

Abstract
Chronic obstructive pulmonary disease (COPD) is a leading cause of mortality worldwide. COPD exacerbation, or episodic worsening of symptoms, often results in hospitalization and increased mortality rates. Airway infections by new bacterial strains, such as nontypeable Haemophilus influenzae (NTHi), are a major cause of COPD exacerbation. NTHi express lipooligosaccharides that contain sialic acids, and may interact with Siglec-14, a sialic acid recognition protein on myeloid cells that serves as an activating signal transduction receptor. A null allele polymorphism in SIGLEC14 may attenuate the inflammatory responses to NTHi by eliminating Siglec-14 expression. We asked if the loss of Siglec-14 attenuates the inflammatory response by myeloid cells against NTHi, and if the SIGLEC14-null polymorphism has any effect on COPD exacerbation. We found that NTHi interacts with Siglec-14 to enhance proinflammatory cytokine production in a tissue culture model. Inhibitors of the Syk tyrosine kinase suppress this response. Loss of Siglec-14, due to SIGLEC14-null allele homozygosity, is associated with a reduced risk of COPD exacerbation in a Japanese patient population. Taken together, Siglec-14 and its downstream signaling pathway facilitate the “infection–inflammation–exacerbation” axis of COPD disease progression, and may represent promising targets for therapeutic intervention.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1311-7) contains supplementary material, which is available to authorized users.
Keywords: Siglec, Sialic acids, Haemophilus influenzae, Chronic obstructive pulmonary disease, Exacerbation
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by airflow limitation that is not fully reversible [1], and is projected to be the third leading cause of mortality in the world by 2030 [2]. Stable COPD is managed by a combination of pharmaceutical treatments, smoking cessation, and pulmonary rehabilitation. However, COPD exacerbation often results in hospitalization and increased patient mortality, and therefore account for a major proportion of the clinical and socioeconomic burden of the disease [3–5]. Acquisition of new bacterial strains that colonize airways often trigger COPD exacerbation, which in turn leads to disease progression [6, 7]. While vaccination can reduce the risk of certain infectious triggers, treatment options for exacerbations are limited, and thus new therapies would be of great value [8].
Studies of genetic polymorphisms and disease susceptibility often yield clues to new treatment approaches [9, 10]. While there are many reports correlating genetic polymorphisms and COPD susceptibility [11–15], those examining exacerbations are more limited [16–20]. A recent study has shown that a subgroup of patients appears to be particularly susceptible to exacerbation, and that this frequent-exacerbation phenotype is relatively stable [21], suggesting the presence of contributing genetic factor(s). Given that bacterial airway infections play a major role in COPD exacerbation [6, 7], one potentially fruitful avenue is to explore correlations between COPD exacerbations and genetic polymorphisms of proteins involved in bacterial recognition.
Nontypeable Haemophilus influenzae (NTHi), a Gram-negative bacterial pathogen considered a major cause of COPD exacerbation [6], expresses lipooligosaccharides that contain sialic acids [22, 23]. Sialic acids are acidic sugars commonly found on terminal positions of vertebrate glycans, but only occasionally in pathogenic bacteria [24]. Siglecs are a family of sialic acid-binding lectins expressed mainly on innate immune cells [25, 26]. Siglec-14, a family member with an activating signaling property, is expressed on granulocytes and monocytes, while Siglec-5, an inhibitory counterpart with extensive sequence identity to Siglec-14 (over 250 amino acids at the N-terminus), is found on granulocytes and B-lymphocytes [27, 28]. In humans, the ancestral (wildtype) SIGLEC gene cluster has both SIGLEC14 and SIGLEC5 genes in tandem, while a derived (SIGLEC14-null) allele harbors a single SIGLEC14/5 fusion gene. The product of this fusion gene is identical to that of the wildtype SIGLEC5 gene at the amino acid level [28]. We speculated that loss of Siglec-14 could result in attenuated immune responses against pathogenic bacteria that express sialic acids [28]. However, this possibility has not been formally tested.
