

Abstract
Introduction
Alström Syndrome (ALMS) is a rare autosomal recessive monogenic disease included in an emerging class of genetic disorders called ‘ciliopathies’ and is likely to impact the central nervous system as well as metabolic and endocrine function. Individuals with ALMS present clinical features resembling a growth hormone deficiency (GHD) condition, but thusfar no study has specifically investigated this aspect in a large population.
Material and Methods
Twenty-three ALMS patients (age 1-52 years, 11 M, 12 F) were evaluated for anthropometric parameters (growth charts and Standard Deviation Score (SDS) of height, weight, BMI), GH secretion by growth-hormone-releasing-hormone + arginine test (GHRH-arg), bone age, and hypothalamic-pituitary magnetic resonance imaging (MRI). A group of 17 healthy subjects served as controls in the GH secretion study. Longitudinal retrospective and prospective data were utilized.
Results
The length-for-age measurements from birth to 36 months showed normal growth with most values falling within -0,67 SDS to +1.28 SDS. A progressive decrease of stature-for-age was observed after 10 years of age, with a low final height in almost all ALMS subjects (> 16-20 years: mean SDS -2.22±1.16). The subset of 12 ALMS patients tested for GHRH-arg showed a significantly shorter stature than age-matched controls (154.7±10.6 cm vs 162.9±4.8 cm, p= 0.009), and a mild increase of BMI (Kg/m2) (27.8±4.8 vs 24.1±2.5, p=0.007). Peak GH after GHRH-arg was significantly lower in ALMS patients in comparison to controls (11.9±6.9 ug/L vs 86.1±33.2 ug/L, p <0,0001). Severe GHD was evident biochemically in 50% of ALMS patients. The 10 adult ALMS patients with GHD showed a reduced height in comparison to those without GHD (149.7±6.2 cm vs 161.9±9.2 cm, p= 0.04). MRIs of the diencephalic and pituitary regions were normal in 11 of 12 patients. Bone age was advanced in 43% of cases.
Conclusions
Our study shows that 50% of non-obese ALMS patients have an inadequate GH reserve to GHRH-arg and may be functionally GH deficient. The short stature reported in ALMS may be at least partially influenced by impairment of GH secretion.
Keywords: Alström Syndrome, ALMS1, GH, arginine, recombinant GH (rGH), fibrosis
INTRODUCTION
Alström Syndrome [ALMS (MIM #203800)] is a rare autosomal recessive monogenic disorder characterized by early retinal photoreceptor degeneration, sensorineural hearing impairment, childhood obesity, and severe insulin resistance followed by diabetes mellitus. Systemic fibrosis and multiple organ involvement including dilated cardiomyopathy (DCM), hepatic and renal failure may occur with an increased morbidity leading to a life expectancy that rarely exceeds 40 years of age (1-4). There is no cure for ALMS; therefore, careful detection of systemic manifestations and prompt intervention are highly recommended to prevent both short and long term complications.
ALMS is caused by mutations in ALMS1, encoding a protein ubiquitously expressed, including in endocrine-related tissues such as the pituitary gland and hypothalamus (5;6). ALMS1 localizes to the centrosomes and basal bodies of ciliated cells and roles in cytoplasmic microtubular organization, intracellular transport, and ciliary function have been suggested (7-10). These observations led to the inclusion of ALMS in an emerging class of human genetic disorders called “ciliopathies” (11;12). Although its precise function is unknown, ALMS1 impacts the central nervous system and both metabolic and endocrine function.
Hypothyroidism, hypergonadotropic hypogonadism in males and irregular menses or amenorrhea in females have been frequently reported in ALMS patients. Despite normal growth rate in childhood, individuals with ALMS present clinical features that resemble a growth hormone deficiency (GHD) condition such as a reduced final height, hypertriglyceridemia, central obesity, and heart failure. Low Insulin-like Growth Factor 1 (IGF-I) levels have been described (13-15), while a reduction of Acid-labile Subunit (ALS) and an increase of Insulin-like Growth Factor Binding Proteins 2 (IGFBP-2) was found in a larger cohort (16). Low GH secretion consistent with GHD has also been reported in two young patients who were treated for one year with recombinant GH (rGH), without side effects (13).
Previous studies have been limited by the small number of subjects - mostly within the same family and not always genetically investigated, a low potency of GH segretagogues to measure GHD, a lack of a control population, and an absence of auxological trends. Considering these limitations, we aimed to investigate growth patterns and the occurrence of the GHD condition in a large group of ALMS patients.
PATIENTS AND METHODS
Patients and controls
We evaluated 23 patients with ALMS (cases 1-23) from 17 unrelated families, over the course of ten years, by means of longitudinal retrospective or prospective study (Table I). Patients were divided into two groups: 12 adult cases (>18 years; cases 1-12; 7 male and 5 female; age range 18-52 years, mean age 29.1±10.2 years) and 11 pediatric cases (cases 13-23; 4 male and 7 female; age range 1-14 years, mean age 6.5±4.6 years). Six sibling pairs were among the patients (cases 3&6, 7&12, 8&10, 13&15, 16&21, and 17&19). Twenty patients were Italian, two sisters were of Moroccan ancestry (cases 17&19) and case 23 was of Albanian ancestry.
Table I. Clinical features of ALMS patients.
Phenotypes of ALMS patients at the time of evaluation.
