No | ASD and PDA | None | None | [9] |
Table 3.
Patients with compound heterozygous G6PC3 deficiency
| Fam | Pt. no. | Eth | Sex | Age | Mutation | Protein | Atypical hemat features | Bone marrow | Vascular features | Cardiac features | Renal and genital defects | Other features | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 39 |
49 |
Cauc |
M |
7 |
c.[131C > T]; [758 G > A] |
p.[P44L]; [R253H] |
None |
Reduced numbers of mature neutrophils |
Prominent veins |
ASD |
Bilateral undescended testis, poor renal cortico-medullary differentiation |
Flat malar region, short philtrum, splenomegaly, right ptosis |
[11] |
| 40 |
50 |
Cauc |
M |
7 |
c.[208insC]; [778G > C] |
p.[I70fsX16]; [G260R] |
None |
Hypocellular bone marrow with mature arrest. |
Prominent veins |
ASD |
Left inguinal hernia |
Triangular face, height and weight below 3rd centile and growth hormone deficiency. |
[11] |
| 41 |
51 |
Hisp |
M |
1 |
c.[210delC]; [348G > A] |
p.[I70fsX46]; [M116I] |
None |
Maturation arrest at promyelocyte/myelocyte stage |
Prominent veins |
ASD |
Ambiguous genitalia. Enlarged prostatic utricle and congenital hydronephrosis. |
Triangular face, prominent upper lip, depressed tip of nose, narrow thorax, inverted nipples and flattening of acetabulum with the sacroiliac notch. |
[11] |
| 42 |
52 |
NA |
F |
18 |
c.[326–1 G > A]; [778G > C] |
p.[?]; [Gly260R] |
T-cell lymphopenia |
Normal haematopoiesis |
Prominent veins |
Mitral valve insufficiency |
None |
Inflammatory bowel disease diagnosed at 8y, hypergammaglobulinemia and growth delay. |
[19] |
| 43 |
53 |
Cauc |
M |
18 |
c.[482G > A]; [565C > T] |
p.[R161Q]; [R189X] |
None |
Not described |
Prominent veins |
ASD and bicuspid aortic valve |
Small kidneys, bilateral undescended testes |
Delayed puberty, Pyloric stenosis neonatally, massive splenomegaly age 14 years requiring removal, growth retardation. |
[11] |
| 44 |
54 |
Cauc |
F |
16 |
c.[677 + 1G > A]; [829C > T] |
p.[?]; [Gln277X] |
None |
Not described |
Prominent veins |
ASD |
None |
None |
[11] |
| 45 |
55 |
White French |
F |
13 |
c.[677 + 1G > A]; [829C > T] |
p.[?]; [Gln277X] |
None |
Not described |
Prominent veins |
None |
None |
Myopathy |
[9] |
| 46 | 56 |
White British |
F |
8 |
c.[757C > T]; [1000_1001del] | p.[R253C]; [M334fs] | None |
Normal cellularity and maturation. |
None |
None |
None |
None |
[13] |
| 57 | White British | F | 18 | None | Not described | None | None | None | None | [13] |
Key for Tables 1, 2 and 3: ASD atrial septal defect, Arm Armenian, Cauc Caucasian, Eth ethnicity, Fam family, Ger German, hemat haematological, Hisp Hispanic, Leb Lebanese, Mor Moroccan, MR mitral regurgitation, NA not available, Pak Pakistani, PFO patent foramen ovale, PHT pulmonary hypertension, PS pulmonary stenosis, Pt. no. – patient number, Ref references, TR tricuspid regurgitation, Turk Turkish, VUR vesico uretric reflux; and * denotes entries where the information is derived or corrected from the original publication. †The c.210delC mutation was described to result in p.I70fsX115 by Xia et al. However, the correct predicted protein with this mutation should be p.I70fsX46. ‡ Please see main text for explanation for how this mutation affects the splice site.
Clinical description
Typical haematological features
The haematological phenotype of G6PC3 deficiency is variable but severe neutropenia in peripheral blood (neutrophils count below 0.5×109/L) is present in all reported patients. Patients with G6PC3 deficiency usually present in the first few months of life with recurrent bacterial infections although a patient presenting with symptoms only in late teenage years has been described [13]. Sino-pulmonary infections, otitis media, urinary tract infections, skin abscesses and sepsis are common. Some patients may also suffer from oral ulcers, periodontitis, stomatitis, gingivitis and fungal infections. Untreated patients remain susceptible to bacterial infections. Nearly two-thirds of patients with G6PC3 deficiency demonstrate intermittent thrombocytopenia [11], but this has not been reported to cause clinical consequences. Importantly, unlike some other constitutional neutropenia syndromes, no cases with myelodysplastic syndrome have been yet reported in untreated or G-CSF treated patients with G6PC3 deficiency. A systematic analysis has shown this to be statistically significant [11].