In this work, we asked if Siglec-14 expression would alter the inflammatory response of myeloid cells elicited by NTHi, and if the SIGLEC14-null polymorphism is associated with altered risk of COPD exacerbation. We demonstrated that NTHi interacts with Siglec-14 to enhance production of proinflammatory cytokines in a tissue culture model, and that the loss of Siglec-14, due to SIGLEC14-null allele homozygosity, is associated with reduced risk of COPD exacerbation. Based on these findings, we propose that Siglec-14 and its downstream signaling pathway may contain promising targets for therapeutic intervention.
Materials and methods
Study subjects
Healthy volunteers were recruited at the University of California, San Diego, for Siglec expression analysis. Protocols for sample collection, genotyping, and related analyses were approved by the Institutional Review Board (IRB) at the University. Written informed consent was obtained from each donor.
For the analysis of correlations between SIGLEC14 genotype and clinical phenotypes, outpatients who visited the Respiratory Care Clinic, Nippon Medical School, and had a diagnosis of COPD according to the global initiative for chronic obstructive lung disease (GOLD) criteria [1] were invited to participate in the study. The patients were current or ex-smokers with current symptoms compatible with COPD (e.g., dyspnea during exertion, prolonged coughing, or sputum) and air-flow limitation assessed by a pulmonary function test (post-bronchodilator forced expiratory volume in 1 s/forced vital capacity, FEV1/FVC, <70 %). Protocols for sample collection, genotyping, and related analyses were approved by the IRB at Nippon Medical School. Written informed consent was obtained from each donor.
Genotyping
Genotyping of the SIGLEC14 locus was performed as described previously [28]. In brief, a set of three PCR reactions, specifically amplifying (1) SIGLEC14, (2) SIGLEC5, and (3) SIGLEC14/5 fusion genes, were run in parallel using genomic DNA from each donor as template. Primer sequences and PCR conditions are provided in the Electronic supplementary material. The genotype of each donor was determined based on the patterns of amplification: genomic DNA from homozygous wildtype individuals yields products in reactions 1 and 2 but not in reaction 3 (+/+/−); that from heterozygous individuals yields products in all three reactions (+/+/+); and that from homozygous null individuals yields product only in reaction (3) (−/−/+).
Siglec expression analysis
Peripheral blood leukocytes were collected from healthy volunteers at the University of California, San Diego. Leukocytes were prepared by erythrocyte lysis using ACK lysing buffer, and stained with allophycocyanin-labeled anti-Siglec-5/14 antibody (FAB10721A) or control antibody (IC002A; both R&D Systems, Minneapolis, MN) and AlexaFluor488-labeled anti-CD19 antibody (557697; BD Biosciences Pharmingen, San Diego, CA), and analyzed with a flow cytometer (FACSCalibur; BD Biosciences Immunocytometry Systems, San Jose, CA).
NTHi binding to Siglecs
NTHi (strain 2019) cultured in minimal medium not supplemented or supplemented with N-acetylneuraminic acid (0.5 mM) was prepared as described previously [29, 30], heat-killed, and stored frozen until use. The bacteria (1.5 × 109 CFU) were labeled with fluorescein isothiocyanate (50 μg/ml in Dulbecco’s phosphate-buffered saline, D-PBS) for 2 h at room temperature, washed, and suspended in 3 ml of assay buffer (1 % bovine serum albumin, BSA, in D-PBS). Recombinant Siglec-Fc fusion proteins were prepared as described previously [27]. A 96-well plate was coated overnight with protein A (0.5 μg/well in 50 mM sodium bicarbonate buffer, pH 9.5) at 4 °C. Subsequent incubations were carried out at room temperature. The protein A-coated plate was blocked with assay buffer, incubated with Siglec-Fc (0.5 μg/well in assay buffer) for 2 h, washed, and incubated with 100 μl (5 × 107 CFU) per well of labeled NTHi for 2 h. The wells were washed, and fluorescence was measured with a fluorescent microplate reader (excitation 490 nm, emission 530 nm; MTP-650FA, Corona Electrics, Hitachinaka, Japan).