| CASE | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 18 | 19 | 21 | 23 | 28 | 33 | 44 | 24 | 25 | 30 | 32 | 52 |
| Gender | M | M | M | M | M | M | M | F | F | F | F | F |
| ALMS1 mutation | / | YES | YES | / | YES | YES | YES | YES | YES | YES | YES | YES |
| Retinopathy | YES | YES§ | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| Hearing impairment | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| Dilated cardiomyopathy | / | YES | / | / | / | / | YES | YES | / | / | / | YES |
| Respiratory disorders | YES | YES | / | / | / | / | / | / | / | / | / | YES |
| Nephropathy | / | YES | / | YES | YES | / | YES | YES | / | / | / | YES |
| Hepatic disorders | YES | YES | / | / | YES | / | YES | YES | YES | YES | YES | / |
| Hypertriglyceridaemia | YES | / | YES | YES | YES | YES | / | YES | / | YES | YES | YES |
| Type 2 diabetes | YES | IGT | / | YES | YES | IGT | IGT | / | YES | / | YES | / |
| Hypothyroidism | / | YES | / | YES | / | / | / | YES | YES | YES | / | / |
| Gonadal dysfunctions | / | YES | YES | / | YES | YES | / | YES | YES | / | / | / |
| Low stature | YES | / | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| State of feeding | OVR | OVR | OVR | OVR | OB | OVR | OVR | OVR | OB | OB | NRM | OVR |
| GHRH + Arginine test | N.A. | N | N | N | N | GHD | N | GHD | N | N.A. | GHD | GHD |
| IGF-I | N.A. | 2.3 | 1.5 | 1.3 | 0.3 | 1.49 | 1.3 | 1.1 | 0.81 | N.A. | 1.62 | 1.22 |
| Hypotalamic-pituitary MRI | N.A. | MRI | MRI | N.A. | MRI | MRI | N.A. | MRI | PES | N.A. | MRI | N.A. |
| CASE | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 |
| Age (years) | 3 | 4 | 6 | 11 | 1 | 2 | 2 | 7 | 10 | 12 | 14 |
| Gender | M | M | M | M | F | F | F | F | F | F | F |
| ALMS1 mutation | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES | / |
| Retinopathy | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| Hearing impairment | / | / | / | YES | / | YES | / | / | YES | YES | YES |
| Dilated cardiomyopathy | / | YES | / | YES | YES | YES | / | YES | YES | YES | YES |
| Respiratory disorders | / | YES | / | YES | / | / | / | YES | / | YES | YES |
| Nephropathy | / | / | / | YES | / | YES | / | / | / | YES | / |
| Hepatic disorders | / | / | YES | YES | / | YES | / | YES | YES | YES | YES |
| Hypertriglyceridaemia | / | / | / | / | / | / | / | / | / | / | YES |
| Type 2 diabetes | / | / | / | YES | IFG | / | / | / | YES | / | YES |
| Hypothyroidism | / | / | / | / | / | / | / | / | / | YES | YES |
| Gonadal dysfunctions | / | / | / | / | / | / | / | / | / | / | / |
| Low stature | / | / | / | / | N.A. | / | / | / | / | YES | / |
| State of feeding | OB | OB | OB | NRM | OB | UDR | OB | OB | OB | OVR | OVR |
| GHRH + Arginine test | N.A. | N.A. | N.A. | GHD | N.A. | N.A. | N.A. | N.A. | N.A. | GHD | N.A. |
| IGF-I | N.A. | N.A. | N.A. | 1.05 | N.A. | N.A. | N.A. | N.A. | N.A. | 0.9 | N.A. |
| Hhypotalamic-pituitary MRI | MRI | N.A. | MRI | MRI | N.A. | N.A. | N.A. | MRI | MRI | N.A. | N.A. |
Cases for whom GH secretion studies were carried out are indicated in bold. M= male; F = female; /= not present; N= normal reponse §= residual visual acuity; n.a. = not available; IGT= impaired glucose tolerance; IFG= impaired fasting glycemia; OVR= overweight; OB= obese; NRM= normal weight; UDR= underweight; GH= Growth Hormone; IGF-I= Insulin-like Growth Factor-I; MRI= magnetic resonance imaging; PES= partial empty sella; GHD= biochemical growth hormone deficiency. IGF-I was calculated as the ratio of patient’s value to the lower normal limit (pathologic when <1). Respiratory disorders included asthma, restrictive ventilatory defect or bronchiectasis with frequent pulmonary infections. Nephropathy is defined by elevation of blood urea nitrogen or creatinine or abnormal glomerular filtration rate assessed by renal scintigraphy. Hepatic disorders consisted of elevation of transaminases or liver steatosis observed by ultrasonography.
A group of 17 (age and gender matched) unaffected subjects selected from patients with normal pituitary function evaluated in our Department (7 males and 10 females; age range 14-33 years, mean age 20.9±5.8 years) served as controls in the GH secretion study. All participants provided written informed consent for genetic (The Jackson Laboratory, Bar Harbor, ME, USA) and laboratory (University Hospital, Padua, Italy) investigations; for pediatric patients, the consent was granted by parents. The research protocol was approved by the Institutional Review Boards from each institution.
Anthropometric parameters and growth charts
Body weight and height were measured using standardized procedures and, retrospectively retrieved from prior clinical records for each patient. The Standard Deviation Score (SDS) for height, weight, and body mass index (BMI) in the age range 2-20 years were established using the SIEDP (Società Italiana di Endocrinologia e Diabetologia Pediatrica) growth calculator (http://www.siedp.it). Growth trends were reconstructed utilizing standard growth charts (www.cdc.gov/growthcharts). Height genetic target was calculated with the following formula: father height + mother height + 13 (male subject) or – 13 (female subject) / 2. Obesity was defined as BMI>30 kg/m2 in adults (underweight ≤18.5 kg/m2; normal weight 18.6-24.9 kg/m2; overweight 25-29.9 kg/m2) and >95th percentile in children (underweight <10th percentile; normal weight 10th-75th percentile; overweight 76th-95th percentile). Height > + 1.88 SDS was defined as tall stature and values < -1.88 SDS as short stature for pediatric patients.
Assays
After overnight fasting, 12 ALMS patients (7 male and 5 female; age range 11-52 years, mean age 27±12 years; Table I bold font) and 17 healthy controls were tested for IGF-I and GH before (-15’, 0’) and after (15’, 30’, 45’, 60’, 90’, 105’, 120’) infusion of GHRH (GHRH29, GEREF, Serono, Italy; 1 ug/kg e.v. at 0 min) + arginine (Arginine hydrochloride, 0.5 g/kg e.v. over 30 min from 0 to +30 min) (GHRH-arg). GH was measured by chemiluminescent immunometric assay (Medical System S.p.a., Genova, Italy). Sensitivity: 0.05 ug/L (normal range: 0.06-5.0 ug/L for males and females). IGF-I was measured by IRMA assay (BioChem Immuno Systems S.p.a., Casalecchio di Reno, Bologna, Italy) or by chemiluminescence immunoassay (Liaison, DiaSorin S.p.a., Saluggia, Italy), using age-gender reference values supplied by each kit. Low GH secretion was defined according to the cut-off levels for GHRH-arg of the Consensus Statement on Adult GH Deficiency (17): GH peak <11 ug/L for BMI <25 kg/m2, GH peak <8 ug/L for BMI 25-30 kg/m2, and GH peak <4 ug/L for BMI >30 kg/m2. In patients 16 and 22, low GH secretion was determined according to the cut-off validated for children (GH peak after GHRH-arg <19.4 ug/L) (18). The insulin tolerance test (ITT) was performed only in case 11 and consisted of a bolus of regular human insulin (0.1 U/kg body weight e.v.). Severe GHD was defined by a peak GH response to hypoglycemia of less than 3 ug/L (19-21). Owing to age, sex and methods variability, IGF-I was calculated as the ratio of patient’s value to the lower limit of reference values indicated by the Laboratory (pathologic when <1). Basal serum prolactin (PRL), Adrenocorticotropic hormone (ACTH), cortisol, Follicle-stimulating hormone (FSH), Luteinizing hormone (LH), testosterone (in males), 17-beta-estradiol, thyroid-stimulating hormone (TSH), Triiodothyronine (FT3), and free thyroxine (FT4) were collected and measured using standard commercial kits. Four male patients (2, 3, 5, 6) were undergoing testosterone therapy when tested with GHRH+Arg.
