

Abstract
Oog1 is an oocyte-specific gene whose expression is turned on in mouse oocytes at embryonic day (E) 15.5, concomitant with the time when most of the female germ cells stop proliferating and enter meiotic prophase. Here, we characterize the Oog1 promoter, and show that transgenic GFP reporter expression driven by the 2.7 kb and 3.9 kb regions upstream of the Oog1 transcription start site recapitulates the intrinsic Oog1 expression pattern. In addition, the 3.9 kb upstream region exhibits stronger transcriptional activity than does the 2.7 kb region, suggesting that regulatory functions might be conserved in the additional 1.2 kb region found within the 3.9 kb promoter. Interestingly, the longer promoter (3.9 kb) also showed strong activity in male germ cells, from late pachytene spermatocytes to elongated spermatids. This is likely due to the aberrant demethylation of two CpG sites in the proximal promoter region. One was highly methylated in the tissues in which GFP expression was suppressed, and another was completely demethylated only in Oog1pro3.9 male and female germ cells. These results suggest that aberrant demethylation of the proximal promoter region induced ectopic expression in male germ cells under the control of 3.9 kb Oog1 promoter. This is the first report indicating that sex-dependent gene expression is altered according to the length and the methylation status of the promoter region. Additionally, our results show that individual CpG sites are differentially methylated and play different roles in regulating promoter activity and gene transcription.
Introduction
Oogenesin1 (Oog1) is an oocyte-specific gene that is expressed after entry into meiosis and during early embryogenesis [1]. The mouse genome contains five copies of Oog1 clustered on chromosomes 4 and 12. All of the copies contain a TATA-box in the proximal upstream region, suggesting that they are transcribed. Oog1 expression begins in oocytes at E15.5 and continues to the 2-cell stage following fertilization [1]. Interestingly, OOG1 protein is localized in the nucleus of late 1-cell to early 2-cell embryos, concomitant with zygotic gene activation and first mitotic division. We previously identified a potential binding partner of OOG1, Ras and Ral guanine nucleotide dissociation stimulator (RalGDS), by yeast two-hybrid screening of a germinal vesicle (GV) oocyte cDNA library [2], but the function of Oog1 remains unknown.
Understanding how oocyte-specific genes are transcriptionally controlled is important not only for uncovering mechanisms of oogenesis and early development, but also for generating useful tools to study gene function in oocytes. For instance, the Gdf9 and Zp3 promoters, which become active in mouse oocytes after birth, are frequently used for oocyte-specific transgenic and conditional KO studies [3–7]. Three other promoters (H1oo, Npm2, and Zar1) were recently shown by injection of luciferase reporter constructs into GV oocytes to drive reporter expression in oocytes [8]. Transgenic studies have shown that a relatively short core promoter region (~100 bp) is sufficient to induce germ cell–specific expression [9,10]. Germ cell–specific isoforms of transcription factors such as TRF2, TRF3, TAF4b, TAF7L, and ALF [11–13] likely bind to this core promoter region and induce germ cell–specific gene expression.
However, some transcription factors that bind proximal or distal regions outside of the core promoter strongly affect the expression of oocyte-specific genes [14,15], suggesting that the core promoter region might be insufficient to regulate spatiotemporal gene expression in oocytes. For example, FIGα is a beta helix-loop-helix transcription factor that binds to an E-box element (CANNTG) and regulates the coordinated expression of mouse zona pellucida genes [16]. FIGα deficiency causes downregulation of several oocyte-expressed genes, including Mater, Dppa3/Stella, and Oct4 [17]. An E-box consensus sequence (CAGCTG) at -182 bp in the Gdf9 promoter is also critical for inducing gene expression in oocytes [18]. Similarly, Nobox, an oocyte-specific homeobox transcription factor expressed as early as E15.5, has been shown by mutant analysis to be required for expression of many oocyte genes, including Oog1, in newborn mouse ovaries [14,19]. A NOBOX DNA binding element (NBE: 5´-TAATTG/A-3´) located at -1796 bp in the Npm2 promoter is crucial for enhancing the basal transcriptional activity [8], indicating that distal regulatory regions are involved in oocyte-specific gene expression. In addition, the methylation status of promoter regions has been shown to control the sex-specific expression of germ cell–specific genes [20].
Here, we analyzed the promoter region of Oog1, by comparing the 5´-flanking sequences of the five Oog1 copies in the mouse genome. We identified long conserved sequences with several gaps, two of which (2.7 kb and 3.9 kb in length) were used to drive expression of a GFP reporter gene in transgenic mice: transgenic mice with either the 2.7 kb (Oog1pro2.7) or 3.9 kb (Oog1pro3.9) Oog1 upstream sequence. Both the 2.7 kb and 3.9 kb sequences showed functional promoter activity in mouse oocytes, possibly as early as E15.5, and will be useful for analyzing the function of genes expressed in oocytes. We also found that the 3.9 kb promoter functioned in male germ cells, and that the methylation status of the proximal promoter region differed between the Oog1pro2.7 and Oog1pro3.9 transgenes in male and female germ cells, suggesting that CpG methylation of the proximal region of the Oog1 promoter may control gene expression in both male and female germ cells.
Materials and Methods
Generation of transgenic mice
Oog1 5'-flanking sequences (2688 bp and 3870 bp long) containing a TATA box on the 3'-end and restriction sites on both ends were amplified from mouse genomic DNA. The PCR products were purified, cleaved with restriction enzyme, and used to replace the CMV promoter in the pAcGFP1-mem vector plasmid (Clontech Laboratories, Mountain View, CA). Linearized transgene fragments were purified and microinjected into the male pronucleus of fertilized C57BL/6J mouse eggs (SLC japan, Shizuoka, Japan). Microinjected eggs were then transferred into oviducts of pseudopregnant ICR females (CLEA japan, Tokyo, Japan). Transgenic mice were identified by PCR genotyping using the following AcGFP primer pair: sense, 5´- CACATGAAGCAGCACGACTT -3´; antisense, 5´- TTGCCATCCTCCTTGAAATC -3´ (176 bp fragment). Four transgenic founders were obtained: one Oog1pro2.7 male, two Oog1pro3.9 males (lines A and C), and one Oog1pro3.9 female (line B). Transgenic female offspring obtained from crossing between transgenic founder animals and wild-type animals were used for studies. Since all three Oog1pro3.9 transgenic lines showed the same reporter expression in the ovary and testis, these lines were used interchangeably in our analyses.