Assays for NTHi-induced proinflammatory cytokine responses
The THP-1 sublines expressing Siglec-5 or Siglec-14 were prepared as described previously [27]. The subline expressing both Siglec-5 and Siglec-14 was prepared by retroviral transduction of a Siglec-14+ subline with Siglec-5/pMSCVpuro packaged using the PLAT-A cell line [31]. The THP-1 sublines (1 × 106/ml, 0.5 ml/well of 24-well plate) in Macrophage-SFM (Life Technologies, Carlsbad, CA) were mixed with 5 × 106 CFU of heat-killed NTHi (multiplicity of infection 10) and incubated for 24 h. Commercial enzyme-linked immunosorbent assay (ELISA) kits were used to quantify TNF-α (human TNF-alpha Quantikine HS ELISA kit; R&D Systems, Minneapolis, MN) and IL-8 (BD OptEIA human IL-8 ELISA set; BD Biosciences Pharmingen, San Diego, CA) in the culture supernatant.
In experiments investigating Syk inhibitors, the cells were preincubated with the inhibitors R406 (Selleck Chemicals, Houston, TX) or BAY61-3606 (Merck KGaA, Darmstadt, Germany) at various concentrations for 2 h before adding NTHi.
COPD patient populations
Outpatients who visited the Respiratory Care Clinic in November 2008 and had a diagnosis of COPD according to the GOLD criteria [1] were invited to participate in this longitudinal study. All subjects with obvious other pulmonary comorbidities were excluded. Written informed consent was obtained from each patient. Enrolled patients were carefully interviewed about the frequency of exacerbations every month for 1 year. We defined exacerbation in the following two ways: (1) using the classic definition of Anthonisen et al. [32], to determine the frequency of relatively severe exacerbations; and (2) using the new definition of Rodriguez-Roisin [33], to determine the frequency of mild and severe exacerbations.
Paired serum samples were collected from 21 COPD patients (age 71.0 ± 8.2 years; 19 men and 2 women; including six current smokers) seen both during a stable state and during exacerbations. A stable state was 8 weeks free from an exacerbation by the criteria of Anthonisen et al. [32]. Exacerbation sampling was only performed in patients who had not received prior oral corticosteroids and/or antibiotics.
Pulmonary function parameters
Post-bronchodilator FEV1, carbon monoxide-diffusing capacity (diffusing capacity divided by alveolar volume), vital capacity (VC), and FVC were measured according to the American Thoracic Society guidelines [34] using equipment for lung function testing (CHESTAC; CHEST Co., Tokyo, Japan). We used the reference values for post-bronchodilator FEV1 and VC specified by the Japanese Respiratory Society [35].
High-resolution computed tomography parameters
The percentage low attenuation area (LAA %) reflects the severity of emphysema. We performed helical high-resolution computed tomography scans at 1.25 mm collimation, 0.8 s scan time (rotation time), 120 kV and 100–600 mA with a LightSpeed Pro16 CT scanner (GE Company, Tokyo, Japan). LAA % was calculated as described previously [36–38].
Enzyme-linked immunosorbent assay for soluble Siglec-14
A mouse monoclonal antibody against human Siglec-14 (clone 40-1) was raised as described previously [28], and was provided by the National Institute of Advanced Industrial Science and Technology (Tsukuba, Ibaraki, Japan). Antibody (1 mg) was labeled with biotin using a commercial kit (biotin protein labeling kit; Roche Applied Science, Mannheim, Germany). All of the following procedures were carried out at room temperature. A 96-well plate was coated overnight with anti-Siglec-5/14 antibody (clone 194128; R&D Systems, Minneapolis, MN; 1 μg/ml in D-PBS, 100 μl/well), washed three times with 400 μl wash buffer (0.05 % Tween 20 in D-PBS) per well, blocked with 300 μl/well of assay buffer (1 % BSA in D-PBS) for 1 h, and washed three times. Serum samples (diluted 20times with assay buffer) were added to the prepared wells and incubated for 2 h, and removed by aspiration. Wells were washed three times, incubated with biotinylated antibody against Siglec-14 for 2 h (0.5 μg/ml in assay buffer, 100 μl/well), washed, further incubated with streptavidin-conjugated horseradish peroxidase for 20 min (RPN1231V; GE Healthcare, Piscataway, NJ; diluted 1:1,000 in assay buffer, 100 μl/well), and washed. Finally, peroxidase substrate (SureBlue Reserve TMB 1-Component Microwell Peroxidase Substrate; Kirkegaard and Perry Laboratories, Gaithersburg, MD; 0.1 ml/well) was added to the wells and incubated for 15 min, and color development was stopped by adding 0.1 ml/well 1 N HCl. Absorbance at 450 nm was measured using a Model 680 microplate reader (Bio-Rad, Hercules, CA).