Bone age and hypothalamic-pituitary magnetic resonance imaging (MRI)
Bone age was determined from several X-rays of the hand, metacarpus or pelvis (cases 2, 5, 6, 9, 14, 16, and 22; range 1-12 years) measured along their developmental age according to standardization determined by Greulich and Pyle (22). Twelve patients underwent hypothalamic-pituitary MRI examination (cases 2-3, 5-6, 8-9, 11, 13, 15-16, 20-21): all patients were scanned along the sagittal and coronal axis with T1W1, before and after the administration of gadolinium.
Histopathologic analysis of the pituitary gland
Pituitary specimens were collected during post-mortem investigation of three ALMS patients who were not included in this study cohort.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). The statistical analyses were performed using the unpaired Student’s t-test, Chi-square, or Fisher test, as appropriate. The SDSs for height, weight and BMI were grouped across different age ranges (2-5, >5-10, >10-16, >16-20 years) and a single SDS was considered for a patient in a single age range. Multiple measurements for the same patient within and age range were replaced by the mean. A linear regression model was used to study the relationship between numerical variables. Significance was set at P < 0.05 for all comparisons.
RESULTS
Clinical features of the ALMS cohort
Twenty-one ALMS patients exhibited the typical age-dependent clinical features of the syndrome (2) (Table 1). Two patients presented an atypical phenotype: case 2 had normal height and mild cone-rod dystrophy with residual visual acuity, and case 18 was underweight and presented with neurological dysfunction manifesting as axial hypotonia, microcephaly, and erratic eye movements.
Mutations in ALMS1 were demonstrated in twenty subjects. ALMS1 genetic investigation for cases 1, 4 and 23 is not yet complete. None of the patients showed overt liver or kidney failure at the time of this study.
