

Abstract
Sarcomas are a key feature of Li-Fraumeni and related syndromes (LFS/LFL), associated with germline TP53 mutations. Current penetrance estimates for TP53 mutations are subject to significant ascertainment bias. The International Sarcoma Kindred Study is a clinic-based, prospective cohort of adult-onset sarcoma cases, without regard to family history. The entire cohort was screened for mutations in TP53 using high-resolution melting analysis and Sanger sequencing, and multiplex-ligation-dependent probe amplification and targeted massively parallel sequencing for copy number changes. Pathogenic TP53 mutations were detected in blood DNA of 20/559 sarcoma probands (3.6%); 17 were germline and 3 appeared to be somatically acquired. Of the germline carriers, one appeared to be mosaic, detectable in the tumor and blood, but not epithelial tissues. Germline mutation carriers were more likely to have multiple cancers (47% vs 15% for non-carriers, P = 3.0×10−3), and earlier cancer onset (33 vs 48 years, P = 1.19×10−3). The median survival of mutation carriers following first cancer diagnosis was not significantly different from non-carriers. Only 10/17 (59%) pedigrees met classical or Chompret criteria for LFS. In summary, germline TP53 mutations are not rare in adult patients with sarcoma, with implications for screening, surveillance, treatment and genetic counselling of carriers and family members.
Introduction
Germline TP53 mutations result in the classical Li-Fraumeni or Li-Fraumeni-like syndromes (LFS/LFL) [1], rare inherited syndromes with a lifetime cancer penetrance up to 73% for males and ∼100% for females[2]–[7]. Historically there has been little enthusiasm in the medical community for germline TP53 testing in LFS/LFL. Reasons include the perceptions of rarity, a lack of proven risk management strategies, and the potential for psychological harm by identifying people with an unmodifiable, extreme cancer risk [8]. However, developments in breast and whole body MRI screening [9], [10], pre-implantation genetic diagnosis for family planning, and the use of genetic information to guide cancer therapy, may influence decision-making for TP53 genetic testing.
Mutation frequencies and penetrance estimates are largely derived from pedigree-ascertained pediatric, cohorts[4]–[6], [8], [11], fraught with ascertainment biases. Moreover, studies of LFS-associated cancers[12]–[16] suggest many germline TP53 mutation carriers have little family history, or will be increasingly identified through genomic screens of cancer populations unselected for family history. Accurate risk counselling to the carriers identified in these ways will require study of the impact of TP53 mutations outside of familial settings.
Sarcomas are the most common cancer type seen in LFS [17]; approximately 90% of sarcomas occur in adults [18]. To determine the incidence and clinical spectrum of germline TP53 mutations in adult-onset sarcoma populations, a systematic screen using multiplexed ligation-dependent probe amplification and Sanger sequencing was undertaken in 559 probands consecutively recruited from adult sarcoma clinics–agnostic to family history–on the Australian arm of the International Sarcoma Kindred Study (ISKS; http://www.anzctr.org.au; http://www.australiansarcomagroup.org/sarcomakindredstudy/index.html).
Results
Pathogenic or putatively pathogenic TP53 events occurred in the peripheral blood DNA of 20/559 probands (3.6%), comprising 18 single nucleotide mutations or indels, and 2 whole gene deletions (Table 1). Pathogenicity was assigned as described in the Materials and Methods, and in Figure S1. Most were previously reported somatically [19], but 10 are reported here for the first time in the germline. Six variants were regarded as putatively pathogenic. The age of sarcoma onset in individuals carrying pathogenic variants was not significantly different from those carrying putatively pathogenic variants (mean±standard deviation: 38±17 years versus 40±19 years, compared to 48±18 years for non-carriers in the ISKS cohort). Seventeen were putative germline events, with the mutant allele also detected in tumor DNA and 8 tumors also demonstrating loss of heterozygosity. The remaining 3 cases suggested somatic origin: Case 18 had clinical evidence of myelodysplasia (MDS), and neither parent carried the TP53 mutation. Both cases 19 and 20 demonstrated heterozygous whole gene deletion. While neither cases 19 or 20 had clinical evidence of MDS, both cases 18 and 19 showed widespread copy number changes in the peripheral blood–including the RB1 locus in case 19–suggestive of somatic tumor changes rather than germline events. Only case 18 had been exposed to chemotherapy prior to blood sampling.