Statistical analyses
Statistical analyses of in vitro assays were performed using GraphPad Prism 4 (GraphPad Software, La Jolla, CA). Values were compared between two groups using an unpaired t test (or by a paired t test for paired samples), and those among three or more groups were compared by analysis of variance (ANOVA) followed by appropriate post-tests (as specified in the figure legends). Statistical analyses on patients were performed using JMP Genomics software version 5.1 (SAS Institute, Cary, NC). The effect of the SIGLEC14 genotype on the incidence of bacterial exacerbation was analyzed by logistic regression, and that on the frequency of all exacerbations was analyzed by a Poisson regression with additive models on the wildtype (SIGLEC14-positive) allele. The generalized linear models that maximized the likelihood, and thus fit best, were considered appropriate. Age and gender were used as covariates, and smoking status, usage of inhaled corticosteroid, and post-bronchodilator FEV1 % predicted were also used as covariates when appropriate. Since separation can occur with relatively small datasets with many covariates, Firth’s bias reduction was applied to the regression [39].
Results
SIGLEC14 genotype determines Siglec-5 and Siglec-14 expression on monocytes
We have previously reported that while Siglec-14 (activating-type signal transduction molecule) is expressed on granulocytes and monocytes, Siglec-5 (inhibitory counterpart) is found on granulocytes and B-lymphocytes [28] (Electronic supplementary material, Fig. 1a). In humans, the ancestral (wildtype) SIGLEC gene cluster has both SIGLEC14 and SIGLEC5 genes in tandem, while a derived (SIGLEC14-null) allele harbors a single SIGLEC14/5 fusion gene, whose product is identical in amino acid sequence to that of wildtype SIGLEC5 [28] (Electronic supplementary material, Fig. 1b). Since the 5′-upstream region of the SIGLEC14/5 fusion gene is derived from SIGLEC14, we speculated that expression of the fusion gene product might parallel that of Siglec-14. Indeed, by analyzing Siglec-14-null individuals who are homozygous for the SIGLEC14/5 fusion gene (i.e., homozygous SIGLEC14-null), we found that the fusion gene product is expressed not only on granulocytes and B-lymphocytes (typical for wildtype Siglec-5), but also on monocytes (typical for wildtype Siglec-14; Fig. 1). Thus, monocytes express different Siglec proteins depending on the genotype: homozygous wildtype, Siglec-14 only; heterozygous, Siglec-14 and Siglec-5; homozygous SIGLEC14-null, the Siglec-5-like fusion protein only (Electronic supplementary material, Fig. 1c).
Fig. 1.

Expression of SIGLEC14/5 fusion gene product. Peripheral blood leukocytes from homozygous SIGLEC14-null individuals were analyzed by flow cytometry. Gray shaded areas cells stained with a negative control antibody (IC002A, R&D Systems). Solid lines cells stained with antibody recognizing both Siglec-5 and Siglec-14 (FAB10721A, R&D Systems). SIGLEC14/5 fusion gene product, which is identical to Siglec-5 in amino acid sequence, is expressed on granulocytes, monocytes, and B lymphocytes
NTHi binds to Siglec-14 in both a sialic acid-dependent manner and a sialic acid-independent manner
Some Siglec family members (e.g., Siglec-9) interact with pathogenic bacteria that express sialic acids [40–42]. To assess if Siglecs on myeloid cells interact with NTHi, a major cause of COPD exacerbation [6], we evaluated binding of fluorescein-labeled NTHi to recombinant Siglec. NTHi bound to Siglec-14 and Siglec-5 in both a sialic acid-dependent and a sialic acid-independent manner, but exhibited only sialic acid-dependent binding to Siglec-9 (Fig. 2a). No appreciable binding of NTHi to Siglec-3 or TREM1 (used as a negative control) was observed. The presence of a sialic acid-independent interaction between NTHi and Siglec-14 was confirmed by the binding of NTHi to the Arg119 → Ala mutant of Siglec-14 (Siglec-14 R119A), which cannot recognize sialic acids [28] (Fig. 2b).