Twelve cases suffered from DCM and congestive heart failure (CHF) (cases 2 and 22 died with complications of CHF subsequent to this study). However, at the time of the pituitary investigation, these patients were in stable hemodynamic condition. Normal ejection fraction was documented by echocardiography in eight of the remaining cases (cases 3, 5, 6, 9, 10, 11, 13, and 15; data not shown) and cases 1, 4, and 19 had a normal chest X-ray, normal physical examination and uneventful medical and pharmacological history related to heart failure.
Anthropometric data, growth charts and SDS calculation
The length-for-age measurements from birth to 36 months (7 M, Fig. 1a and 8 F, Fig 1b), showed a regular rising trend with most values falling within – 0,67 SDS to +1.28 SDS. In contrast, a marked elevation of weight-for-age values from birth to 36 months was observed in both age groups without gender differences. At 36 months the majority of patients presented a weight SDS > +1.28. A progressive decrease of stature-for-age trend was observed across the different age groups from 10 years of age with a low final height (>16-20 years: mean SDS -2.22±1.16) in almost all ALMS subjects (Fig. 1c, 1d, Fig. 2). The weight-for-age trend after 36 months showed a progressive normalization, especially in males (Fig 1c, 1d). The weight SDS and BMI SDS confirmed this trend beginning at 10 years of age across the different age groups (Fig. 2).
Figure 1. Growth curves of ALMS patients (longitudinal).
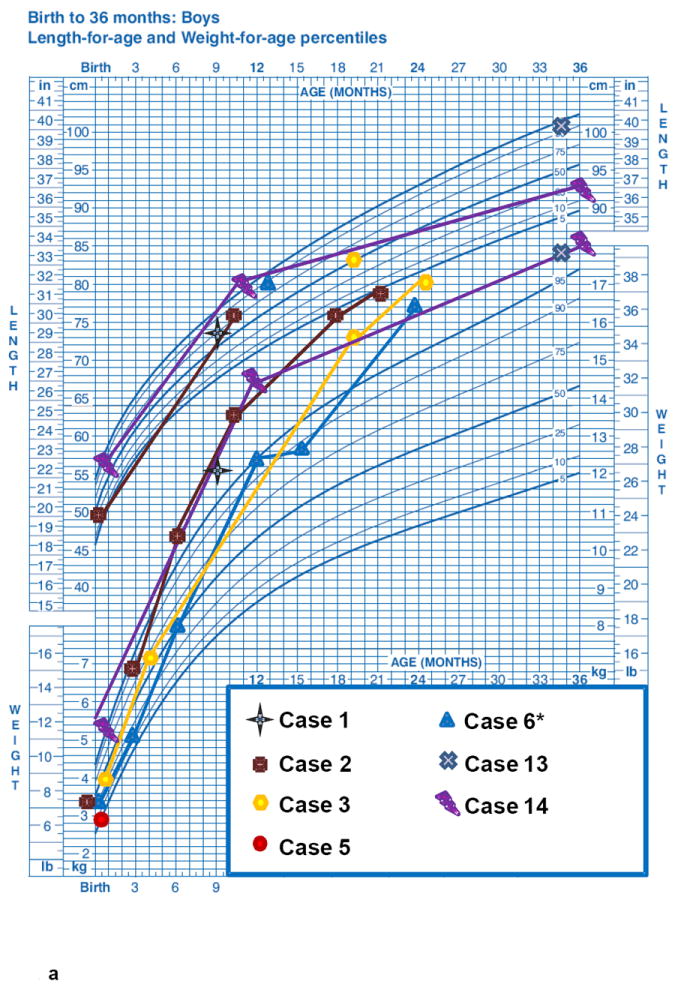
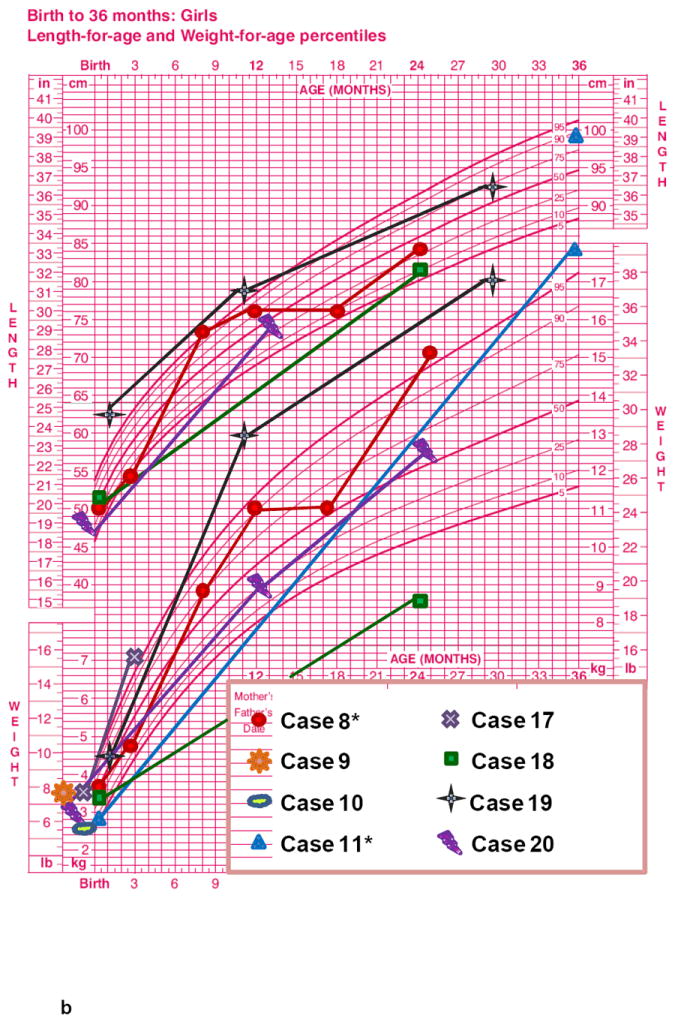
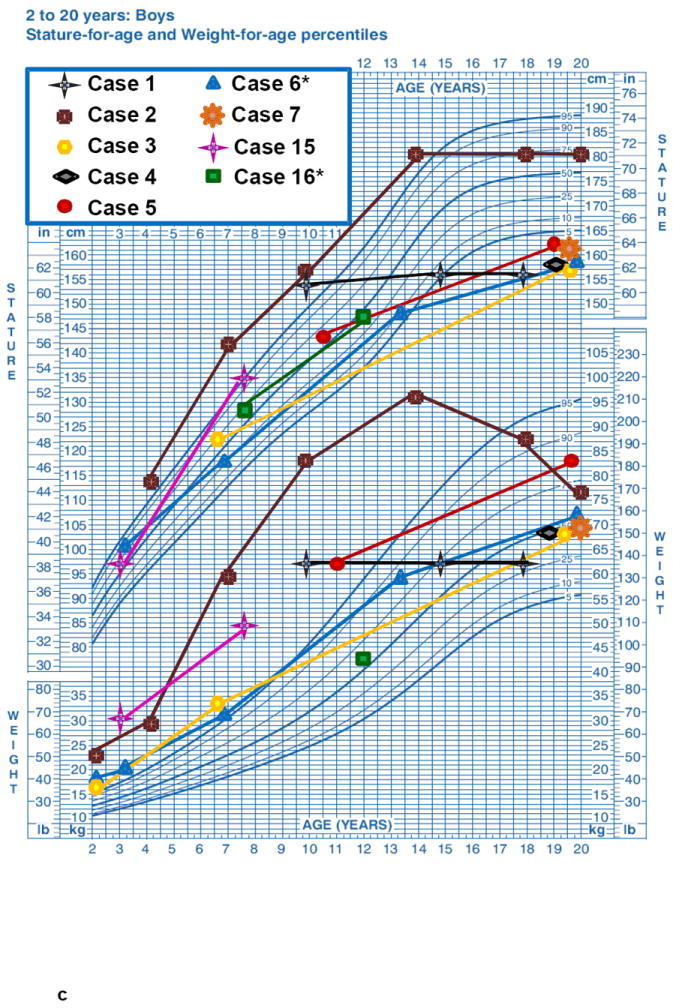
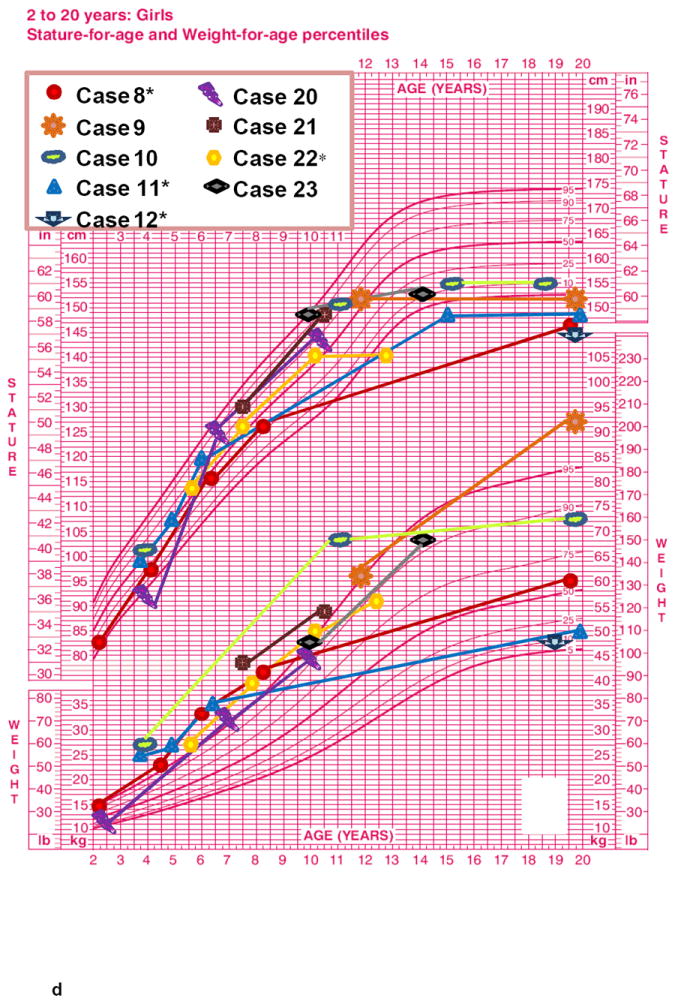
Height-age and weight-age curves were constructed utilizing the charts developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion, for boys (blue) and girls (pink), in the period from birth to 36 months (a - b) and from 2 to 20 years (c - d). Each patient is represented by a specific symbol as designated in the legend. * = patients with biochemical growth hormone deficiency when tested with GHRH+Arginine.