Table 1. Genetic events detected in the peripheral blood of ISKS probands.
| Case | Genetic variant TP53 | Amino acid change | Mutation type | Condel23,24 | Reported somatic cases22 * | Reported germline cases22 * | Mutant allele present in tumour | Heterozygosity in tumour | Pathogenic |
| Putative germline | |||||||||
| 1 | c.72del | p.Lys24AsnfsX20 | frameshift | NA | 0 | 0 | yes | LOH | yes |
| 2 | c.586C>T | p.Arg196X | nonsense | NA | 241 | 13 | yes | LOH | yes |
| 3 | c.559+1G>T | exon skipping | splice site | NA | 10 | 0 | yes | LOH | yes |
| 4 | c.329G>C | p.Arg110Pro | missense | del | 15 | 0 | yes | LOH | putative |
| 5 | c.783-1G>A | exon skipping | splice site | NA | 7 | 0 | yes | No LOH | yes |
| 6 | c.700T>C | p.Tyr234His | missense | del | 33 | 0 | yes | LOH | putative |
| 7 | c.853G>A | p.Glu285Lys | missense | del | 186 | 5 | yes | LOH | yes |
| 8 | c.997C>T | p.Arg333Cys | missense | del | 0 | 0 | yes | unknown | putative |
| 9 | c.473G>A | p.Arg158His | missense | del | 113 | 9 | yes | LOH | yes |
| 10 | c.877G>T | p.Gly293Trp | missense | del | 6 | 2 | yes | LOH | yes |
| 11 | c.847C>T | p.Arg283Cys | missense | del | 29 | 10 | yes | No LOH | yes |
| 12 | c.586C>T | p.Arg196X | nonsense | NA | 241 | 13 | yes | No LOH | yes |
| 13 | c.826_840del | p.Ala276_Arg280del | frameshift | NA | 1 | 0 | unknown | unknown | yes |
| 14 | c.843C>A | p.Asp281Glu | missense | del | 28 | 0 | yes | No LOH | putative |
| 15 | c.841G>A | p.Asp281Asn | missense | del | 37 | 4 | yes | No LOH | yes |
| 16 | c.469G>A | p.Val157Ile | missense | del/neut | 19 | 0 | yes | No LOH | putative |
| 17 | c.835G>A | p.Gly279Arg | missense | del | 8 | 0 | yes | No LOH | putative |
| Putative somatic | |||||||||
| 18 | c.532C>G | p.His178Asp | missense | del | 9 | 0 | yes | LOH | yes |
| 19 | whole gene del | - | deletion | NA | 0 | 1 | - | - | yes |
| 20 | whole gene del | - | deletion | NA | 0 | 1 | - | - | yes |
Database version R16 November, 2012; del: deleterious; neut: neutral; FS: frame shift; LOH: loss of heterozygosity; NA: not applicable. The assignment of pathogenicity was performed as outlined in supplementary Figure 3.
Ten of 17 (59%) germline carriers had classical LFS or Chompret pedigrees that would have prompted genetic testing (Table 2). Case 14 showed somatic mosaicism, with 20–25% of mutant alleles (estimated by both Sanger sequencing and HRM analysis) in the peripheral blood, heterozygosity in the tumor, and absent in adjacent buccal mucosa (Figure 1). Somatic mosaicism for TP53 mutations has been previously reported [20]. Of the putatively pathogenic variants, 1 occurred in a family fitting classical Li-Fraumeni criteria (case 4); two occurred in individuals fitting Chompret criteria (cases 6 & 8), and three did not demonstrate an unusual family history of cancer (cases 14, 16 & 17). This pattern is not meaningfully different than carriers of pathogenic variants.