Cryosectioning
Tissue was fixed in cold 4% paraformaldehyde in PBS (pH 7.4) for 2 h. Ovaries were dehydrated in 20% sucrose in PBS overnight, and then 30% for 1–3 h until the tissue sank. Testes were fixed overnight in 4% paraformaldehyde in PBS containing 20% sucrose, and then were sunk in a 30% sucrose solution. Dehydrated samples were then placed in Tissue Tek O.C.T. compound (Sakura Finetechnical, Tokyo, Japan) and frozen for sectioning. Five µm thick sections on glass slides were placed in cold PBS in the dark and examined for GFP expression. After that, slides were mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA) and examined again for nuclear staining. GFP and DAPI signals were examined under a fluorescence microscope (BX50, OLYMPUS, Tokyo, Japan) equipped with appropriate filters (OLYMPUS filter sets U-MWIB and U-MWU).
RNA isolation, cDNA synthesis, and reverse transcription polymerase chain reaction (RT-PCR)
Ovaries were obtained from different ages of transgenic mice; E15.5 fetus, newborn, 1-week-old, and 5-week-old female mice. Testis, liver, kidney, spleen, heart, lung, and brain were obtained from 5-week-old female and 10-week-old male mice. For oocytes samples, 3- to 5-week-old female mice were superovulated with intraperitoneal injections of 5 IU equine chorionic gonadotropin (eCG) (ASUKA Pharmaceutical, Tokyo, Japan). After 48 h from hormonal treatment, GV oocytes were recovered from ovaries. Oocytes were freed from the surrounding cumulus cells and used for RNA isolation. Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) from each tissue. After treatment with RNase-free DNase I (Roche, Indianapolis, IN), reverse transcription was performed on isolated RNA in a 20 µl reaction volume using ReverTra Ace (TOYOBO, Osaka, Japan) with random primers (Invitrogen). The following RT-PCR primers were used: AcGFP1-mem: sense, 5´- TGTTCACCGGCATCGTGCCC -3´; antisense, 5´- CTCGGCGCGCGACTTGTAGT -3´ (314 bp); Oog1, sense, 5´- AGGAGGCCTTCACTGATGGA -3´; antisense, 5´-GTCCTTCGCATGAAGGGCAG -3´ (346 bp); β-actin (internal control): sense, 5´- ATGAGCTGCGTGTGGCCCCT -3´; antisense, 5´- CGGAACCGCTCGTTGCCAAT -3´ (494 bp). For quantification, relative band intensities of PCR products were determined with a model 4.0 Atto densitograph (Atto, Tokyo, Japan). Intensities were normalized to the intensity of the Actin band, and the averages from two independent experiments were analyzed.
Embryo culture
Four-to five-week-old female mice were superovulated with intraperitoneal injections of 5 IU eCG followed 48 h later by 5 IU human chorionic gonadotropin (hCG) (ASUKA Pharmaceutical). hCG injected mice were crossed with male mice, and 32 h after hCG injection, 2-cell embryos were flushed from the oviduct and cultured in modified KSOM medium. GFP fluorescence was observed after culture in vitro for 0 h (2-cell), 24 h (4-cell), 48 h (morula), and 72 h (blastocyst) using an inverted microscope (TMD300, Nikon, Tokyo, Japan) with a B2 filter set (Nikon).
Bisulfite sequencing
Genomic DNA from transgenic testes and oocytes was processed with the MethylCode Bisulfite Conversion Kit (Invitrogen), according to the manufacturer’s instructions. Growing oocytes were collected from ovaries of 2-week-old female mice by dissection in HTF medium containing 0.1% hyaluronidase, 0.2% collagenase, and 0.25% DNase I. Ovulated MII oocytes were collected from ovaries of 4- to 5-week-old superovulated females 16 h after hCG injection. Converted DNA was amplified by hemi-nested PCR. The first round of PCR was performed using EpiTaq HS (Takara Bio Inc., Otsu, Japan) as follows: 30 cycles of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 120 s using 5´- AGGGTATATGAGGGAAATGAATTATAGG -3´ and 5´- TTTCAACCTATTTAATTCTTCTCATACAACACAAC -3´ (for the promoter region of the transgenes), 5´- ACTATAACTCCAAACTCCAAAAAACCTAAT -3´ (for intrinsic Oog1 promoter), as primers. Then, a second round of PCR was performed using KOD -Plus- (TOYOBO, Japan), with the nested primer set of 5´- GAGAGTATTTGGGTGGAGTTTGTAG -3´ and 5´- ACCAAACAAATCAACTTAATTTCACC -3´. Amplified fragments were cloned into the pCR2.1-TOPO vector (Invitrogen) and sequenced. Sequence identity and methylation status of obtained sequences were analyzed using QUantification tool for Methylation Analysis (QUMA) (http://quma.cdb.riken.jp/).
Statistical analyses
Differences in GFP mRNA expression levels between the transgenic mouse lines were analyzed using the Student’s t-test. Differences in the methylation status of each CpG or in the overall methylation status between Oog1pro2.7 and Oog1pro3.9 transgenic lines was analyzed statistically with the QUMA program, using the Fisher’s exact test for individual CpGs and the Mann–Whitney U test for overall methylation. For all analyses, the difference was considered significant when p<0.05.
Ethical approval for the use of animals
All animal experiments were approved by the Animal Research Committee of Kyoto University (Permit Number: 24-17), and were performed in accordance with the committee’s guidelines.