Figure 2. SDS height, weight and BMI of ALMS patients (cross-sectional).
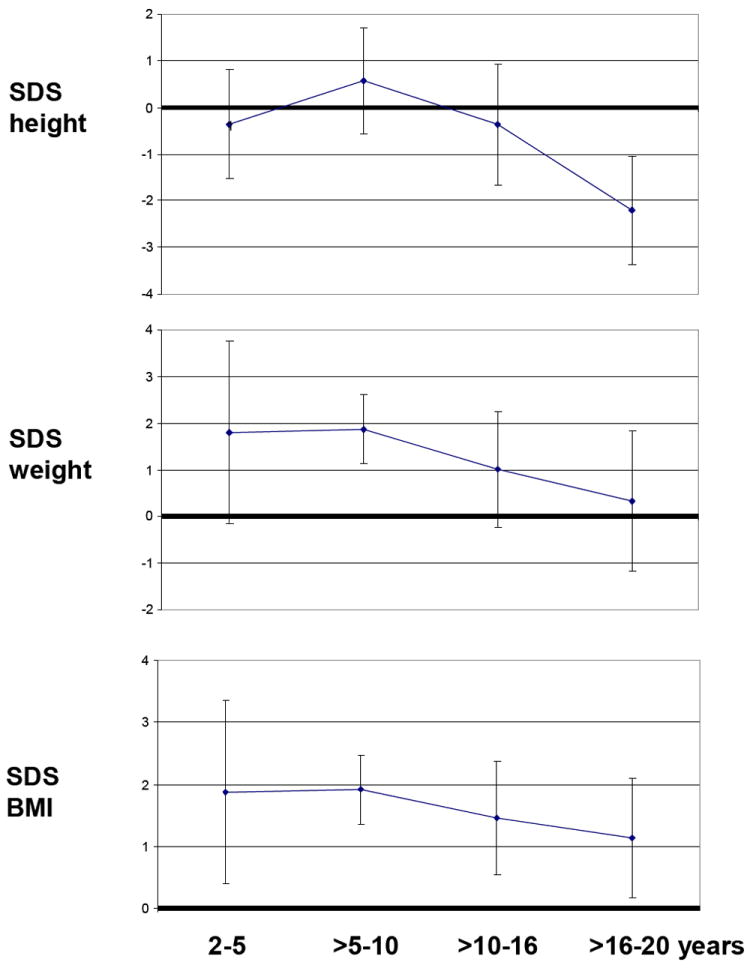
Graphic representation of SDS (Standard Deviation Score) height, weight and body mass index of ALMS patients across the different age groups. Results are expressed as mean values ± SD.
GH-IGF-I axis in ALMS patients
The subset of 12 ALMS patients tested for GHRH-arg (Table I, Bold), showed a significantly shorter stature than age-matched controls (154.7±10.6 cm vs 162.9±4.8 cm, p= 0.009), and a mild increase of BMI (27.8±4.8 vs 24.1±2.5, p=0.007). Hormone values including IGF-I, baseline and peak GH after GHRH-arg stimulation, are summarized in Table I and Fig. 3. There was no significant association between the peak GH response after GHRH-arg in ALMS patients and age (ρ= -0.25, p=0.43), BMI (ρ= -0.13, p= 0.68), height (ρ= 0.38, p=0.22), or SDS height (ρ= 0.14, p=0.66). Biochemically severe GHD was observed in 50% of ALMS patients interpreted according to Consensus Guidelines in adults (17) and the validated cut-off for children (18).
Figure 3. GHRH-arginine test in ALMS patients.
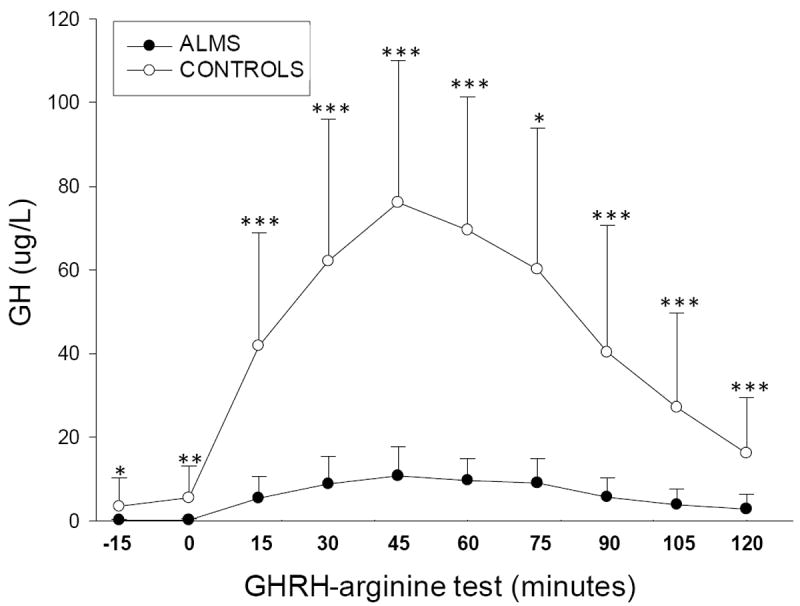
GH values after GHRH-arginine test (ordinate) at specific times (abscissa) in patients with ALMS (n= 12) versus healthy controls (n= 17). Results are expressed as mean values ± SD. * p< 0.05; ** p< 0.001; *** p<0.0001.