Table 2. Proband cancers and clinical classification.
| Case | Sex | Proband primary cancers, age at diagnosis (yrs) | Clinical classification |
| Putative germline | |||
| 1 | M | rhabdomyosarcoma 33 | LFS |
| 2 | M | osteosarcoma 20 | LFS |
| 3 | M | chondrosarcoma 24; liposarcoma 39 | LFS |
| 4 | M | sarcoma NOS 37; liposarcoma 44 | LFS |
| 5 | F | angiosarcoma 25 | Chomp LFL |
| 6 | F | breast 33; leiomyosarcoma 48 | Chomp LFL |
| 7 | F | breast 38; leiomyosarcoma 45; thyroid 46 | Chomp LFL |
| 8 | F | ALL 10; Ewing sarcoma 16 | Chomp LFL |
| 9 | F | breast 26; sarcoma NOS 36; pheochromocytoma 37 | Chomp LFL |
| 10 | M | Hodgkin’s lymphoma 34; melanoma 47; sarcoma NOS 60 | Chomp LFL |
| 11 | M | DSRCT 21 | Negative |
| 12 | M | testis 36; rectum 69; leiomyosarcoma 69 | Negative |
| 13 | F | chondrosarcoma 57 | Negative |
| 14 | M | osteosarcoma 19 | Negative |
| 15 | M | osteosarcoma 31 | Negative |
| 16 | F | leiomyosarcoma 58 | Negative |
| 17 | F | liposarcoma 62 | Negative |
| Putative somatic | |||
| 18 | M | mediastinal GCT with rhabdomyosarcomatous differentiation 19 | Chomp LFL |
| 19 | M | GIST 65; melanoma 69; sarcoma NOS 76; mycosis fungoides 76 | Negative |
| 20 | F | sarcoma NOS 80 | Negative |
ALL, acute lymphoblastic leukemia; DSRCT, desmoplastic small round cell tumour; GCT, germ cell tumour; GIST, gastrointestinal stromal tumour; Chomp, Chompret; M, male; F, female.
Figure 1. Somatic mosaicism demonstrated in an ISKS proband.

Case 14 presented with an osteosarcoma of the mandible at age 19yrs. HRM analysis of the peripheral blood DNA estimated that 20–25% of alleles were mutant. The mutation was detected in tumour DNA and found to be heterozygous, but was absent in multiple other non-tumour tissues of the mouth.
Regardless of family history, carriers of TP53 mutations appeared at increased personal risk for cancer. The median age of onset (±standard deviation) of first cancer in the germline TP53 probands was 33±14yrs compared to 48±18yrs in non-carriers (Student’s unpaired 2-tailed t-test P = 1.19×10−3), and the median age at first sarcoma was 36±17yrs vs 50±18yrs (P = 4.33×10−3) (Table 3). Eight of 17 mutation carriers had multiple primary cancers (Table 2), three occurring within prior radiation fields. Mutation carriers had an increased incidence of multiple cancers (47% versus 15% of non-carriers, Fisher’s exact test 2-tailed P = 3.0×10−3). With a short median follow up of 20 months, the survival of carriers in the ISKS cohort from first cancer diagnosis was not significantly different from non-carriers (Hazard ratio 1.175, 95% CI 0.44–3.12, Mantel-Cox P = 0.75). To investigate a possible survival bias, we also analysed those cases newly diagnosed with sarcoma during the period of recruitment from 2007 onwards (Figure 2). Again, the survival of carriers again was not significantly different to non-carriers (Hazard ratio 1.02, 95% CI 0.36–2.89, Mantel-Cox P = 0.97). Excluding probands, the median age at first cancer onset in TP53 mutation positive families was 54±22yrs versus 60±18yrs in TP53 negative families (P = 9.3×10−3). As recently reported in the IARC TP53 database [18], more leiomyosarcoma and undifferentiated pleomorphic sarcomas were seen compared to previous reports in pediatric populations [3], [21].