Fig. 2.
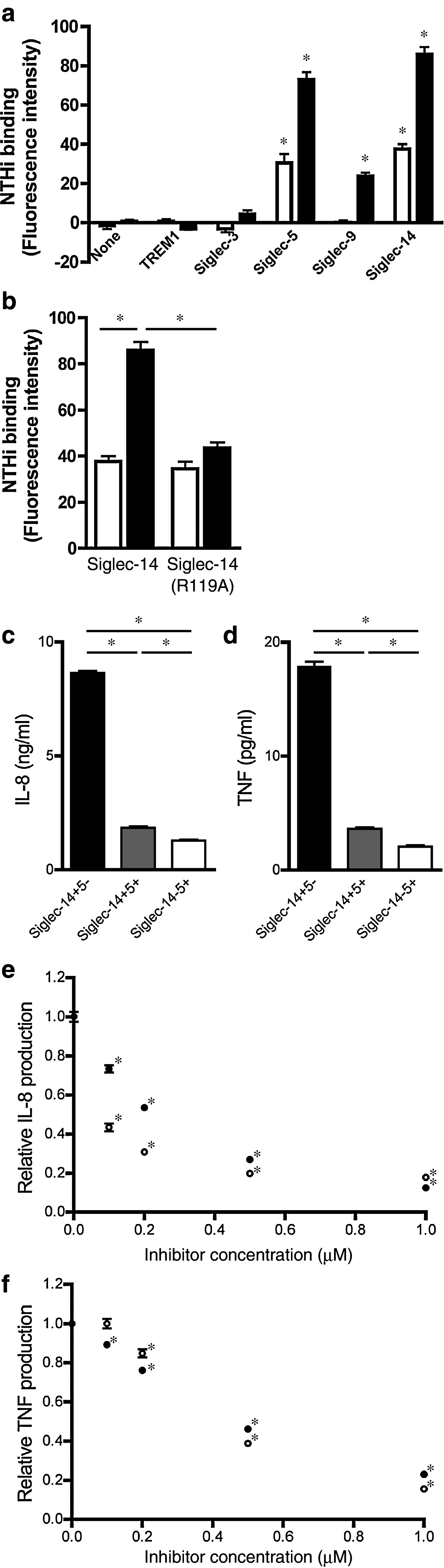
Influence of Siglec-14 and Siglec-5 expression on inflammatory responses. a Binding of NTHi to human myeloid Siglecs. Both sialic acid-positive (solid bars) and sialic acid-negative (open bars) NTHi bound to Siglec-5 and Siglec-14, whereas only sialic acid-positive NTHi bound to Siglec-9. Background fluorescence (autofluorescence of assay buffer) was subtracted. *p < 0.05 by one-way ANOVA followed by Dunnett’s multiple comparison test, vs. “none” within each group. b Binding of NTHi to Siglec-14 wildtype and R119A mutant. Both sialic acid-positive NTHi (solid bars) and sialic acid-negative NTHi (open bars) showed binding to both wildtype Siglec-14 and the R119A mutant. Background fluorescence (autofluorescence of assay buffer) was subtracted. *p < 0.05 by two-way ANOVA followed by Bonferroni multiple comparison test (four pairwise comparisons: Siglec-14 wildtype, sialic acid-negative vs. sialic acid-positive NTHi; Siglec-14 R119A, sialic acid-negative vs. sialic acid-positive NTHi; sialic acid-negative NTHi, Siglec-14 wildtype vs. R119A; sialic acid-positive NTHi, Siglec-14 wildtype vs. R119A). a, b Measurements performed in triplicate wells and repeated at least three times independently. Consistent results were obtained, and representative data are shown. Values shown are means ± standard error of the mean (SEM). c, d IL-8 production (c) and TNF-α production (d) from THP-1 sublines stimulated with NTHi (solid bars THP-1 cells mimicking homozygous wildtype monocytes, gray bars THP-1 cells mimicking heterozygous monocytes, open bars THP-1 cells mimicking homozygous SIGLEC14-null monocytes). *p < 0.05 by one-way ANOVA followed by Bonferroni’s multiple comparison test (three pairwise comparisons). e, f IL-8 production (e) and TNF-α production (f) from Siglec-14+5− THP-1 subline stimulated with NTHi in the presence of R406 (open circles) or BAY61-3606 (closed circles). *p < 0.05 by one-way ANOVA followed by Dunnett’s multiple comparison test (vs. vehicle control = “0 μM”, within each treatment group). c–f Measurements were performed in quadruplicate wells and repeated four times independently with consistent results, and representative data are shown. All values shown are means ± SEM
Siglec-14 enhances proinflammatory cytokine responses elicited by NTHi