Atypical haematological features
In addition to the key haematological features described above, some patients with G6PC3 deficiency can present with additional haematological abnormalities. For example, a sibling pair who were originally described as the first cases of Dursun syndrome and later shown to have G6PC3 deficiency, had severe lymphopenia and thymic hypoplasia [12,18]. Another patient recently described by Bégin et al. had persistent lymphopaenia [19]. Interestingly, this patient had low naïve CD4+ counts suggesting a defect in thymic structure or function. G6PC3 deficiency, therefore, is in the differential diagnosis for Di-George (velo-cardio-facial) syndrome.
Bone marrow examination may show maturation arrest in the myeloid lineage but some patients with G6PC3 deficiency may have a hyper- or normo-cellular bone marrow [20,21]. The reason for this variability in the phenotype of G6PC3 deficiency is not clear [21] but it emphasises the importance of considering G6PC3 deficiency as a possible cause of neutropenia even if the bone marrow does not show the maturation arrest of the myeloid cell line.
Frequent non-haematological features
The majority of patients described with G6PC3 deficiency have additional non-haematological features that, in a clinical setting, help to distinguish G6PC3 deficiency from other causes of neutropenia. Tables 1, 2 and 3 summarize important clinical features of all 57 patients with G6PC3 deficiency described in the medical literature.
A prominent superficial venous pattern is the most easily clinically recognizable non-haematological feature of G6PC3 deficiency. At least 38/57 (66.6%) patients in the medical literature have been described to demonstrate this. We have shown that the superficial veins may be less evident in infancy in some patients and tend to gradually become more prominent with age [10]. In adults these vascular changes can develop into varicose veins and result in venous ulcers.
Forty four (77.1%) patients with G6PC3 deficiency have been reported to have a congenital cardiac anomaly, making it the most common non-haematological feature of the condition. Of these 44 patients, 37 had an atrial septal defect. Other rare heart anomalies include patent foramen ovale, cor triatriatum, patent ductus arteriosus, critical pulmonary stenosis and hypoplastic left ventricle. A range of valvular anomalies, including mitral valve prolapse, insufficiency or regurgitation, tricuspid insufficiency, bicuspid aortic valves and pulmonary valve stenosis has been described.
Twenty five (43.8%) patients with G6PC3 deficiency have been described to have renal system malformation or genital anomalies. These are significantly more common in male patients (16/31) than in females (4/25). The commonest problem in males is cryptorchidism (15/25). Other features in males include inguinal hernia, ambiguous genitalia, genital dysplasia, micropenis, poor renal cortico-medullary differentiation, hydronephrosis, small kidneys and vesico-uretric reflux. In females, vesico-uretric reflux, urachal fistula and genital dysplasia have been described.
Rare non-haematological features
At least five patients, including two sibling pairs, with G6PC3 deficiency have been described with inflammatory bowel disease (IBD) [14,19,22,23]. This is in the context of a background prevalence of IBD of one in 250 in the UK [24]. IBD in G6PC3 deficiency most closely resembles Crohn’s disease and the intestinal pathology of GSD1b [19]. IBD is more commonly present in individuals with other causes of neutropenia and Fernandez et al. therefore suggested that this feature may be secondary to the neutropenia [22].
Pulmonary hypertension (PHT) has been described in 5/57 patients from three families. The diagnosis of increased pulmonary arterial pressures, unrelated to congenital heart defect, was made in infancy in one patients with G6PC3 deficiency [10] and in a sib-pair who were initially described as Dursun syndrome [12,18]. In two other patients, PHT was noted only in later life [20,22]. Interestingly, PHT is also a well recognized feature of GSD1 [25].
Endocrine abnormalities have been described in 8/57 patients with G6PC3 deficiency. These include growth hormone deficiency in two patients, including one with an enlarged anterior pituitary lobe [11]. Delayed puberty, hypogonadotropic hypogonadism or poorly developed secondary sexual characteristics have been noted in six patients [10,11,26,27]. Additionally, hypothyroidism was noted in an affected brother and sister [10].
Intrauterine growth retardation, failure to thrive and poor postnatal growth have been described in a number of children. It is not clear if the postnatal growth retardation is part of the primary phenotype or secondary to repeated infections. Notably, female g6pc3−/− mice exhibit growth retardation [8].
Some children have also been described with minor facial dysmorphism, mainly with a triangular face and a depressed nasal bridge. Experience with adult phenotype is limited but fullness of face may be an additional feature [10,23]. Minor skeletal and integument anomalies have been described in a number of patients [10]– [12]. These include scoliosis, pectus carinatum, ligamentous laxity, loose skin or cutis laxa, palmar erythema, small and/or inverted nipples. Neurological and muscular features such as microcephaly [9,20,26], sensori-neural hearing loss [11,20,28], myopathy [9], myositis [14], muscle weakness [20], and congenital ptosis [11] have been described in some patients. Cleft palate or high palate has also been described rarely [9].
One family included four patients with G6PC3 deficiency with mild to moderate developmental delay and learning difficulties [10]. Intellectual deficits have not been observed in any other patients with G6PC3 deficiency and could have been coincidental to the G6PC3 deficiency in this family. In the same family one child had raised gamma-glutamyl transferase, liver calcifications and choledocholithiasis and one adult patient had a low HDL levels and increased amylase activity. Raised serum cholesterol and LDL levels were recently noted in one patient [27].