Results
In silico analysis of the upstream sequences of Oog1 gene
Oog1 is a multi-copy gene, with two copies on chromosome 4 [GenBank: NM_001007077, GenBank: NM_001177542] and three copies on chromosome 12 [GenBank: NM_178657 (Oog1), GenBank: XM_003085569, GenBank: NM_001105254]. Since all copies have a TATA box at -31 bp from the predicted transcription start site, they are likely all functional. Thus, we compared the upstream regions of all five copies of Oog1 to identify the promoter region. Genomic sequence information for the 20 kb region upstream of each copy of Oog1, including the TATA box, was obtained from the NCBI (National Center for Biotechnology Information) database (http://www.ncbi.nlm.nih.gov/projects/mapview/). A homology comparison revealed that about 3.9 kb of the upstream sequence shared high homology between copies (Figure 1A). These sequences were then scanned for known transcription factor binding sites and annotated using the JASPAR CORE database (http://jaspar.genereg.net/). We found two Nobox binding sites (NOBOX DNA binding elements/NBEs; -2829 bp and -3490 bp) and one SP1 binding site (-1369 bp). In addition, we found eight E-boxes (or sequences similar to E-boxes) in the 3.9 kb promoter. However, only two of the E-boxes (-118 bp and -3457 bp from the transcriptional start site) retained perfect homology among the five sequences (Figure 1B). Furthermore, when the five promoters were compared with one another, inserted sequences at -0.7 kb, -2.7 kb, and -3.2 kb from the TATA box element were observed in some of the promoters (Figure 1A). The largest gap was seen -2.7 kb from the TATA box. Based on the above results, two candidate promoter sequences (2.7 kb and 3.9 kb in length) were isolated from the 5´-flanking sequence of Oog1 (Gene ID: 193322; plus strand of chromosome 12) and were further analyzed for activity.
Figure 1. Schematic diagrams of the 5’-flanking sequences of the Oog1 coding regions.
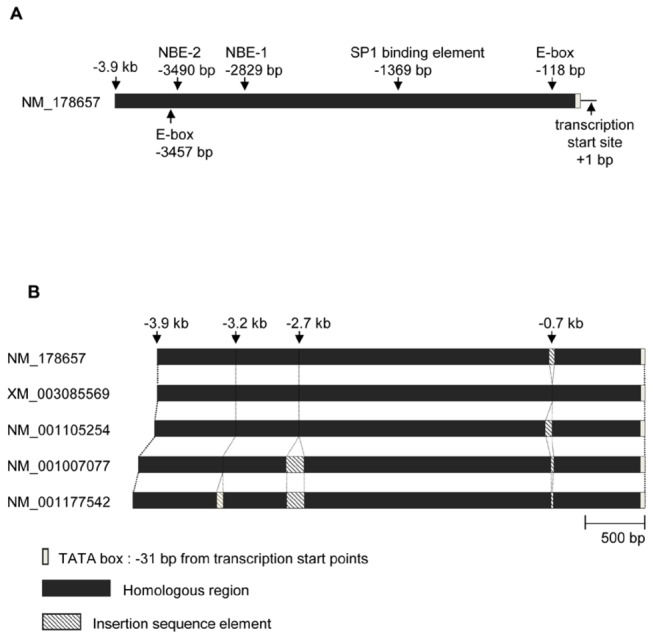
A. Locations of putative transcription factor binding sites in the 3.9 kb Oog1 promoter region (on chromosome 12, NT_039551) are shown by arrows. E-box (-188 bp), SP1 binding element (-1369 bp), and NBEs (-2829 bp and -3490 bp) are conserved among upstream regions of the five copies of Oog1. B. Promoter regions of the five copies of Oog1. All five sequences share a ~3.9 kb long, highly homologous region including a TATA box. Some sequences have sequence insertions at -0.7 kb, -2.7 kb, and -3.2 kb from the TATA box. The sequences on chromosome 4 (the upstream regions of NM_001007077 and NM_001177542) have the largest gaps at -2.7 kb.
The 2.7 kb and 3.9 kb promoter regions function specifically in oocytes of transgenic ovaries
We generated transgenic mice with either the 2.7 kb (Oog1pro2.7) or 3.9 kb (Oog1pro3.9) Oog1 upstream sequence driving expression of a GFP reporter gene (Figure 2). Oocyte-specific GFP expression was observed in both Oog1pro2.7 and Oog1pro3.9 transgenic ovaries (Figure 3A). The intensity of the GFP signal in Oog1pro3.9 transgenic oocytes was stronger than that observed in Oog1pro2.7 transgenic oocytes. We confirmed by semi-quantitative RT-PCR that the promoter activity is about three times stronger in the Oog1pro3.9 ovary than in the Oog1prp2.7 ovary (Figure 3B). In addition, while GFP fluorescence in Oog1pro2.7 ovaries was observed in oocytes of type 4 secondary follicles and in subsequent stages of development, GFP signal was found in oocytes of type 2 primordial follicles in Oog1pro3.9 ovaries (Figure 3C). This difference may also be due to the difference in strength of transcriptional activity of each promoter.
Figure 2. Transgene constructs for generating transgenic mice.
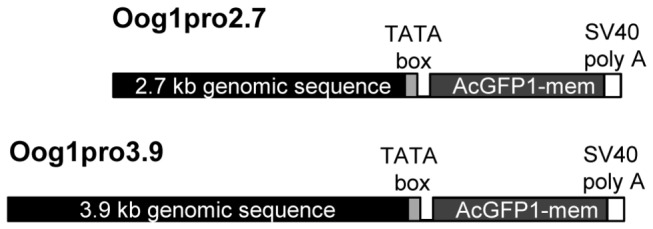
Two constructs (Oog1pro2.7 and Oog1pro3.9) were used to generate transgenic mice.
Figure 3. Oocyte-specific activities of Oog1 promoters in transgenic ovaries.