Since monocytes and macrophages are thought to play pivotal roles in COPD exacerbation [43, 44], we next evaluated the effect of Siglec-14 and Siglec-5 expression on monocytes, using as a model the THP-1 human monoblastoid cell line that expresses very low levels of endogenous Siglec-14 and Siglec-5. We introduced cDNAs for Siglec-14 and/or Siglec-5 and selected separate Siglec-14+5−, Siglec-14+5+ and Siglec-14−5+ THP-1 sublines, mimicking monocytes from homozygous wildtype, heterozygous and homozygous SIGLEC14-null individuals, respectively (Electronic supplementary material, Fig. 1c). Exposure to NTHi in Siglec-14+5− THP-1 cells (mimicking homozygous wildtype monocytes) elicited strong release of tumor necrosis factor-α (TNF-α) and interleukin-8 (IL-8), two cytokines with an established connection to COPD exacerbation [45] (Fig. 2c, d). Progressively weaker TNF-α and IL-8 responses were seen in Siglec-14+5+ THP-1 cells (mimicking heterozygous monocytes) and Siglec-14−5+ THP-1 cells (mimicking homozygous SIGLEC14-null monocytes), indicating that the relative amounts of Siglec-14 and Siglec-5 influence the magnitude of proinflammatory responses.
Syk inhibitor attenuates proinflammatory cytokine responses elicited by NTHi
Since Siglec-14 associates with adapter protein DAP12 [27], downstream signaling is predicted to proceed through recruitment of the protein tyrosine kinase Syk. Consistent with this prediction, incubation of Siglec-14+5− THP-1 cells (mimicking homozygous wildtype monocytes) with R406 or BAY61-3606 (Syk-specific inhibitor) indeed blocked cytokine responses to NTHi in a dose-dependent manner (Fig. 2e, f).
SIGLEC14-null allele is associated with reduced risk of COPD exacerbation
Based on these data, we hypothesized that the SIGLEC14 genotype may influence the occurrence of COPD exacerbations secondary to NTHi infection. To evaluate this hypothesis, we genotyped 135 Japanese COPD patients with a 1-year follow-up record for exacerbations (Table 1). We defined exacerbation in the following two ways: (1) using the classic definition of Anthonisen et al. [32], to determine the incidence of exacerbations putatively caused by bacterial infection; and (2) using the new definition of Rodriguez-Roisin [33], to determine the frequency of all exacerbations. We found that SIGLEC14 gene dosage was correlated positively with the incidence of bacterial exacerbations and the frequency of all exacerbations (p = 0.0015 and 0.0002, respectively; Fig. 3a, b). Since smoking cessation, milder airflow limitation (post-bronchodilator FEV1 % predicted), and medication (inhaled corticosteroid) have each been reported to reduce the rate of COPD exacerbations [21, 46, 47], we repeated the same analyses with adjustment for each factor. Once again, we identified a significant association between SIGLEC14 gene dosage and the incidence of bacterial exacerbations and the frequency of all exacerbations (p = 0.0034 and 0.0044, respectively).
Table 1.