Non-syndromic congenital neutropenia
At least six individuals with G6CP3 deficiency have been described with isolated neutropenia without any other manifestations of the disease. This suggests that more than 10% cases of G6CP3 deficiency could be apparently non-syndromic, although even this could be an underestimate due to ascertainment bias. This observation demonstrated that G6PC3 deficiency should be considered as part of the differential diagnoses in any patient with unexplained congenital neutropenia.
Aetiology and disease mechanism
Mutations in G6PC3 result in significantly reduced enzyme activity [9]. Bone marrow promyelocytes of patients with G6PC3 deficiency show increased expression of BiP, suggesting ER stress (Figure 2) [9]. Consistent with this, activation of the protein kinase-like ER kinase (PERK) pathway has been demonstrated in neutrophils of g6pc3−/− mice [29]. Neutrophils and skin fibroblasts of patients with G6PC3 deficiency have increased rates of apoptosis after induction with TNF-α or agents that induce ER stress [9]. The apoptosis is mediated, in part, by the intrinsic mitochondrial pathway [29]. G-CSF modulates these apoptotic mediators and leads to increased glucose uptake, elevated intracellular levels of glucose-6-phosphate, lactate, and adenosine-5’-triphosphate in neutrophils. Additionally, decreased intracellular glucose concentration could activate GSK-3β, leading to phosphorylation of Mcl-1 and induction of apoptosis (Figure 2).
Figure 2.

A flow diagram summarising proposed mechanisms of haematological features of G6PC3 deficiency. G6PC3 deficiency leads to decreased cytoplasmic glucose and glucose-6-phosphate levels [31] and ER stress and activation of protein like ER-kinase (PERK) [9,29]. The lower levels of glucose possibly lead to activation of GSK-3β and phosphorylation of Mcl-1. Activation of these pathways contributes to apoptosis of the cells (this part of the pathway is shown in blue boxes). G6PC3 deficiency also results in aberrant glycosylation of a NADPH oxidase subunit, gp91phox (shown in green box). The precise mechanism of aberrant glycosylation is not clear but may be mediated by perturbation of the Leloir pathway of galactose metabolism (shown in faded green). The final effect of these dysfunctions is maturation arrest of neutrophils, neutropenia and diminished respiratory burst (shown in red boxes).
In addition to the decreased neutrophil numbers, dysfunction of neutrophil activity has also been demonstrated in some patients. Hayee et al. showed severe defects in the synthesis of N- and O- glycans in patients with G6PC3 deficiency (Figure 2) [30]. Notably, they showed truncated N-glycosylation of gp91phox, a component of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase found in secondary granules of neutrophils. They proposed that aberrant glycosylation might underlie the diminished respiratory burst seen in G6PC3 deficient granulocytes and hypothesized that the glycosylation defects may also contribute towards ER stress and apoptosis. The mechanism of aberrant glycosylation is unclear. Hayee et al. postulated that accumulation of glucose-6-phosphate in G6PC3 deficiency might inhibit formation of UDP-galactose through the Leloir pathway of galactose metabolism. However, Jun et al. have shown lower levels of glucose-6-phosphate and lower glucose uptake in cells of g6pc3−/− mice [31].
Some patients with G6PC3 deficiency paradoxically demonstrate normal or hypercellularity of the bone marrow. McDermott et al. studied such a sibling pair and showed increased expression of CXCR4 in neutrophils [20]. They proposed that in such patients, stress induces overexpression of CXCR4 in patients leading to reduced egression or premature return of granulocytes to the bone marrow for destruction. They also showed in g6pc3−/− mice that a specific CXCR4 antagonist, AMD3100, rapidly reversed neutropenia.
The immune problems of G6PC3 deficiency extend beyond neutrophil dysfunction. Jun et al. demonstrated impaired macrophage respiratory burst, chemotaxis, calcium flux and phagocytic activities with down-regulation of reduced NADPH oxidase subunits and membrane translocation of p47phox, [31].
Molecular diagnosis and the mutational spectrum of G6PC3
Summary of mutations
G6PC3 maps to 17q21.31, consists of six exons and encodes the G6PC3 protein that is comprised of 346 residues and is anchored in the ER by nine transmembrane helices in a way that keeps the active site inside the ER lumen [5,6]. Histidine at position 167 is the phosphate acceptor site and arginine at 79 and histidine at 114 are proton donors [32]. The signature phosphatase motif, K-X6-RP-(X12-54)-PSGH-(X31-54)-SR-X5-H-X3-D, spans between residues 66 and 171 of G6PC3.
G6PC3 deficiency can be diagnosed by sequencing all the six exons of the gene (NM_138387.3). There are now at least 57 patients with genetically proven G6PC3 deficiency described in the literature (Tables 1, 2 and 3). Of the 114 mutated alleles, 66 are missense (Figure 3). Fourteen G6PC3 missense mutations resulting in amino acid substitutions in nine different codons have been identified. These missense mutations are spread all across the gene but no missense mutation has been described in exon 2. The largest number of mutated missense alleles have been described in exon 6. The other 48/114 mutated G6PC3 alleles result from four nonsense mutations, seven frameshift deletions or insertions and four splice-site mutations.