A. Frozen sections of ovaries obtained from 5-week-old transgenic mice. GFP signal was detected in the oocytes of both Oog1pro2.7 and Oog1pro3.9 transgenic mice. Because we employed membrane targeted GFP as the reporter gene, the green fluorescence signals were observed to concentrate around the plasma membrane of the oocytes. Scale bar: 100 µm. B. Quantification of GFP mRNA in 5-week ovaries of each transgenic line. The bar graph indicates the average value of the trials. RT-PCR was conducted twice using ovary cDNA obtained from two different animals per line, and similar results were obtained in each trial (* p<0.05, t-test). C. Magnified images of oocytes at various stages of folliculogenesis in transgenic ovaries. GFP signal was detected in the oocytes of secondary to preovulatory follicles in Oog1pro2.7 transgenic ovaries, but in the oocytes of primordial to preovulatory follicles in Oog1pro3.9 transgenic ovaries. Analysis of type 2 follicles was performed on sectioned 2-week old ovaries, and the remaining analyses were done using sectioned 5-week old ovaries. Arrows indicate primordial follicles; Arrowheads indicate primary follicles. Scale bars: primordial and primary follicles: 10 µm, secondary and antral follicles: 100 µm. D. RT-PCR analysis of transgenic ovaries using AcGFP1-mem and Oog1 primers. Fetal (E15.5), neonatal (day 0), juvenile (day 7), and adult (5-week-old) ovary cDNA were obtained from transgenic (+) and non-transgenic (-) animals. AcGFP1-mem mRNA was detected in ovaries of all stages in Oog1pro2.7 and Oog1pro3.9 transgenic mice, similar to the pattern of Oog1 mRNA expression.
Although these data suggest that the 2.7 kb and 3.9 kb sequences functioned specifically in oocytes within the ovary, GFP fluorescence was not observed in the oocytes within ovarian cysts (data not shown). Moreover, by Western blotting, GFP protein was not detected in newborn or fetal ovaries containing primordial follicles or ovarian cysts, respectively (data not shown). On the other hand, by RT-PCR, GFP transcripts were detected in E15.5 fetal transgenic ovaries, suggesting that both 2.7 kb and 3.9 kb promoters could function to produce mRNA in oocytes within the fetal ovary (Figure 3D). Indeed, the expression profiles of GFP mRNA in transgenic ovaries obtained at various stages from E15.5 to adult were similar to those of Oog1.
The 2.7 kb and 3.9 kb promoters do not function in early embryos
Since Oog1 mRNA and protein are detected in early embryos until the late 2-cell stage [1], we examined the promoter activities during early preimplantation development (Figure 4). Appreciable fluorescence was observed in all zygotes derived from transgenic females crossed with wild-type males. The GFP signal decreased markedly at around morula stage, but was still detectable until the blastocyst stage. On the other hand, in embryos derived from non-transgenic females crossed with transgenic males, no GFP fluorescence was observed during preimplantation development. Because all transgenic males used for the experiments were confirmed to carry the transgenes and to successfully pass the transgenes on to their offspring, about half of the embryos collected in this experiment should carry the transgenic allele in their genome. In addition, the genes introduced into embryos by sperm will be activated at the time of zygotic gene activation (ZGA) and this occurs at the late 1-cell stage in the mouse. Thus, neither the 2.7 kb nor the 3.9 kb promoter appears to function after fertilization. These data suggest that Oog1 transcript observed in early preimplantation embryos are of maternal origin.
Figure 4. GFP signal in embryos derived from transgenic females.

GFP signal was detected only in embryos obtained from transgenic females crossed with wild-type males. No signal was detected in embryos obtained from wild-type females crossed with transgenic males. Embryos were recovered at 1.5 days after hCG injection, and then were cultured for 3 days in vitro. Embryos at the 2-cell, 4-cell, morula, and blastocyst stages were observed at 1.5 days, 2.5 days, 3.5 days, and 4.5 days after hCG injection, respectively. M: Male, F: Female.
The longer promoter is active in male germ cells as well as in oocytes
Although the transcriptional activities of the 2.7 kb and 3.9 kb promoters differed, as shown by the transcript levels, both promoters functioned specifically in oocytes of ovarian cysts in transgenic ovaries. Unexpectedly, we also found by RT-PCR analysis of somatic tissues that the 3.9 kb promoter has strong transcriptional activity in the testis (Figure 5A). Whereas GFP mRNA was detected predominantly in the ovary in Oog1pro2.7 transgenic mice, it was detected in both female and male gonads in Oog1pro3.9 transgenic lines. GFP fluorescence was detected in male germ cells, from late pachytene spermatocytes to elongated spermatids, in Oog1pro3.9 transgenic testes (Figure 5B).
Figure 5. Activities of Oog1 promoters in various tissues, including the testis.

A. RT-PCR for GFP transcript in various somatic tissues of transgenic mice. Abundant GFP mRNA was detected in the ovaries of Oog1pro2.7 and Oog1pro3.9 transgenic mice, and in the testis of Oog1pro3.9 transgenic mice. Faint GFP mRNA expression was also detected in the brain in both transgenic lines, as well as in the testis of Oog1pro2.7 transgenic mice. Non-transgenic (NTG) ovary cDNA was used for controls. B. Frozen sections of the testis obtained from an Oog1pr3.9 transgenic male. (a) Seminiferous tubule at stage VI. GFP signal was detected in the round and elongated spermatids but not in mid-pachytene spermatocytes. Arrow: Mid pachytene spermatocyte; Arrow head: step 6 spermatid. (b) Seminiferous tubule at stage VIII. Late pachytene spermatocytes show the visible GFP signal. Arrow: Late pachytene spermatocyte; Arrow head: step 8 spermatid. (c) Seminiferous tubule of non-transgenic testis at stage VI is shown as a control. Scale bar: 10 µm.