Basic characteristics and SIGLEC14 genotypes of subjects
| SIGLEC14 genotype | |||
|---|---|---|---|
| Wildtype (n = 33) | Heterozygote (n = 71) | Null (n = 31) | |
| Age | 71.8 ± 8.2 | 72.3 ± 7.5 | 73.7 ± 8.8 |
| Gender (n) | |||
| Male | 30 | 69 | 29 |
| Female | 3 | 2 | 2 |
| Smoking status at the time of enrollment | |||
| Ex (n) | 31 | 70 | 29 |
| Current (n) | 2 | 1 | 2 |
| Pack-years | 79.9 ± 60.7 | 74.7 ± 44.8 | 68.0 ± 37.2 |
| Pulmonary function tests | |||
| VC (l) | 3.11 ± 0.91 | 3.32 ± 0.91 | 3.10 ± 0.88 |
| % VC (%) | 90.8 ± 23.0 | 96.1 ± 19.5 | 91.2 ± 18.5 |
| FEV1 (l) | 1.49 ± 0.68 | 1.56 ± 0.67 | 1.58 ± 0.54 |
| FEV1% (%) | 47.3 ± 11.7 | 47.5 ± 12.4 | 52.9 ± 9.3 |
| FEV1 % predicted (%) | 55.2 ± 21.5 | 57.8 ± 21.6 | 60.3 ± 16.6 |
| RV/TLC (%) | 51.2 ± 10.9 | 47.9 ± 10.6 | 47.9 ± 8.1 |
| % DLCO/VA (%) | 58.3 ± 27.8 | 56.6 ± 20.3 | 68.0 ± 23.2 |
| Computed tomography | |||
| Low attenuation area (%) | 32.8 ± 13.1 | 35.1 ± 15.0 | 30.1 ± 11.2 |
| Therapy (%) | |||
| Long-acting beta agonist | 88.0 | 84.6 | 77.8 |
| Long-acting muscarinic antagonist | 84.0 | 92.3 | 81.5 |
| Inhaled corticosteroid | 68.0 | 73.9 | 69.2 |
| Systemic corticosteroid | 0 | 0 | 0 |
The values shown are means ± standard deviation. There were no significant differences between the groups regarding any of the parameters shown.VC vital capacity; %VC actual/predicted VC ratio, in percentage; FEV1% FEV1/FVC ratio, in percentage; FEV1 % predicted actual/predicted FEV1 ratio, in percentage; RV/TLC residual volume/total lung capacity; %DLCO/VA diffusing capacity for carbon monoxide/alveolar volume, in percentage
Fig. 3.
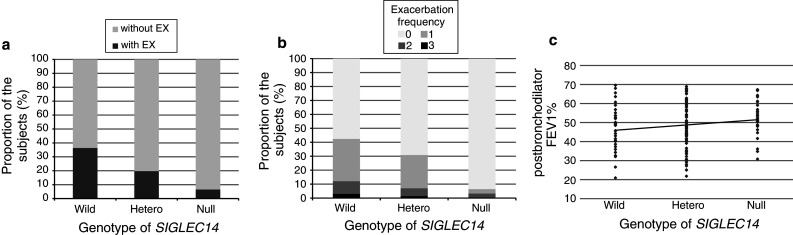
Effect of SIGLEC14 genotype on the frequency of COPD exacerbations. a SIGLEC14 genotype and the incidence of bacterial exacerbations using the definition of Anthonisen et al. [32] (without EX patients without bacterial exacerbations, with EX patients with one or more incidences). b SIGLEC14 genotype and the frequency of all exacerbations using the definition of Rodriguez-Roisin. Values are means ± standard deviation. c SIGLEC14 genotype and severity of airflow obstruction (post-bronchodilator FEV1 %). Each point represents an individual patient (Wild homozygous wildtype, Hetero heterozygous, Null homozygous SIGLEC14-null)
Possible correlation between SIGLEC14 genotype and airflow limitation
We also found a negative correlation between the number of wildtype (SIGLEC14-positive) alleles and post-bronchodilator FEV1 % (p = 0.0249; Fig. 3c). This correlation was significant (p = 0.0322) even after correcting for age, gender, and smoking history (pack-years) as covariates. These findings imply that more frequent exacerbations in homozygous wildtype and heterozygous individuals would lead to more severe airflow limitation. Taken together, these findings support the hypothesis that infection-induced exacerbation leads to faster reduction in lung function in individuals expressing Siglec-14.