Table 3. Characteristics of the ISKS cohort.
| Probands | ||
| TP53 | non-TP53 | |
| n | 17 | 539 |
| Gender | ||
| Male | 9 (53%) | 291 (54%) |
| Female | 8 (47%) | 248 (46%) |
| Median age at diagnosis (yrs±SD) | ||
| First cancer | 33±14 | 48±18 |
| Range | (10–59) | (3–93) |
| Sarcoma | 36±17 | 50±18 |
| Range | (16–68) | (3–93) |
| Individuals with multiple primaries | 8 (47%) | 83 (15%) |
| Number of tumours/individual (mean±SD) | 2.5±0.5 | 2.2±0.5 |
| Sarcoma subtypes | ||
| n | 19* | 546* |
| Bone | ||
| Osteosarcoma | 3 (16%) | 52 (10%) |
| Chondrosarcoma | 2 (11%) | 48 (9%) |
| Ewing/Primitive neuroectodermal tumour | 1 (5%) | 47 (9%) |
| Other | – | 3 (1%) |
| Soft tissue | ||
| Undifferentiated pleomorphic sarcoma | 3 (16%) | 67 (12%) |
| Leiomyosarcoma | 4 (21%) | 61 (11%) |
| Fibromyxosarcoma | – | 51 (9%) |
| Well differentiated/Dedifferentiate LPS | 1 (5%) | 43 (8%) |
| Myxoid LPS | – | 27 (5%) |
| LPS not otherwise specified | 2 (11%) | 14 (3%) |
| Synovial sarcoma | – | 38 (7%) |
| Angiosarcoma | 1 (5%) | 8 (1%) |
| Epithelioid sarcoma | – | 8 (1%) |
| Malignant peripheral nerve sheath tumour | – | 8 (1%) |
| Rhabdomyosarcoma | 1 (5%) | 7 (1%) |
| Desmoplastic small round cell tumour | 1 (5%) | – |
| Other | – | 64 (12%) |
| Family history | ||
| Classic Li Fraumeni Syndrome | 4 (24%) | 4 (<1%) |
| Chompret Li Fraumeni Like | 6 (35%) | 44 (8%) |
| Other familial cancer syndrome | – | 8 (1%) |
| No family history | 7 (41%) | 458 (85%) |
| Uninformative | – | 25 (5%) |
some probands have >1 sarcoma; SD, standard deviation; LPS, liposarcoma.
Figure 2. Kaplan-Meier overall survival analysis comparing TP53 mutation carriers to non-carriers.
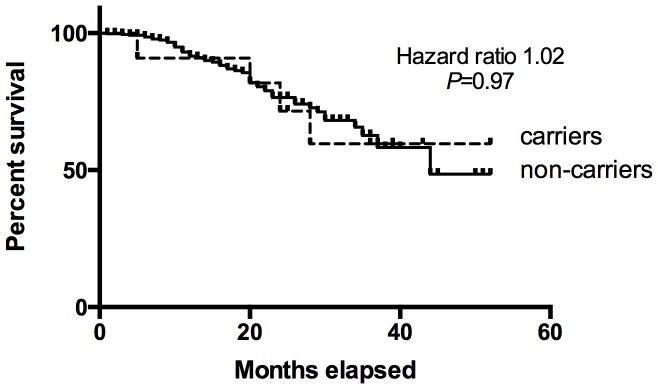
To correct for survival bias, this analysis was limited to ISKS participants prospectively recruited from 2007 onwards (TP53 mutation carriers, n = 11; Non-carriers, n = 420).
Discussion
The germline mutation rate observed in the ISKS cohort (3%) matches the 2–4% [12] , [21] in childhood osteosarcoma, 2–3% reported in early onset breast cancer [15], [22], but is less than reported for choroid plexus carcinomas (44%) [14]. Only 60% of carriers had a family history potentially recognisable as associated with germline TP53 mutations. One mutation is clearly due to somatic mosaicism. Assuming a 20% new mutation rate [3], [6], the penetrance of the remaining mutations may account for the lack of a strong family history [3].
The identification of somatic and mosaic mutations remind us that peripheral blood is only a surrogate for the germline. Somatic TP53 mutations are common in hematologic cancers, and cancer-prone individuals may harbour preclinical genetic evidence of dysplasia even in apparently normal blood. This differentiates TP53 testing from genes such as BRCA1/2 or the mismatch repair genes, which appear rarely somatically mutated in hematologic malignancy. Prior mutagen exposure, including chemotherapy, may be important. It is clinically important to confirm the presence of putative germline TP53 mutations in more than one tissue, including the tumor tissue.