Methylation status of the proximal region of the promoters affects sex-dependent gene expression
Since we observed differences in the tissue specificity of transgene expression between Oog1pro2.7 and Oog1pro3.9, we next analyzed the CpG methylation status of the promoter regions of Oog1pro2.7 and Oog1pro3.9. Bisulfite-sequencing of the proximal promoter region revealed that the cytosine of the CpG at -597 bp is highly methylated in tissues in which GFP expression was suppressed (Figure 6A, B), whereas the methylation ratio of the same cytosine was significantly reduced in tissues and cells in which GFP was expressed. Moreover, in both males and females, the cytosine of the CpG at -698 bp is highly methylated in the Oog1pro2.7 transgene and the endogenous Oog1 promoter, but is completely demethylated in the Oog1pro3.9 transgene, suggesting that the methylation of this cytosine is involved in repressing promoter activity only in male germ cells. Furthermore, the proximal promoter regions of the transgenes were highly methylated in somatic cells (Figure S1). These data suggest that the aberrant cytosine demethylation of two CpGs (at -587 bp and -698 bp) results in activation of the 3.9 kb promoter in Oog1pro3.9 male germ cells. Specifically, the cytosine methylation of a single CpG at -587 bp controls the basal promoter activity in both male and female germ cells, while cytosine methylation of the CpG at -698 bp is involved in suppressing aberrant expression in male germ cells.
Figure 6. Bisulfite-sequencing analysis of the methylation status of the Oog1 promoters.

A. Location of CpGs and amplification primers on the Oog1 promoter. The conserved 2.7 kb promoter region was analyzed with a program to predict DNA methylation (methylator, http://bio.dfci.harvard.edu/Methylator/index.html) and the indicated region (-508 bp to -985 bp) was selected for analysis of methylation status. Primers specifically recognizing the transgene were used for amplification of bisulfite-converted genomes (see materials and methods). B. DNA methylation status of individual CpGs is shown. Open circles indicate unmethylated, and filled circles indicate methylated CpG dinucleotides. Total methylation ratios were indicated under the diagrams. Asterisks indicate a significant difference in methylation status of individual CpGs between GFP expressed and non-expressed cells/tissues (p<0.05, Fisher’s exact test).
Discussion
Several germ cell–specific promoters have been isolated and used for the genetic analysis of germ cells [5]. So far, Zp3 and Gdf9 promoters are the only promoters known to have activity specifically in female germ cells, but they function only after birth [7]. Here, we isolated two Oog1 promoter fragments (2.7 kb and 3.9 kb) and demonstrated that they function specifically in oocytes in the mouse ovary as early as E15.5. Our in silico analysis of the 3.9 kb promoter identified two E-box elements conserved perfectly among the five Oog1 copies in the mouse genome. E-boxes are known to mediate differential gene expression by binding to homodimeric or heterodimeric complexes of beta helix-loop-helix transcription factors [21–24], such as FIGα [16], and play a key role in the regulation of oocyte-specific promoter activity [8,16,18,25]. Thus, the conserved E-box at -118 bp of both the 2.7 kb and 3.9 kb promoters may induce oocyte-specific transgenic expression.
We also found that Oog1pro3.9 drives stronger expression in oocytes than Oog1pro2.7. One possible explanation for this is that Oog1pro3.9 includes two NOBOX DNA binding elements (NBEs) (Figure 1A). Nobox is a homeobox transcription factor expressed in oocytes and can enhance the expression of oocyte-specific genes by binding to NBEs [8,14,19,26]. In Nobox-null newborn ovaries, the expression level of Oog1 was dramatically reduced [19]. By contrast, there was little difference in GFP expression in GV oocytes between Oog1pro2.7 and Oog1pro3.9 (Figure S2). What might account for this discrepancy? During oogenesis, the rate of transcription decreases sharply when oocytes grow to their full size [27]. Concomitant with the decreased rate of transcription is a decrease in the concentrations of major transcription factors (TBP2 and SP1) during oocyte growth [13,28,29]. TBP2 (also known as TRF3) is preferentially expressed in germ cells in frogs and mice, and is replaced by TBP in growing oocytes [13,30]. SP1 binds to proximal binding sites, but can also interact with distal enhancer binding complexes to activate transcription, as shown for the IFN-β locus [31,32]. The fact that the Oog1 promoter has a TATA-box and an SP1 binding site raises the possibility that enhancer complexes that bind to the 3.9 kb promoter function only during oocyte growth, and that this enhancer activity ceases in fully grown GV-stage oocytes, when the expression of TBP2 and SP1 decline significantly (Figure S3). This difference is also corroborated by the fact that the number of fully grown oocytes in the ovary is reduced by an order of magnitude as compared to that found in growing or non-growing oocytes [33].
Interestingly, Oog1pro3.9 also showed strong promoter activity in male germ cells during meiotic stages. GFP expression in Oog1pro3.9 males was detected in late pachytene spermatocytes (at stage VIII of spermatogenesis) and later on in elongated spermatids. Though this does not reflect the intrinsic Oog1 expression pattern [1], it is intriguing that this ectopic expression in male germ cells is observed during meiotic stages. Since Oog1 is normally expressed in female germ cells starting from stage E15.5, concomitant with the onset of the pachytene stage of meiotic prophase [34], this raises the possibility that Oog1 plays a role in meiosis. However, this hypothesis needs to be tested directly.
The strong expression of Oog1pro3.9 in male germ cells cannot be explained by the presence of NBEs. Thus, there may be other regulatory functions in 1.2 kb sequence beyond the 2.7 kb promoter region, because 3.9 kb promoter drives germ cell–specific expression in both females and males. Thus, the expression of intrinsic Oog1 might be controlled by multi-regulatory pathways, in which one pathway, such as the Nobox pathway, functions only in females, whereas the other functions in both sexes.
Methylation analysis of the Oog1pro2.7 and Oog1pro3.9 transgenes revealed that there is a significant difference in the methylation status of two CpGs (at -597 bp and -698 bp) in male and female germ cells. Thus, aberrant cytosine demethylation of these two CpGs in Oog1pro3.9 might cause GFP expression in male germ cells. A similar relationship between CpG methylation status and gene transcription was observed in the endogenous Oog1 promoter in the testis and oocytes. Promoter methylation and gene expression are known to be correlated [35–39], and tissue-specific differentially methylated regions (TDMs) are involved in the regulation of germ cell–specific gene expression [20,35]. Since the proximal promoter region of Oog1pro2.7 in testis has a similar methylation pattern to the endogenous Oog1 promoter, it is possible that the regulatory elements in the distal promoter region in Oog1pro3.9 induced demethylation of the proximal promoter region, resulting in ectopic gene expression in the testis. The promoter region of Oog1pro3.9 has binding elements for two transcription factors, SP1 (5´-CCCCTTCCCC-3´) at -0.9 kb and NF-κB (5´-GGGAAATTCT-3´) at -3 kb. NF-κB can induce selective demethylation adjacent to its binding region [40] and can interact with SP1, which binds the proximal promoter region [41–43]. Because SP1 can mediate chromatin looping by interacting with distal enhancer complexes [31,32], it is possible that SP1 binds to the Oog1 promoter and interacts with NF-κB as a trans-acting factor to demethylate the proximal promoter region, resulting in expression in Oog1pro3.9 male germ cells. Although the details remain to be investigated, our results suggest that CpG methylation of the proximal promoter region is involved in regulating the differential expression of Oog1 in female germ cells.