Figure 3.
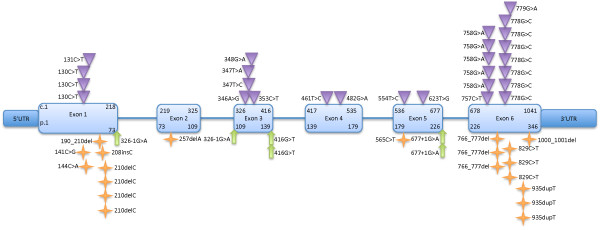
A summary of G6PC3 mutations. A schematic representation of the G6PC3 gene. The starting and ending nucleotide numbers of the cDNA and their corresponding amino acid residue numbers are provided within each exon. Each mutation is represented once for every family in which it was detected. The inverted triangles represent missense mutations, the block-arrows represent splice-site mutations and stars represent truncating or frame-shift mutations. UTR is untranslated region.
Of note, the c.416G > T mutation has been identified in two patients and predicted to be a missense change (p.S139I) [11,27]. However, the codon for residue S139 is determined by the last two nucleotides of exon 3 and the first nucleotide of exon 4. The nucleotide change responsible for p.S139I is c.416G > T and is the last nucleotide of exon 3. Hence, it is possible that this change, in fact, affects splicing. The resultant protein has not been characterized and NNsplice software predicts the spice donor site in the wild-type sequence is lost with c.416G > T [33]. We have, therefore, considered c.416G > T as a splice-site mutation in our analysis.
Founder mutations
To date, a number of G6PC3 mutations have been identified more frequently in particular ethnic groups. The homozygous p.P44S mutation has been described in three unrelated Pakistani patients and in two of these patients it was confirmed to be present on a common haplotype [14]. Ten patients, from four unrelated families, with the homozygous p.R253H mutation have been ascertained from different countries in the Middle East, suggesting that p.R253H may be a founder mutation in this population. Seven patients from six families with the homozygous p.G260R mutation have a White European ancestry. Two patients with compound heterozygous mutations also have p.G260R as one of the changes suggesting that it is an ancient European founder mutation in Caucasians. Other mutations described in multiple unrelated patients from particular ancestry include p.G277X in Europeans, p.N313fs in Persians and p.I70fsX46 in Hispanic patients.
Functional prediction of G6PC3 mutations
The mechanism by which G6PC3 missense mutations lead to loss of enzyme activity remains unclear. Notably, the protein structure of G6PC3 has not been defined. G6PC3 is a paralog of G6PC and the two genes, along with G6PC2, are thought to have arisen from ancient duplication events [34]. The structure of liver glucose-6-phosphatase (coded by G6PC) is better characterized [35]. Due to their similarities, G6PC may serve as a useful model for studying G6PC3 [5,6]. The G6PC molecule is constituted from 357 amino acids (in contrast with 346 for G6PC3) and 120 of 346 (~35%) amino acids are identical between the two molecules.
Out of the nine sites substituted in patients with G6PC3 deficiency, five amino acids (P44, M116, T118, R161 and G260) demonstrate complete conservation between G6PC3 and G6PC amino acid sequences (Figure 4). One residue, R253, is semi-conserved and three are not conserved (L154, L185 and L208). Furthermore, three G6PC3 mutants with reduced enzyme activity that have not been demonstrated in patients, but were created in the laboratory (R79A, H114A and H167A), also affect fully conserved sites [32]. Six out the nine residues affected by missense G6PC3 mutations are fully conserved in orthologs till the level of Xenopus (P44, M116, T118, R161, R253 and G260) (Figure 5). L154 is conserved till Drosophila and L185 and L208 are identical in human and mouse. All the active site residues are fully conserved.
Figure 4.
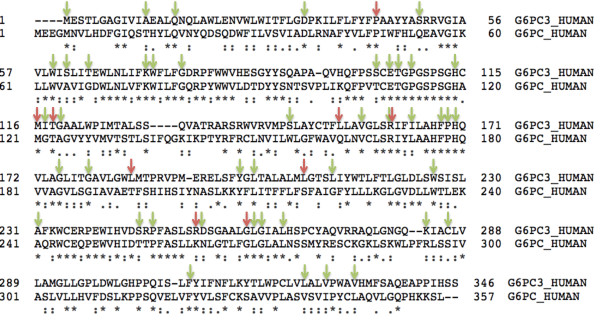
G6PC3 and G6PC alignment and known missense mutations. A ClustalW alignment of G6PC3 and G6PC amino acid sequences. Identical residues are marked by *. Sites where missense mutation has been identified in G6PC3 are highlighted by red arrows. Green arrows highlight missense mutations in GSD1a. The conserved residues are enriched in missense mutations described in two diseases.
Figure 5.

G6PC3 multi-species alignment. Multi-species ClustalW alignment of G6PC3. Identical residues are marked by *. Sites where missense mutation has been identified in G6PC3 are highlighted by red arrows. Amino acids in trans-membrane regions are shown under lines of different colours. Note that all the missense mutations affect trans-membrane residues.