In the 6 ALMS patients with GHD the height was significantly reduced in comparison to the 6 patients without GHD (ALMS GHD 147.6±6.2 cm vs ALMS non GHD 161.9±9.2 cm, p= 0.0098; control subjects vs ALMS non GHD, p= 0.745; control subjects vs ALMS GHD, p< 0.0001). Considering the 10 adult ALMS patients only (6M and 4 F), those with GHD showed a reduced height in comparison to those without GHD (149.7±6.2 cm vs 161.9±9.2 cm, p= 0.04). Although the predicted height genetic target of adult patients was still lower in ALMS GHD (163.6 ± 12.3 cm) in comparison to ALMS non GHD (172.5 ± 9.8 cm), this difference was not significant (p= 0.24). Considering the adult patients only, we could speculate that the predicted height genetic target was reduced by 13.9 cm in ALMS GHD and 10,6 cm in ALMS non GHD patients. All control subjects presented normal baseline IGF-I values and normal peak GH in response to GHRH-arg (range 33.6-144 ug/L).
All of the other pituitary hormones were within normal limits in the ALMS patients taking part in the GH secretion study, apart from 5 Cases (cases 2, 4, 8-9, and 22) with a subclinical hypothyroidism. All of them had normal FT3 and FT4 baseline values at the time of the GH investigation, with (case 8) or without (cases 2, 4, 9, and 22) L-thyroxine replacement therapy. Some male patients showed primary hypogonadism with mild LH/FSH elevation and low testosterone values (cases 2, 3, 5, and 6). There was no significant difference in GH secretion after GHRH-arg in patients 2 and 3 before and after six months of testosterone replacement therapy and normalization of FSH and LH (data not shown). Finally, cases 8 and 9 were hyperandrogenic.
Bone age, hypothalamic-pituitary MRI, histopathologic analysis of the pituitary gland
We investigated bone age by X-ray in 7 patients. In 4 cases (5, 6, 9, and 14 evaluated at 11, 7, 9, and 1 year, respectively), the bone age corresponded to chronologic age. In case 2, the bone age corresponded to chronologic age when investigated at 2 years but when tested at 12 years bone age was advanced by 2 years. Cases 16 and 22 also showed a 1-2 year advanced bone age. The MRIs of the diencephalic and pituitary regions were normal in 11 of 12 patients analysed (cases 2, 3, 5, 6, 8, 11, 13, 15, 16, 20, 21). Partial empty sella was observed in case 9. Pituitary specimens from 3 patients were examined microscopically (Figure 4). In 2 out of 3 patients, a marked and diffuse fibrosis was observed in the adenohypophysis, while the neurohypophysis was not affected. Immunohistochemical staining of GH, LH, FSH, ACTH, TSH and PRL secreting cells showed normal localization of proteins (data not shown).
Figure 4. Pituitary specimen.
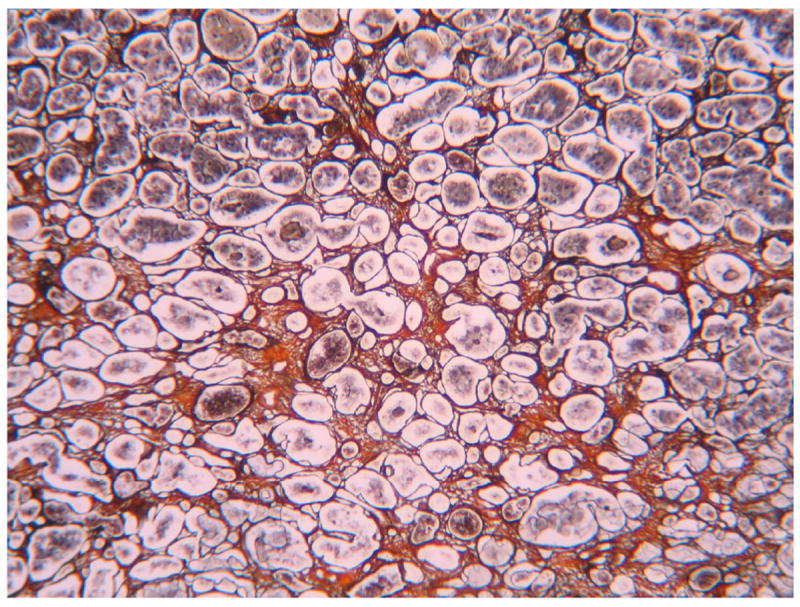
A marked and diffused fibrosis was observed in the adenohypophysis of two ALMS patients (Van Gieson staining).
DISCUSSION
We demonstrated by longitudinal auxological assessment that ALMS subjects presented, after a period of normal or somewhat increased growth, a reduced final height which is associated with an impaired GH reserve. These findings were not influenced by peripheral organ damage and are consistent with a GHD condition.
At present, auxological information on the GHD condition can be obtained only from clinical studies. In three Alms1 mouse models, the only report related to GHD showed an increase of nasal-anal length in female ALMS1 foz/foz mice compared to controls (23). In one cross sectional cohort, one third of patients had a stature below the 25th percentile (-0.67 SDS) (24). In another cross sectional study of a large series of patients, most children had a rapid growth above the 50th percentile (0 SDS) before puberty although the final height was below the 5th percentile (-1.64 SDS); furthermore, skeletal age was 1 to 3 years advanced in comparison to chronologic age in some children (2). In our longitudinal study, advanced bone age was confirmed in several young subjects, possibly explaining their reduced final height.
The relationship between low GH secretion and short stature in ALMS has been explored in very few reports. Alter & Moshang (13) described normal growth early in life and advanced bone age in two siblings with clinical features of ALMS. A low average height since infancy was seen in the male sibling and, at the age of 11 years, his height was in the 25th percentile (-0.67 SDS), whereas the height of his 10-year-old sister was below the 5th percentile (-1.64 SDS). They reported low GH concentrations after arginine and clonidine administration and low serum GH concentrations in the pooled 12-hour overnight samples. Zumsteg et al. (14) observed blunted GH concentrations following an arginine stimulation test in three affected siblings. These sibs had a normal growth velocity in early childhood, and a marked reduction in final height at the age of 15 years. Tai et al. (15) described a 15-year-old Taiwanese boy whose height was below the 3rd percentile (-1.88 SDS) and low GH secretion was documented by insulin-induced hypoglycemic stimulation and clonidine test. Recently, three subjects presenting with short stature described by Catrinoiu et al. (25) were diagnosed with severe GHD based on the results of the provocative tests with insulin (ITT). However, in most of these reports, a clinical diagnosis of ALMS was made without molecular genetic confirmation, and no control group was available. In addition, both false positive and negative results from these tests can occur because the somatotrope response to arginine or clonidine shows a great intra and inter-individual variability (19;26;27). Also, the evaluation of spontaneous GH secretion over 24 hours has no diagnostic value because of the overlap between normal and GHD subjects (28;29). Conversely, ITT is an unpleasant test for patients and complex to administer, requiring the achievement of adequate hypoglycemia under close medical supervision. Furthermore, the relevance of ITT in ALMS, which is characterized by diabetes mellitus and marked insulin resistance, is questionable.