Kido and Lau [44] showed that the promoter region that controls testis-specific protein Y-encoded (Tspy) gene can also function in female germ cells in transgenic mice expressing the Cre gene under the control of the Tspy promoter. In the case of Tspy, expression in females is avoided because the gene is located on the Y chromosome. In the case of Oog1, additional suppression mechanisms might be required to repress ectopic expression in male germ cells, since endogenous expression is restricted to female germ cells. Yan et al. reported that factors that interact with the Gdf9 gene body are necessary to suppress Gdf9 gene expression in male germ cells [18]. Thus, it is possible that Oog1 expression in male germ cells is suppressed by factors interacting with the Oog1 gene body.
We also confirmed that the 2.7 kb and 3.9 kb Oog1 promoters do not function in preimplantation embryos. There was no GFP signal in embryos produced by crossing transgenic males with non-transgenic females. It has been reported that the expression of the gene derived from transgenic male is first detected at the late 1-cell stage in the mouse [45]. These results indicate that Oog1 promoters are not activated after fertilization; rather, Oog1 protein observed in early embryos is likely translated from maternally inherited mRNA. Combined with its nuclear localization in the late 1-cell and early 2-cell stages, the possibility that Oog1 plays a role in zygotic transcription at the 1- to 2-cell stage and/or in chromosome segregation of the first mitotic cell division as a maternal effect gene cannot be ruled out. Because oocyte-specific genes have been reported to be involved in oogenesis, fertilization, and early embryogenesis, characterizing the function of Oog1 should help to elucidate the mechanisms of these biological phenomena in vivo.
Despite their importance, oocyte-expressed genes are difficult to study in vitro because there are no oocyte cell lines [9,18,46]. Although knockout (KO) and conditional KO animals are often used to analyze the functions of genes in vivo, this approach is not suitable for multi-copy genes such as Oog1. However, RNA interference (RNAi) constructs have been successfully used to study gene function in mouse oocytes [6,47,48] and could be effective for analyzing the function of multi-copy genes. Since this approach will likely require tight spatiotemporal control of RNAi constructs, the Oog1 promoter will be very useful for future studies. We are currently generating transgenic mice using these newly identified promoters in order to knock down Oog1 function during the first meiotic prophase in oocytes.
Supporting Information
(TIF)
RT-PCR was conducted twice using different samples; each trial included 20 oocytes per sample. Similar results were obtained in each trial. The bar graph indicates the average value obtained from both trials. No significant differences were observed between Oog1pro2.7 and Oog1pro3.9 (n = 2, t-test).
(TIF)
(A) In growing oocytes, the oocyte-specific core transcription factor TBP2 is involved in maintaining basal transcription of the 2.7 kb and 3.9 kb Oog1 promoters. SP1, which is also abundant in growing oocytes, interacts with the distal enhancer complex and upregulates transcription in the case of the 3.9 kb Oog1 promoter. (B) In fully grown oocytes, the concentrations of TBP2 and SP1 proteins in oocytes are dramatically reduced, and the promoter activity is abrogated.
(TIF)
Funding Statement
This work was supported by Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science to NM (3380164, 19380158). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Minami N, Aizawa A, Ihara R, Miyamoto M, Ohashi A et al. (2003) Oogenesin is a novel mouse protein expressed in oocytes and early cleavage-stage embryos. Biol Reprod 69: 1736-1742. doi:10.1095/biolreprod.103.018051. PubMed: 12890732. [DOI] [PubMed] [Google Scholar]
- 2. Tsukamoto S, Ihara R, Aizawa A, Kishida S, Kikuchi A et al. (2006) Oog1, an oocyte-specific protein, interacts with Ras and Ras-signaling proteins during early embryogenesis. Biochem Biophys Res Commun 343: 1105-1112. doi:10.1016/j.bbrc.2006.03.063. PubMed: 16580637. [DOI] [PubMed] [Google Scholar]
- 3. Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K et al. (2010) MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLOS Biol;8(8): ii: e1000453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Audouard C, Le Masson F, Charry C, Li Z, Christians ES (2011) Oocyte-targeted deletion reveals that hsp90b1 is needed for the completion of first mitosis in mouse zygotes. PLOS ONE 6: e17109. doi:10.1371/journal.pone.0017109. PubMed: 21358806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hammond SS, Matin A (2009) Tools for the genetic analysis of germ cells. Genesis 47: 617-627. doi:10.1002/dvg.20539. PubMed: 19548313. [DOI] [PubMed] [Google Scholar]
- 6. Stein P, Svoboda P, Schultz RM (2003) Transgenic RNAi in mouse oocytes: a simple and fast approach to study gene function. Dev Biol 256: 187-193. PubMed: 12654301. [DOI] [PubMed] [Google Scholar]
- 7. Lan ZJ, Xu X, Cooney AJ (2004) Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod 71: 1469-1474. doi:10.1095/biolreprod.104.031757. PubMed: 15215191. [DOI] [PubMed] [Google Scholar]
- 8. Tsunemoto K, Anzai M, Matsuoka T, Tokoro M, Shin SW et al. (2008) Cis-acting elements (E-box and NBE) in the promoter region of three maternal genes (Histone H1oo, Nucleoplasmin 2, and Zygote Arrest 1) are required for oocyte-specific gene expression in the mouse. Mol Reprod Dev 75: 1104-1108. doi:10.1002/mrd.20863. PubMed: 18324673. [DOI] [PubMed] [Google Scholar]