These results challenge nihilistic perceptions regarding germline TP53 mutation incidence, cancer risk and survival [23], [24]. As genomic technologies are increasingly applied to cancer cohorts, regardless of clinical or family history, more TP53 mutation carriers will be identified and require counselling and care from their medical supports. Interpretation of purely genotypic information is difficult using data mostly ascertained on clinical or familial criteria. While more common than expected, the outlook for TP53 mutation carriers with sarcoma appears comparable to non-carriers, and options are emerging for cancer screening [10], family planning [25], and the selection of less carcinogenic cancer treatments. Continued research into germline TP53 mutations is critical to understanding the impact of these mutations on cancer treatments and outcomes, and to develop effective cancer screening strategies for our patients and their families.
Materials and Methods
International Sarcoma Kindred Study (ISKS)
ISKS is a clinic-based prospective cohort of adult-onset sarcoma cases and families aimed to investigate the hereditary aspects of this disease. Probands (n = 559, 54% male) were consecutively recruited from 6 major sarcoma treatment centres across Australia. Probands over 14 years of age with a histologically confirmed sarcoma (64% soft tissue, 36% bone subtypes) were consented to donation and use of biospecimens and provided family history information. Medical history and treatment records were obtained for each proband where possible. All reported cancer diagnoses were independently verified by reference to the medical records, Australian and New Zealand cancer registries or death certificates. Study questionnaires containing demographic, medical, epidemiological and psychosocial information were completed, including personal history of cancer or exposure to known risk factors for sarcoma. Self-reported ethnicity was 84% Caucasian, 4% Chinese or South East Asian, 3% unknown, with the remainder from diverse ethnic backgrounds.
Clinical Classification of Families
Pedigrees to at least second degree relatives of the proband were classified according to a recognised set of clinical criteria[1], [26]–[29]. A family history was considered positive if the classical LFS or Chompret LFS criteria were met.
Biospecimen Processing
Anti-coagulated blood was processed using a Ficoll gradient. DNA was extracted from the nucleated cell product using QIAamp DNA blood kit (Qiagen, Germany).
After micro-dissection of tumour material from formalin fixed paraffin embedded tissue, DNA was extracted using DNeasy tissue kit (Qiagen, Germany) as described previously [30].
TP53 HRM Screening
High resolution melt (HRM) analysis was used to screen for mutations in exons 2–11 of the TP53 gene. PCR and HRM were performed on the LightCycler 480 (Roche Diagnostics, Australia). The reaction mixture included 1× PCR buffer, 2.5 mM MgCl2, 200 nM of each primer, 200 µM of dNTPs, 5 µM of Syto 9 (Invitrogen, Carlsbad, CA), 0.5 U of HotStarTaq polymerase (Qiagen, Valencia, CA), 10 ng DNA and PCR grade water in a total volume of 10 µL. PCR conditions included an activation step of 15 minutes at 95°C followed by 55 cycles of 95°C for 10 seconds, annealing for 10 seconds comprising 10 cycles of a touchdown from 65 to 55°C at 1°C/cycle followed by 35 cycles at 55°C, and extension at 72°C for 30 seconds; one cycle of 95°C for 1 minute, 45°C for 1 minute and a HRM step from 72 to 95°C rising at 0.02°C per second. Primers for all exons are shown in Table S1. The primers used for HRM analysis for exons 6–8 were those published in Krypuy et al [31] with the exception of the exon 6 reverse primer. For both exons 4 and 5, a set of three amplicons were designed to span the coding region of each exon.
All analyses were performed in duplicate. At least five different normal controls were included in each run. Where possible, a positive control sample was included for each amplicon. Samples showing an aberrant melt profile compared to normal controls via HRM were directly sequenced from a 1/10 dilution of the HRM product using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions.
Variants were triaged for pathogenicity as follows (see Figure S1). Variants were defined as pathogenic if previously reported to be associated with Li-Fraumeni syndrome in the IARC TP53 database (R16, accessed February 2013), or they resulted in a frameshift, premature stop codon, or affected an essential splice site. Variants were considered putatively pathogenic those variants which have either been reported somatically mutated more than 5 times in the IARC database, or which were predicted to be pathogenic by Condel, or both. In all but one case (R333C), both criteria were satisfied. R333C was included because a known pathogenic variant (R337H) is located in the same region (the tetramerization domain of TP53), and has been reported to result in loss of function by Kato et al [32].