In GSD1a, 54 G6PC missense mutations resulting in substitution of 48 residues have been reported [36]. Out of these 48 residues, 29 are completely conserved between G6PC and G6PC3 (Figure 4). Combining the G6PC and G6PC3 missense mutations data, 33 of 120 highly conserved and only 20 of 226 semi-conserved or non-conserved residues are substituted due to a missense mutation in either GSD1a or G6PC3 deficiency. This difference is statistically significant (p < 0.0001, calculated by Fisher’s exact test). Hence, it is likely that the protein structure, the mutation spectrum and the molecular mechanisms underlying missense mutations in the two diseases are comparable.
G6PC missense mutations have been classified into those affecting active site, helical and non-helical depending on their predicted topological position [36]. Studies have demonstrated that active site mutations abolish the G6PC enzyme activity without affecting its stability. The majority of the helical substitutions destabilize the mutant protein because the integrity of trans-membrane helices is critical for correct folding and stability. Moreover, the non-helical regions of G6PC have been shown to be less important for enzyme stability and activity.
Mapping known missense G6PC3 mutations on the predicted protein structure shows that all nine missense mutations are predicted to be helical (Figure 6). Hence analogous to findings in G6PC, we predict the nine known G6PC3 mutations result in decreased enzyme stability and loss of function.
Figure 6.
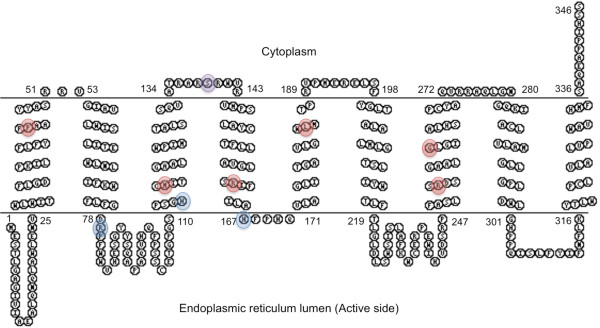
Predicted topology of G6PC3 with missense mutations. Residues substituted by missense mutation are highlighted by red circles and the laboratory generated substitutions by blue circles. The S139 residue, predicted in previous publications to be substituted by c.416G > T mutation is highlighted by purple circle. Our analysis suggests that the c.416G > T might be a splice-site mutation (see main text for discussion).
The mutation data additionally highlights the importance of certain critical G6PC3 residues. For example, four different substitutions affecting residue number 116 (p.M116V, p.M116K, p.M116T and p.M116I) have been reported in five patients with G6PC3 deficiency (Figure 3). Similarly, two different substitutions have been detected affecting residue 44 in three patients (p.P44S and p.P44L), with p.P44S detected in two unrelated Pakistani patients.
Truncating mutations can result in nonsense-mediated decay of the transcript, or in formation of a non-functional aberrant protein. In autosomal recessive disorders, therefore, patients homozygous for truncating mutations are predicted to have a complete absence of the protein encoded by the mutated gene. At least 19 patients with bi-allelic truncating G6PC3 mutations (excluding splice-site mutations) have been described to date (Table 2). If mutations in these patients result in complete absence of G6PC3 enzyme activity, then clearly it is compatible with life in humans. However, studies of enzyme activity have not yet been reported in any patients with truncating G6PC3 mutations. Moreover, current evidence does not indicate that patients with bi-allelic truncating G6PC3 mutations have a more severe phenotype than most patients with missense G6PC3 mutations. Another possibility is that an alternative in-frame translation start site could be used in patients with truncating mutations. In this context, it is interesting to note that apart from p.E86fs, all truncating G6PC3 mutations cluster in in exons 1, 5 or 6, which flank the exons coding for the phosphatase activity. This is in contrast with G6PC3 missense mutations that are spread across all exons. Without evidence from functional work it is impossible to conclude if this observation has a functional basis or if it is an incidental finding.
The truncating mutation closest to the carboxy-terminal of the G6PC3 protein is p.M334fs. This mutation has not been characterised functionally but it is predicted to result in read through of additional amino acids and not cause premature termination of the protein. Exploration of the mechanism in this mutation could provide insight about the role of terminal residues of the protein.
Genotype-phenotype correlation in G6PC3 deficiency
We have shown absence of a genotype–phenotype relationship for the bone marrow phenotype in patients with G6PC3 deficiency [21]. However, our recent work has suggested that the extended phenotypic spectrum may depend on the genotype of the patients [13]. Specifically, certain mutations e.g. p.P44S seem to be more often associated with non-syndromic neutropenia with absence of other systemic features. This hypothesis is based on a small number of patients and additional patients need to be identified to test it.
Differential diagnosis
Congenital neutropenia is a genetically and phenotypically heterogeneous group of conditions. Broadly, these conditions can be divided into those where neutropenia is the predominating feature, such as in ELANE, GFI1, HAX1, CSF3R and WAS mutations and those where neutropenia is part of a multi-system disorder. The later group includes glycogen storage disorder type 1b, Schwachman-Diamond syndrome, WHIM syndrome, Barth’s disease, Chediak-Higashi syndrome, Hermansky-Pudlak syndrome type 2, Griscelli syndrome type 2, P14 deficiency, Clericuzio poikiloderma with neutropenia, Pearson syndrome, Cohen syndrome, cartilage-hair hypoplasia and Charcot-Marie-Tooth disease type 2 M [16].