- 9. DeJong J (2006) Basic mechanisms for the control of germ cell gene expression. Gene 366: 39-50. doi:10.1016/j.gene.2005.10.012. PubMed: 16326034. [DOI] [PubMed] [Google Scholar]
- 10. Han S, Xie W, Kim SH, Yue L, DeJong J (2004) A short core promoter drives expression of the ALF transcription factor in reproductive tissues of male and female mice. Biol Reprod 71: 933-941. doi:10.1095/biolreprod.104.030247. PubMed: 15151936. [DOI] [PubMed] [Google Scholar]
- 11. Goodrich JA, Tjian R (2010) Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation. Nat Rev Genet 11: 549-558. doi:10.1038/ni0710-549. PubMed: 20628347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han SY, Zhou L, Upadhyaya A, Lee SH, Parker KL et al. (2001) TFIIAalpha/beta-like factor is encoded by a germ cell-specific gene whose expression is up-regulated with other general transcription factors during spermatogenesis in the mouse. Biol Reprod 64: 507-517. doi:10.1095/biolreprod64.2.507. PubMed: 11159353. [DOI] [PubMed] [Google Scholar]
- 13. Xiao L, Kim M, DeJong J (2006) Developmental and cell type-specific regulation of core promoter transcription factors in germ cells of frogs and mice. Gene Expr Patterns 6: 409-419. doi:10.1016/j.modgep.2005.09.005. PubMed: 16412700. [DOI] [PubMed] [Google Scholar]
- 14. Rajkovic A, Pangas SA, Ballow D, Suzumori N, Matzuk MM (2004) NOBOX deficiency disrupts early folliculogenesis and oocyte-specific gene expression. Science 305: 1157-1159. doi:10.1126/science.1099755. PubMed: 15326356. [DOI] [PubMed] [Google Scholar]
- 15. Soyal SM, Amleh A, Dean J (2000) FIGalpha, a germ cell-specific transcription factor required for ovarian follicle formation. Development 127: 4645-4654. PubMed: 11023867. [DOI] [PubMed] [Google Scholar]
- 16. Liang L, Soyal SM, Dean J (1997) FIGalpha, a germ cell specific transcription factor involved in the coordinate expression of the zona pellucida genes. Development 124: 4939-4947. PubMed: 9362457. [DOI] [PubMed] [Google Scholar]
- 17. Joshi S, Davies H, Sims LP, Levy SE, Dean J (2007) Ovarian gene expression in the absence of FIGLA, an oocyte-specific transcription factor. BMC Dev Biol 7: 67. doi:10.1186/1471-213X-7-67. PubMed: 17567914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan C, Elvin JA, Lin YN, Hadsell LA, Wang J et al. (2006) Regulation of growth differentiation factor 9 expression in oocytes in vivo: a key role of the E-box. Biol Reprod 74: 999-1006. doi:10.1095/biolreprod.105.050013. PubMed: 16495478. [DOI] [PubMed] [Google Scholar]
- 19. Choi Y, Qin Y, Berger MF, Ballow DJ, Bulyk ML et al. (2007) Microarray analyses of newborn mouse ovaries lacking Nobox. Biol Reprod 77: 312-319. doi:10.1095/biolreprod.107.060459. PubMed: 17494914. [DOI] [PubMed] [Google Scholar]
- 20. Pan B, Chao H, Chen B, Zhang L, Li L et al. (2011) DNA methylation of germ-cell-specific basic helix-loop-helix (HLH) transcription factors, Sohlh2 and Figlalpha during gametogenesis. Mol Hum Reprod 17: 550-561. doi:10.1093/molehr/gar017. PubMed: 21427160. [DOI] [PubMed] [Google Scholar]
- 21. Eilers M, Eisenman RN (2008) Myc’s broad reach. Genes Dev 22: 2755-2766. doi:10.1101/gad.1712408. PubMed: 18923074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naya FJ, Stellrecht CM, Tsai MJ (1995) Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev 9: 1009-1019. doi:10.1101/gad.9.8.1009. PubMed: 7774807. [DOI] [PubMed] [Google Scholar]
- 23. Weintraub H, Genetta T, Kadesch T (1994) Tissue-specific gene activation by MyoD: determination of specificity by cis-acting repression elements. Genes Dev 8: 2203-2211. doi:10.1101/gad.8.18.2203. PubMed: 7958889. [DOI] [PubMed] [Google Scholar]
- 24. Ye R, Selby CP, Ozturk N, Annayev Y, Sancar A (2011) Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem 286: 25891-25902. doi:10.1074/jbc.M111.254680. PubMed: 21613214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Millar SE, Lader E, Liang LF, Dean J (1991) Oocyte-specific factors bind a conserved upstream sequence required for mouse zona pellucida promoter activity. Mol Cell Biol 11: 6197-6204. PubMed: 1944285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Choi Y, Rajkovic A (2006) Characterization of NOBOX DNA binding specificity and its regulation of Gdf9 and Pou5f1 promoters. J Biol Chem 281: 35747-35756. doi:10.1074/jbc.M604008200. PubMed: 16997917. [DOI] [PubMed] [Google Scholar]
- 27. Picton H, Briggs D, Gosden R (1998) The molecular basis of oocyte growth and development. Mol Cell Endocrinol 145: 27-37. doi:10.1016/S0303-7207(98)00166-X. PubMed: 9922096. [DOI] [PubMed] [Google Scholar]
- 28. Worrad DM, Ram PT, Schultz RM (1994) Regulation of gene expression in the mouse oocyte and early preimplantation embryo: developmental changes in Sp1 and TATA box-binding protein, TBP. Development 120: 2347-2357. PubMed: 7925035. [DOI] [PubMed] [Google Scholar]
- 29. Gazdag E, Rajkovic A, Torres-Padilla ME, Tora L (2007) Analysis of TATA-binding protein 2 (TBP2) and TBP expression suggests different roles for the two proteins in regulation of gene expression during oogenesis and early mouse development. Reproduction 134: 51-62. doi:10.1530/REP-06-0337. PubMed: 17641088. [DOI] [PubMed] [Google Scholar]