TP53 LOH Testing
For LOH analysis, tumour and matching normal DNA were amplified and sequenced. Evidence for any LOH was only inferred if the strength of the WT allele was reduced by at least 50% on the sequencing trace in tumour DNA compared to matching normal DNA.
Mutilplex Ligation-dependent Probe Amplification (MLPA)
Large deletions or genomic rearrangements of patients were analyzed by a commercial MLPA kit (SALSA MLPA probemix P056-B1 TP53, MRC Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions. DNA (100 ng) extracted from peripheral blood was used with a normal and positive control sample included in every assay. MLPA PCR products were separated on the ABI3730 instrument (Applied Biosystems) and peak heights for each PCR product were compared to a normal sample using GeneMarker software to determine gene dosage for each individual exon. Every positive result was repeated at least twice.
The two cases with whole gene deletion were verified by using custom exon capture using Haloplex reagents (Agilent Technologies, Inc., Santa Clara, CA). Custom capture reagents were designed including the entire gene for TP53, as well as coding exons for 84 additional genes including RB1. Copy number states for all loci were inferred from SNP allelic ratios and the CONTRA algorithm for identifying copy-number gains and losses using fluctuations in sequencing read depth [33]. Target regions were divided into 100 bp bins for the CONTRA analysis, to adjust for the limited number of target regions and deep read coverage of the custom capture.
Statistical Analyses
Statistical analyses of age at cancer diagnosis were performed using a Student’s unpaired two-tailed t-test with unequal variance, and where appropriate a Fisher’s exact test. Survival analyses used a Log-rank (Mantel-Cox) test for survival comparisons.
Ethics Statement
This project was conducted under the auspices of the human research ethics committee of the Peter MacCallum Cancer Centre (HREC approval number 09/11). Participants over the age of 15 gave written consent to the study. For participants below 18 years written consent was obtained from their legal guardians. For participants between 16 and 18 years, written consent was obtained from both participants as well as their legal guardians.
Supporting Information
Assignment of pathogenicity for TP53 variants.
(TIFF)
TP53 HRM and Sequencing Primers.
(XLSX)
Acknowledgments
The authors acknowledge and thank the families who have contributed to the ISKS cohort, as well as the ISKS Australia site investigators; Sandro Porceddu (Princess Alexandra Hospital, Brisbane), Michael Gattas (Genetics Health Queensland), Susan Neuhaus and Graeme Suthers (Royal Adelaide Hospital, Adelaide), Martin Tattersall (Royal Prince Alfred Hospital, Sydney), Kathy Tucker and Craig Lewis (Prince of Wales Hospital, Sydney), and Richard Carey-Smith (Hollywood Private Hospital, Perth). We thank Eveline Niedermayr for data management and Allison Wicht, Vicki Fennelly, Belinda Zielony, Kim Riddell, Jasmine Mar and Jessica MacDonald for co-ordination of ISKS recruitment.