Management
Neutropenia of a number of patients with G6PC3 deficiency has been treated with G-CSF. It leads to improvement in neutrophil numbers, prevents infections and improves quality of life [9,11,20]. However, in some patients G-CSF may fail to control infections even in large doses [14]. On the other hand, mildly affected patients may not necessarily require G-CSF treatment and can be managed with prophylactic antibiotics. T-cell subsets may be examined if accompanying cellular immunodeficiency is suspected.
Echocardiogram, renal and pelvic ultrasound scans should be performed in all cases of suspected or confirmed G6PC3 deficiency. Expert advice in management of congenital heart defects and genito-urinary problems must be sought. Patients, especially adults should be monitored for development of varicose veins and venous ulcers. Growth parameters should be monitored in all children with G6PC3 deficiency. If indicated, evaluation of growth hormone levels in children and pubertal development in adolescents should be considered. Biochemical profile monitoring could be part of routine part of the follow-up. Of note, raised total serum cholesterol and LDL levels were reported to normalize with statins in one patient [27]. Rare manifestations such as PHT and IBD should be managed according to standard guidelines. One patient with G6PC3 deficiency and IBD required a right hemi-colectomy and showed good response to anti-TNF treatment, infliximab [19]. The authors suggested considering bone marrow transplantation for patients with G6PC3-related IBD refractory to anti-TNFα therapy.
Prognosis
Prognosis of most patients with G6PC3 deficiency is generally good on G-CSF treatment or prophylactic antibiotics in mildly affected individuals. However, untreated G6PC3 deficiency can be fatal. Both patients with Dursun syndrome died at 18 months due to severe respiratory distress [12,18]. The oldest patient ever described with G6PC3 deficiency died at 37 years of age due to infective endocarditis [22]. Of note this patient was non-compliant with his treatment. The youngest reported death in a patient with G6PC3 deficiency was at nine months due to severe lung infection [37].
Unresolved questions
A number of questions remain to be answered in G6PC3 deficiency. Identification of more patients is required to clarify the extended phenotypic spectrum of the disease and the possible genotype-phenotype relationship. Although no cases of myelodysplastic syndrome have yet been noted in G6PC3 deficiency, long-term follow-up will be required to confirm this observation. At present, the reason for variability of the bone marrow phenotype in patients with mutations in G6PC3 is uncertain and further characterisation is warranted. Understanding the molecular basis of mutations leading to loss of enzyme activity may help to explore the therapeutic potential of small molecule chaperones in this disease, especially because missense mutations affect the majority of patients with G6PC3 deficiency. Furthermore, a similar approach may also benefit patients with GSD1a. Much work has been undertaken to understand the effect of G6PC3 deficiency on neutrophils, but mechanisms underpinning the developmental defects of other organs and other haematological lineages in the condition remain to be studied.
Conclusions
In this review we have compiled the clinical features of all the 57 patients with G6PC3 deficiency described in the literature demonstrating the variability of the condition. This will help in management of patients and in genetic counselling of families. G6PC3 deficiency should be part of the diagnostic screen of any patient with severe congenital neutropenia. Analysis of mutational spectrum provides insights into the disease mechanism and reveals a possible genotype-phenotype relationship.
Endnote
aNote – G6PC3 gene is incorrectly mapped to ORPHA486 (autosomal dominant severe congenital neutropenia) on http://www.orpha.net.
Abbreviations
ER: Endoplasmic reticulum; IBD: Inflammatory bowel disease; G6PC3: Ubiquitous glucose-6-phosphatase; G-CSF: Granulocyte-colony stimulating factor; NADPH: Nicotinamide adenosine dinucleotide phosphate; PHT: Pulmonary hypertension.
Competing interests
The authors have no competing interests to declare.
Authors’ contributions
Both authors planned the study. SB compiled and reviewed the data and wrote the manuscript. WGN reviewed the manuscript. Both authors read and approved the final manuscript.
Disease name/synonyms
G6PC3 deficiencya
Ubiquitous glucose-6-phosphatase deficiency
Severe congenital neutropenia type 4
Dursun syndrome (ORPHA178503)
Contributor Information
Siddharth Banka, Email: Siddharth.Banka@manchester.ac.uk.
William G Newman, Email: william.newman@manchester.ac.uk.