In the present study, we assessed GH secretion, by evaluation of serum GH response following stimulation with the high potency secretagogue GHRH-arg. An impaired GH reserve was observed in 50% of patients (6/12) and a low-normal reserve in the remaining six. It is well established that GHRH-arg administration is safe and as reliable as ITT (17;18)., Within an appropriate clinical context, GHD in adults can be demonstrated biochemically by single provocative testing (26;28;30). We can speculate that elevated body weight, as well as elevated insulin levels which are a frequently reported in ALMS (2-4;31), support the normal or accelerated growth in the first years of life. Subsequently, this mechanism cannot compensate for the failure of the pituitary to produce GH or other unknown genetic factors which could affect final height. In fact, growth failure appeared after 10 years of age, thus suggesting that auxological evaluation and detection of GH pituitary reserve should be done from early puberty.
In ALMS patients, we noticed other hormonal deficiencies, particularly of testosterone in males and of thyroid function. However, male hypogonadism does not explain the low GH reserve, since two ALMS males were tested before and after testosterone replacement therapy with no differences in GH response to GHRH-arg. Age was not a major determinant of impairment of GH reserve because we observed the presence of GHD both in older as well as in younger subjects. Severe GH secretion impairment does not seem to be the major determinant of heart failure in ALMS because patients with similar GH reserve had different cardiac phenotypes. In fact, two patients (cases 2 & 22) died as a result of severe cardiomyopathy with several events of biventricular cardiac failure in the absence of GHD, while other patients (cases 6 & 11) had a normal ejection fraction and left ventricular diameters.
Alterations of GH response and IGF-I plasma levels related to liver and kidney failure can be excluded because all of the ALMS patients in this cohort had a normal or only slightly altered liver and kidney function. It is well known that obesity might be a confounding factor because it is correlated with an impairment of GH response after several provocative tests (32). However, it should be noted that the two obese subjects (cases 5 & 9) taking part in the present GH secretion study had no alterations in GH response. Furthermore, low GH reserve was defined according to a BMI cut-off, and peak GH was not associated with weight or BMI values. No association between diabetes mellitus or hypertriglyceridemia and GHD were observed. Finally, GH bioactivity is unlikely to be affected since, in the majority of ALMS subjects, serum IGF-I levels were in the normal range.
No macroscopic alterations of hypothalamus and pituitary gland were evidenced by MRI investigation in 92 % of patients, with the exception of a partial empty sella (case 9). Empty sella has also been reported by Catrinoiu et al et al. (25) in a patient with ALMS and interfamilial presence of Bardet-Biedl phenotype. Interestingly, empty sella has also been described in Bardet-Biedl syndrome, another ciliopathy with some overlapping features with ALMS (33). We describe histopathological alterations in the pituitary gland consisting of marked and diffuse fibrosis of adenohypophysis in some subjects for the first time. It is well known that severe systemic fibrosis is a typical finding of ALMS (2) but its pathogenesis remains unclear. The localization of ALMS1 to centrosomes (7;9) suggests a role in regulation of cell cycle. Alterations of ciliary function has been found to be associated with proliferative disorders and fibrosis (34;35). Unfortunately, murine models for ALMS do not consistently reproduce this pathological feature and no data on pituitary function or pathology are available.
This study has some limitations that we want to underline: we collected a mix of retrospective and prospective measurements, GH reserve was investigated primarily in the adult population, and measurements were not complete across different age groups for every patient.
In conclusion, our study shows that 50% of non-obese ALMS patients have an inadequate GH reserve to GHRH-arg and may be functionally GH deficient. Therefore, the short stature reported in ALMS may be at least partially correlated to GH secretion impairment. Further prospective studies are needed, especially in the pediatric population, to identify ALMS patients with decreased GH secretion and to assess whether such patients might benefit from GH replacement in order to enhance their final height and potentially improve some clinical and metabolic complications.
Acknowledgments
We would like to thank the ALMS patients for their willing participation in this study. This work has been supported by Euro-WABB project (agreement number 2010 12 05 PM, GM, NS, VB, RV) and a grant from NIH HD036878 (JDM, JKN, GBC).