- 30. Gazdag E, Santenard A, Ziegler-Birling C, Altobelli G, Poch O et al. (2009) TBP2 is essential for germ cell development by regulating transcription and chromatin condensation in the oocyte. Genes Dev 23: 2210-2223. doi:10.1101/gad.535209. PubMed: 19759265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deshane J, Kim J, Bolisetty S, Hock TD, Hill-Kapturczak N et al. (2010) Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J Biol Chem 285: 16476-16486. doi:10.1074/jbc.M109.058586. PubMed: 20351094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nolis IK, McKay DJ, Mantouvalou E, Lomvardas S, Merika M et al. (2009) Transcription factors mediate long-range enhancer-promoter interactions. Proc Natl Acad Sci U S A 106: 20222-20227. doi:10.1073/pnas.0902454106. PubMed: 19923429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peters H (1969) The development of the mouse ovary from birth to maturity. Acta Endocrinol 62: 98-116. PubMed: 5394354. [DOI] [PubMed] [Google Scholar]
- 34. Kolas NK, Marcon E, Crackower MA, Höög C, Penninger JM et al. (2005) Mutant meiotic chromosome core components in mice can cause apparent sexual dimorphic endpoints at prophase or X–Y defective male-specific sterility. Chromosoma 114: 92-102. doi:10.1007/s00412-005-0334-8. PubMed: 15983832. [DOI] [PubMed] [Google Scholar]
- 35. Singal R, vanWert J, Bashambu M, Wolfe SA, Wilkerson DC et al. (2000) Testis-specific histone H1t gene is hypermethylated in nongerminal cells in the mouse. Biol Reprod 63: 1237-1244. doi:10.1095/biolreprod63.5.1237. PubMed: 11058525. [DOI] [PubMed] [Google Scholar]
- 36. Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T et al. (2005) Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci U S A 102: 3336-3341. doi:10.1073/pnas.0408436102. PubMed: 15728362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kitamura E, Igarashi J, Morohashi A, Hida N, Oinuma T et al. (2007) Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics 89: 326-337. doi:10.1016/j.ygeno.2006.11.006. PubMed: 17188838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y et al. (2008) DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome Res 18: 1969-1978. doi:10.1101/gr.074070.107. PubMed: 18971312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merbs SL, Khan MA, Hackler L Jr., Oliver VF, Wan J et al. (2012) Cell-specific DNA methylation patterns of retina-specific genes. PLOS ONE 7: e32602. doi:10.1371/journal.pone.0032602. PubMed: 22403679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T et al. (1996) A role for nuclear NF-kappaB in B-cell-specific demethylation of the Igkappa locus. Nat Genet 13: 435-441. doi:10.1038/ng0895-435. PubMed: 8696338. [DOI] [PubMed] [Google Scholar]
- 41. Liu S, Liu Z, Xie Z, Pang J, Yu J et al. (2008) Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 111: 2364-2373. doi:10.1182/blood-2007-08-110171. PubMed: 18083845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilkerson DC, Wolfe SA, Grimes SR (2002) H1t/GC-box and H1t/TE1 element are essential for promoter activity of the testis-specific histone H1t gene. Biol Reprod 67: 1157-1164. doi:10.1095/biolreprod67.4.1157. PubMed: 12297531. [DOI] [PubMed] [Google Scholar]
- 43. Panigrahi SK, Vasileva A, Wolgemuth DJ (2012) Sp1 transcription factor and GATA1 cis-acting elements modulate testis-specific expression of mouse cyclin A1. PLOS ONE 7: e47862. doi:10.1371/journal.pone.0047862. PubMed: 23112860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kido T, Lau YF (2005) A Cre gene directed by a human TSPY promoter is specific for germ cells and neurons. Genesis 42: 263-275. doi:10.1002/gene.20147. PubMed: 16035036. [DOI] [PubMed] [Google Scholar]
- 45. Matsumoto K, Anzai M, Nakagata N, Takahashi A, Takahashi Y et al. (1994) Onset of paternal gene activation in early mouse embryos fertilized with transgenic mouse sperm. Mol Reprod Dev 39: 136-140. doi:10.1002/mrd.1080390203. PubMed: 7826613. [DOI] [PubMed] [Google Scholar]
- 46. Zambrowicz BP, Harendza CJ, Zimmermann JW, Brinster RL, Palmiter RD (1993) Analysis of the mouse protamine 1 promoter in transgenic mice. Proc Natl Acad Sci U S A 90: 5071-5075. doi:10.1073/pnas.90.11.5071. PubMed: 8389466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stein P, Zeng F, Pan H, Schultz RM (2005) Absence of non-specific effects of RNA interference triggered by long double-stranded RNA in mouse oocytes. Dev Biol 286: 464-471. doi:10.1016/j.ydbio.2005.08.015. PubMed: 16154556. [DOI] [PubMed] [Google Scholar]
- 48. Svoboda P, Stein P, Schultz RM (2001) RNAi in mouse oocytes and preimplantation embryos: effectiveness of hairpin dsRNA. Biochem Biophys Res Commun 287: 1099-1104. doi:10.1006/bbrc.2001.5707. PubMed: 11587535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
RT-PCR was conducted twice using different samples; each trial included 20 oocytes per sample. Similar results were obtained in each trial. The bar graph indicates the average value obtained from both trials. No significant differences were observed between Oog1pro2.7 and Oog1pro3.9 (n = 2, t-test).
(TIF)
(A) In growing oocytes, the oocyte-specific core transcription factor TBP2 is involved in maintaining basal transcription of the 2.7 kb and 3.9 kb Oog1 promoters. SP1, which is also abundant in growing oocytes, interacts with the distal enhancer complex and upregulates transcription in the case of the 3.9 kb Oog1 promoter. (B) In fully grown oocytes, the concentrations of TBP2 and SP1 proteins in oocytes are dramatically reduced, and the promoter activity is abrogated.
(TIF)