References
- Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem. 2009;284:29241–29245. doi: 10.1074/jbc.R109.025544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei KJ, Shelly LL, Pan CJ, Sidbury JB, Chou JY. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science. 1993;262:580–583. doi: 10.1126/science.8211187. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Mayatepek E, Utermann G. Molecular genetics of type 1 glycogen storage disease. Mol Genet Metab. 2001;73:117–125. doi: 10.1006/mgme.2001.3179. [DOI] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Achouri Y, Collet J-F, Van Schaftingen E. Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type lb. FEBS Letters. 1997;419:235–238. doi: 10.1016/S0014-5793(97)01463-4. [DOI] [PubMed] [Google Scholar]
- Guionie O, Clottes E, Stafford K, Burchell A. Identification and characterisation of a new human glucose-6-phosphatase isoform. FEBS Letters. 2003;551:159–164. doi: 10.1016/S0014-5793(03)00903-7. [DOI] [PubMed] [Google Scholar]
- Shieh J-J, Pan C-J, Mansfield BC, Chou JY. A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. Journal of Biological Chemistry. 2003;278:47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, Lee EJ, Westphal H, Mansfield BC, Chou JY. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest. 2007;117:784–793. doi: 10.1172/JCI30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Oeser JK, Yang C, Sarkar S, Hackl SI, Hasty AH, McGuinness OP, Paradee W, Hutton JC, Powell DR, O’Brien RM. Deletion of the gene encoding the ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein (UGRP)/glucose-6-phosphatase catalytic subunit-β results in lowered plasma cholesterol and elevated glucagon. J Biol Chem. 2006;281:39982–39989. doi: 10.1074/jbc.M605858200. [DOI] [PubMed] [Google Scholar]
- Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J, Germeshausen M, Brandes G, Lee-Gossler J, Noyan F. et al. A Syndrome with Congenital Neutropenia and Mutations in G6PC3. N Engl J Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banka S, Chervinsky E, Newman WG, Crow YJ, Yeganeh S, Yacobovich J, Donnai D, Shalev S. Further delineation of the phenotype of severe congenital neutropenia type 4 due to mutations in G6PC3. Eur J Hum Genet. 2011;19:18–22. doi: 10.1038/ejhg.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boztug K, Rosenberg PS, Dorda M, Banka S, Moulton T, Curtin J, Rezaei N, Corns J, Innis JW, Avci Z, Tran HC, Pellier I, Pierani P, Fruge R, Parvaneh N, Mamishi S, Mody R, Darbyshire P, Motwani J, Murray J, Buchanan GR, Newman WG, Alter BP, Boxer LA, Donadieu J, Welte K, Klein C. Extended spectrum of human glucose-6-phosphatase catalytic subunit 3 deficiency: novel genotypes and phenotypic variability in severe congenital neutropenia. J Pediatr. 2012;160:679–683. doi: 10.1016/j.jpeds.2011.09.019. [DOI] [PubMed] [Google Scholar]
- Banka S, Newman WG, Özgül RK, Dursun A. Mutations in the G6PC3 gene cause Dursun syndrome. Am J Med Genet. 2010;152A:2609–2611. doi: 10.1002/ajmg.a.33615. [DOI] [PubMed] [Google Scholar]
- Banka S, Wynn R, Byers H, Arkwright PD, Newman WG. G6PC3 mutations cause non-syndromic severe congenital neutropenia. Mol Genet Metab. 2013;108:138–141. doi: 10.1016/j.ymgme.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Smith BN, Evans C, Ali A, Ancliff PJ, Hayee B, Segal AW, Hall G, Kaya Z, Shakoori AR, Linch DC, Gale RE. Phenotypic heterogeneity and evidence of a founder effect associated with G6PC3 mutations in patients with severe congenital neutropenia. Br J Haematol. 2012;158:146–149. doi: 10.1111/j.1365-2141.2012.09110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Bellanne Chantelot C. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011;6:26. doi: 10.1186/1750-1172-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei N, Moazzami K, Aghamohammadi A, Klein C. Neutropenia and Primary Immunodeficiency Diseases. Int Rev Immunol. 2009;28:335–366. doi: 10.1080/08830180902995645. [DOI] [PubMed] [Google Scholar]
- Xia J, Bolyard AA, Rodger E, Stein S, Aprikyan AA, Dale DC, Link DC. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol. 2009;147:535–542. doi: 10.1111/j.1365-2141.2009.07888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dursun A, Ozgul RK, Soydas A, Tugrul T, Gurgey A, Celiker A, Barst RJ, Knowles JA, Mahesh M, Morse JH. Familial pulmonary arterial hypertension, leucopenia, and atrial septal defect: a probable new familial syndrome with multisystem involvement. Clin Dysmorphol. 2009;18:19–23. doi: 10.1097/MCD.0b013e32831841f7. [DOI] [PubMed] [Google Scholar]
- Bégin P, Patey N, Mueller P, Rasquin A, Sirard A, Klein C, Haddad E, Drouin E, Deist FL. Inflammatory Bowel Disease and T cell Lymphopenia in G6PC3 Deficiency. J Clin Immunol. 2012;33:520–525. doi: 10.1007/s10875-012-9833-6. [DOI] [PubMed] [Google Scholar]