References
- 1.Marshall JD, Ludman MD, Shea SE, Salisbury S, Willi SM, LaRoche RG, Nishina PM. Genealogy, natural history, and phenotype features of Alström syndrome in a large Acadian kindred and three additional families. Am J Med Genet. 1997;73:150–161. doi: 10.1002/(sici)1096-8628(19971212)73:2<150::aid-ajmg9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 2.Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM. New Alström syndrome phenotypes based on the evaluation of 182 cases. Archives of Internal Medicine. 2005;165:675–683. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- 3.Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. Eur J Hum Genet. 2007;15:1193–1202. doi: 10.1038/sj.ejhg.5201933. [DOI] [PubMed] [Google Scholar]
- 4.Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225–35. doi: 10.2174/138920211795677912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, Nishina PM, Naggert JK. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74–78. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 6.Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly W, Taylor JF, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI. Mutations of ALMS1, a large gene with tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31:79–83. doi: 10.1038/ng874. [DOI] [PubMed] [Google Scholar]
- 7.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 8.Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, Naggert JK. Alms1-disrupted mice recapitulate human Alström syndrome. Hum Mol Genet. 2005;14:2323–2333. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, Wilson DI. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–1587. doi: 10.2337/diabetes.54.5.1581. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong NA, Glynne R. A role for Alström syndrome protein, Alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3(e8):0001–0012. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;22:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 12.Blacque OE, Leroux MR. Bardet-Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell Mol Life Sci. 2006;63:2145–2161. doi: 10.1007/s00018-006-6180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter CA, Moshang TJ. Growth hormone deficiency in two siblings with Alström syndrome. Am J Dis Child. 1993;147:97–99. doi: 10.1001/archpedi.1993.02160250099030. [DOI] [PubMed] [Google Scholar]
- 14.Zumsteg U, Muller PY, Miserez AR. Alstrom syndrome: confirmation of linkage to chromosome 2p 12-13 and phenotypic heterogeneity in three affected sibs. J Med Genet. 2000;37:E8. doi: 10.1136/jmg.37.7.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tai TS, Lin SY, Sheu WH. Metabolic effects of growth hormone therapy in an Alström syndrome patient. Horm Res. 2003;60:297–301. doi: 10.1159/000074248. [DOI] [PubMed] [Google Scholar]
- 16.Maffei P, Boschetti M, Marshall JD, Paisey RB, Beck S, Resmini E, Collin GB, Naggert JK, Milan G, Vettor R, Minuto F, Sicolo N, Barreca A. Characterization of the IGF system in 15 patients with Alström syndrome. Clin Endocrinol. 2007;66:269–275. doi: 10.1111/j.1365-2265.2007.02721.x. [DOI] [PubMed] [Google Scholar]
- 17.Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. 2007;157:695–700. doi: 10.1530/EJE-07-0631. [DOI] [PubMed] [Google Scholar]
- 18.Ghigo E, Bellone J, Aimaretti G, Bellone S, Loche S, Cappa M, Bartolotta E, Dammacco F, Camanni F. Reliability of provocative tests to assess growth hormone secretory status. Study in 472 normally growing children. J Clin Endocrinol Metab. 1996;81:3323–3327. doi: 10.1210/jcem.81.9.8784091. [DOI] [PubMed] [Google Scholar]
- 19.Hoeck HC, Vestergaard P, Jakobsen PE, Laurberg P. Test of growth hormone secretion in adults: poor reproducibility of the insulin tolerance test. Eur J Endocrinol. 1995;133(3):305–312. doi: 10.1530/eje.0.1330305. [DOI] [PubMed] [Google Scholar]
- 20.Jones SL, Trainer PJ, Perry L, Wass JA, Bessser GM, Grossman A. An audit of the insulin tolerance test in adult subjects in an acute investigation unit over one year. Clin Endocrinol. 1994;41(1):123–128. doi: 10.1111/j.1365-2265.1994.tb03793.x. [DOI] [PubMed] [Google Scholar]
- 21.Vestergaard P, Hoeck HC, Jakobsen PE, Laurberg P. Reproducibility of growth hormone and cortisol responses to the insulin tolerance test and the short ACTH test in normal adults. Horm Metab Res. 1997;29(3):106–110. doi: 10.1055/s-2007-979000. [DOI] [PubMed] [Google Scholar]
- 22.Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. 2. Stanford, CA: Stanford University Press; 1959. [Google Scholar]
- 23.Arsov T, Silva DG, O’Bryan MK, Sainsbury A, Lee NJ, Kennedy C, Manji SSM, Nelms K, Liu C, Vinuesa CG, de Kretser DM, Goodnow CC, Petrovsky N. Fat aussie. A new Alström syndrome mouse. Mol Endocrinol. 2006;20(7):1610–1622. doi: 10.1210/me.2005-0494. [DOI] [PubMed] [Google Scholar]
- 24.Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alstrom syndrome. Reoport of 22 cases and literature review. Ophtalmology. 1998;105:1274–1280. doi: 10.1016/S0161-6420(98)97033-6. [DOI] [PubMed] [Google Scholar]
- 25.Catrinoiu D, Mihai CM, Tuta L, Stoicescu R, Simpetru A. Rare case of Alstrom syndrome with empty sella and interfamilial presence of Bardet-Biedl phenotype. J Med Life. 2009;2(1):98–103. [PMC free article] [PubMed] [Google Scholar]
- 26.Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab. 2000;85:3990–3993. doi: 10.1210/jcem.85.11.6984. [DOI] [PubMed] [Google Scholar]
- 27.Shalet SM, Toogood A, Rahim A, Brennan BM. The diagnosis of growth hormone deficiency in children and adults. Endocr Rev. 1998;19:203–223. doi: 10.1210/edrv.19.2.0329. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman DM, O’Sullivan AJ, Baxter RC, Ho KK. Diagnosis of growth hormone deficiency in adults. Lancet. 1994;343:1064–1068. doi: 10.1016/s0140-6736(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 29.Reutens AT, Hoffman DM, Leung K, Ho KK. Evaluation and application of a highly sensitive assay for serum growth hormone (GH) in the study of adult GH deficiency. J Clin Endocrinol Metab. 1995;80:480–485. doi: 10.1210/jcem.80.2.7852508. [DOI] [PubMed] [Google Scholar]
- 30.Ghigo E, Aimaretti G, Gianotti L, Bellone J, Arvat E, Camanni F. New approach to the diagnosis of growth hormone deficiency in adults. Eur J Endocrinol. 1996;134:352–356. doi: 10.1530/eje.0.1340352. [DOI] [PubMed] [Google Scholar]
- 31.Bettini V, Maffei P, pagano C, Romano S, Milan G, Favaretto F, Marshall JD, Paisey R, Scolari F, Greggio NA, Tosetto I, Naggert JK, Sicolo N, Vettor R. The progression from obesity to type 2 diabetes in Alstrom syndrome. Pediatr Diabetes. 2012;13(1):59–67. doi: 10.1111/j.1399-5448.2011.00789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocr Rev. 1994;15:80–101. doi: 10.1210/edrv-15-1-80. [DOI] [PubMed] [Google Scholar]
- 33.Soliman AT, Rajab A, AlSalmi I, Asfour MG. Empty sella, impaired testosterone secretion, and defective hypothalamic-pituitary growth and gonadal axes in children with Bardet-Biedl syndrome. Metabolism. 1996;45(10):1230–1234. doi: 10.1016/s0026-0495(96)90240-1. [DOI] [PubMed] [Google Scholar]
- 34.Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137(1):32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zulato E, Favaretto F, Veronese C, Campanaro S, Marshall JD, Romano S, Cabrelle A, Collin GB, Zavan B, Belloni AS, Rampazzo E, Naggert JK, Abatangelo G, Sicolo N, Maffei P, Milan G, Vettor R. ALMS1-deficient fibroblasts over-express extra-cellular matrix components, display cell cycle delay and are resistant to apoptosis. PLoS One. 2011;6(4):e19081. doi: 10.1371/journal.pone.0019081. [DOI] [PMC free article] [PubMed] [Google Scholar]