Funding Statement
This work was supported by a Johanna Sewell Research Grant, the Rainbows for Kate Foundation, the Victorian Cancer Agency, and the National Health and Medical Research Council (Project Grant 1004017). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Li FP, Fraumeni JF Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, et al. (1988) A cancer family syndrome in twenty-four kindreds. Cancer Res 48: 5358–5362. [PubMed] [Google Scholar]
- 2. Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, et al. (1998) Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene 17: 1061–1068. [DOI] [PubMed] [Google Scholar]
- 3. Chompret A, Brugieres L, Ronsin M, Gardes M, Dessarps-Freichey F, et al. (2000) P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 82: 1932–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, et al. (2001) Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 20: 4621–4628. [DOI] [PubMed] [Google Scholar]
- 5. Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, et al. (2003) Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 63: 6643–6650. [PubMed] [Google Scholar]
- 6. Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, et al. (2009) Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27: 1250–1256. [DOI] [PubMed] [Google Scholar]
- 7. Nichols KE, Malkin D, Garber JE, Fraumeni JF Jr, Li FP (2001) Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev 10: 83–87. [PubMed] [Google Scholar]
- 8. Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, et al. (2010) TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet 47: 421–428. [DOI] [PubMed] [Google Scholar]
- 9. Leach MO, Boggis CR, Dixon AK, Easton DF, Eeles RA, et al. (2005) Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: a prospective multicentre cohort study (MARIBS). Lancet 365: 1769–1778. [DOI] [PubMed] [Google Scholar]
- 10. Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, et al. (2011) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 12: 559–567. [DOI] [PubMed] [Google Scholar]
- 11. Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, et al. (2008) Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45: 535–538. [DOI] [PubMed] [Google Scholar]
- 12. Toguchida J, Yamaguchi T, Dayton SH, Beauchamp RL, Herrera GE, et al. (1992) Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 13. Malkin D, Jolly KW, Barbier N, Look AT, Friend SH, et al. (1992) Germline mutations of the p53 tumor-suppressor gene in children and young adults with second malignant neoplasms. N Engl J Med 326: 1309–1315. [DOI] [PubMed] [Google Scholar]
- 14. Tabori U, Shlien A, Baskin B, Levitt S, Ray P, et al. (2010) TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol 28: 1995–2001. [DOI] [PubMed] [Google Scholar]
- 15. Lalloo F, Varley J, Moran A, Ellis D, O’Dair L, et al. (2006) BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur J Cancer 42: 1143–1150. [DOI] [PubMed] [Google Scholar]
- 16. Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, et al. (2001) An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 98: 9330–9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ognjanovic S, Olivier M, Bergemann TL, Hainaut P (2012) Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118: 1387–1396. [DOI] [PubMed] [Google Scholar]
- 19. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, et al. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629. [DOI] [PubMed] [Google Scholar]
- 20. Prochazkova K, Pavlikova K, Minarik M, Sumerauer D, Kodet R, et al. (2009) Somatic TP53 mutation mosaicism in a patient with Li-Fraumeni syndrome. Am J Med Genet A 149A: 206–211. [DOI] [PubMed] [Google Scholar]
- 21. McIntyre JF, Smith-Sorensen B, Friend SH, Kassell J, Borresen AL, et al. (1994) Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12: 925–930. [DOI] [PubMed] [Google Scholar]
- 22. Mouchawar J, Korch C, Byers T, Pitts TM, Li E, et al. (2010) Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer: Australian Breast Cancer Family Study. Cancer Res 70: 4795–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans DG, Lunt P, Clancy T, Eeles R (2010) Childhood predictive genetic testing for Li-Fraumeni syndrome. Fam Cancer 9: 65–69. [DOI] [PubMed] [Google Scholar]
- 24. Tischkowitz M, Rosser E (2004) Inherited cancer in children: practical/ethical problems and challenges. Eur J Cancer 40: 2459–2470. [DOI] [PubMed] [Google Scholar]
- 25. Offit K, Kohut K, Clagett B, Wadsworth EA, Lafaro KJ, et al. (2006) Cancer genetic testing and assisted reproduction. J Clin Oncol 24: 4775–4782. [DOI] [PubMed] [Google Scholar]
- 26. Chompret A, Abel A, Stoppa-Lyonnet D, Brugieres L, Pages S, et al. (2001) Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 38: 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, et al.. (2009) 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 27: e108–109; author reply e110. [DOI] [PubMed] [Google Scholar]
- 28. Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, et al. (1994) Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 54: 1298–1304. [PubMed] [Google Scholar]
- 29. Eeles RA (1995) Germline mutations in the TP53 gene. Cancer Surv 25: 101–124. [PubMed] [Google Scholar]
- 30. Wu L, Patten N, Yamashiro CT, Chui B (2002) Extraction and amplification of DNA from formalin-fixed, paraffin-embedded tissues. Appl Immunohistochem Mol Morphol 10: 269–274. [DOI] [PubMed] [Google Scholar]
- 31. Krypuy M, Ahmed AA, Etemadmoghadam D, Hyland SJ, DeFazio A, et al. (2007) High resolution melting for mutation scanning of TP53 exons 5–8. BMC Cancer 7: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kato S, Han SY, Liu W, Otsuka K, Shibata H, et al. (2003) Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A 100: 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, et al. (2012) CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28: 1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assignment of pathogenicity for TP53 variants.
(TIFF)
TP53 HRM and Sequencing Primers.
(XLSX)