- McDermott DH, De Ravin SS, Jun HS, Liu Q, Priel DAL, Noel P, Takemoto CM, Ojode T, Paul SM, Dunsmore KP, Hilligoss D, Marquesen M, Ulrick J, Kuhns DB, Chou JY, Malech HL, Murphy PM. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood. 2010;116:2793–2802. doi: 10.1182/blood-2010-01-265942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banka S, Wynn R, Newman WG. Variability of bone marrow morphology in G6PC3 mutations: Is there a genotype-phenotype correlation or age-dependent relationship? Am J Hematol. 2011;86:235–237. doi: 10.1002/ajh.21930. [DOI] [PubMed] [Google Scholar]
- Fernandez BA, Green JS, Bursey F, Barrett B, MacMillan A, McColl S, Fernandez S, Rahman P, Mahoney K, Pereira SL, Scherer SW, Boycott KM, Woods MO. Adult siblings with homozygous G6PC3 mutations expand our understanding of the severe congenital neutropenia type 4 (SCN4) phenotype. BMC Med Genet. 2012;13:111. doi: 10.1186/1471-2350-13-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinane AR, Vilboux T, O’Brien K, Curry JA, Maynard DM, Carlson-Donohoe H, Ciccone C, Markello TC, Gunay-Aygun M, Huizing M, Gahl WA. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J Invest Dermatol. 2011;131:2017–2025. doi: 10.1038/jid.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GP, Hungin APS, Kelly PJ, Ling J. Inflammatory bowel disease: epidemiology and management in an English general practice population. Aliment Pharmacol Ther. 2000;14:1553–1559. doi: 10.1046/j.1365-2036.2000.00886.x. [DOI] [PubMed] [Google Scholar]
- Humbert M, Labrune P, Simonneau G. Severe pulmonary arterial hypertension in type 1 glycogen storage disease. Eur J Pediatr. 2002;161:S93–S96. doi: 10.1007/s00431-002-1012-y. [DOI] [PubMed] [Google Scholar]
- Germeshausen M, Zeidler C, Stuhrmann M, Lanciotti M, Ballmaier M, Welte K. Digenic mutations in severe congenital neutropenia. Haematologica. 2010;95:1207–1210. doi: 10.3324/haematol.2009.017665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytekin C, Germeshausen M, Tuygun N, Dogu F, Ikinciogullari A. A Novel G6PC3 Gene Mutation in a Patient With Severe Congenital Neutropenia. J Pediatr Hematol Oncol. 2013;35:e81–e83. doi: 10.1097/MPH.0b013e3182679000. [DOI] [PubMed] [Google Scholar]
- Gatti S, Boztug K, Pedini A, Pasqualini C, Albano V, Klein C, Pierani P. A Case of syndromic neutropenia and mutation in G6PC3. J Pediatr Hematol Oncol. 2011;33:138. doi: 10.1097/MPH.0b013e3181f46bf4. [DOI] [PubMed] [Google Scholar]
- Jun HS, Lee YM, Song KD, Mansfield BC, Chou JY. G-CSF improves murine G6PC3-deficient neutrophil function by modulating apoptosis and energy homeostasis. Blood. 2011;117:3881–3892. doi: 10.1182/blood-2010-08-302059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayee BH, Antonopoulos A, Murphy EJ, Rahman FZ, Sewell G, Smith BN, McCartney S, Furman M, Hall G, Bloom SL, Haslam SM, Morris HR, Boztug K, Klein C, Winchester B, Pick E, Linch DC, Gale RE, Smith AM, Dell A, Segal AW. G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction. Glycobiology. 2011;21:914–924. doi: 10.1093/glycob/cwr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HS, Cheung YY, Lee YM, Mansfield BC, Chou JY. Glucose-6-phosphatase-β , implicated in a congenital neutropenia syndrome, is essential for macrophage energy homeostasis and functionality. Blood. 2012;119:4047–4055. doi: 10.1182/blood-2011-09-377820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shieh J-J, Pan C-J, Chou JY. Histidine 167 is the phosphate acceptor in glucose-6-phosphatase-β forming a phosphohistidine enzyme intermediate during catalysis. J Biol Chem. 2004;279:12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-O. [DOI] [PubMed] [Google Scholar]
- Sundström G, Larsson TA, Larhammar D. Phylogenetic and chromosomal analyses of multiple gene families syntenic with vertebrate Hox clusters. BMC Evol Biol. 2008;8:254. doi: 10.1186/1471-2148-8-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C-J, Lei K-J, Annabi B, Hemrika W, Chou JY. Transmembrane topology of glucose-6-phosphatase. J Biol Chem. 1998;273:6144–6148. doi: 10.1074/jbc.273.11.6144. [DOI] [PubMed] [Google Scholar]
- Chou JY, Mansfield BC. Mutations in the glucose-6-phosphatase-α (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat. 2008;29:921–930. doi: 10.1002/humu.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh Z, Fazlollahi MR, Eshghi P, Hamidieh AA, Ghadami M, Pourpak Z. Two cases of syndromic neutropenia with a report of novel mutation in G6PC3. Iran J Allergy Asthma Immunol. 2011;10:227–230. [PubMed] [Google Scholar]
- Aróstegui JI, De Toledo JS, Pascal M, García C, Yagüe J, Díaz de Heredia C. A novel G6PC3 homozygous 1-bp deletion as a cause of severe congenital neutropenia. Blood. 2009;114:1718–1719. doi: 10.1182/blood-2009-04-219451. [DOI] [PubMed] [Google Scholar]