Serum concentration of soluble Siglec-14 is associated with SIGLEC14 genotype and susceptibility to COPD exacerbation
We next examined if soluble circulating Siglec-14 can be used as a marker for COPD exacerbations. To this end, we developed a sandwich ELISA that detects Siglec-14 but not Siglec-5 (Fig. 4a), and applied this assay for quantitative measurement of soluble Siglec-14 in matched serum samples of COPD patients before and during exacerbations. The amount of serum Siglec-14 was correlated with the number (n = 0, 1 or 2) of wildtype alleles (p < 0.0001; Fig. 4b). However, we did not observe consistent changes in the concentration of serum Siglec-14 between serum samples collected before and during exacerbations (p = 0.2 by paired t test, and p = 0.0784 by sign test; Fig. 4c). These results suggest that soluble Siglec-14 is present in the serum of homozygous wildtype and heterozygous individuals and its concentration reflects the number of wildtype alleles in these individuals. However, in individual patients, the abundance of soluble Siglec-14 does not change in a strong or consistent fashion with the presence/absence of ongoing exacerbations. Nevertheless, we found that the overall serum concentration of soluble Siglec-14 was positively correlated with exacerbation frequency using the definition of Rodriguez-Roisin (p < 0.0001; Fig. 4d). This result reflects the fact that the SIGLEC14 genotype is positively correlated with both Siglec-14 serum concentration and COPD exacerbation frequency.
Fig. 4.
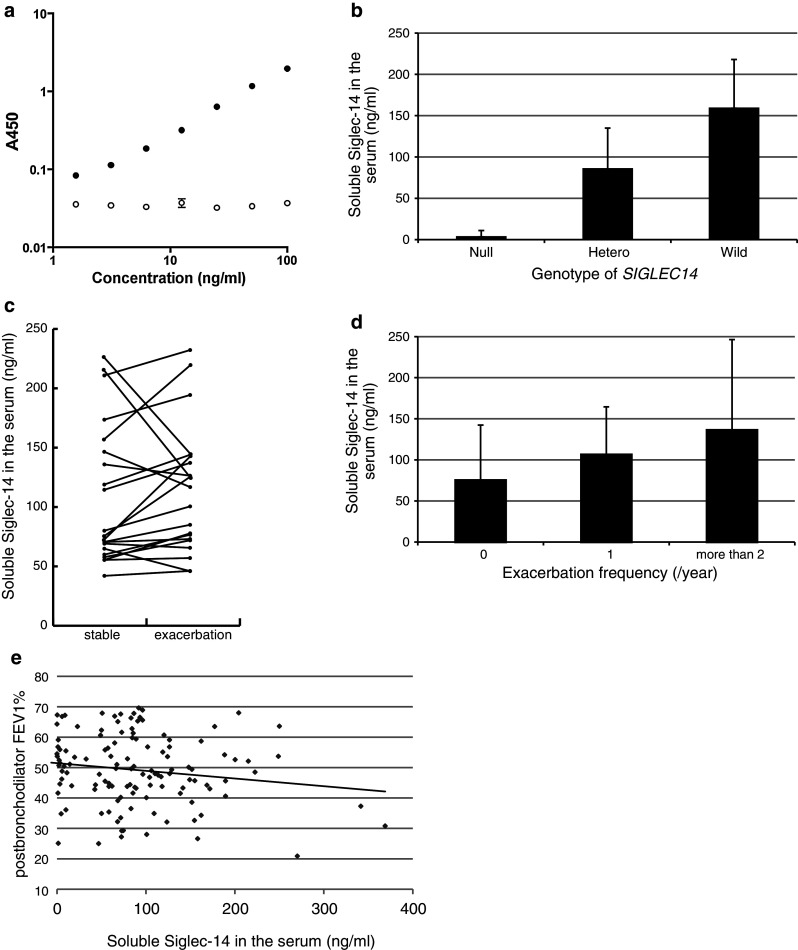
Correlation between SIGLEC14 genotype, serum Siglec-14, and COPD exacerbations. a Siglec-14-Fc (solid circles) and Siglec-5-Fc (open circles), serially diluted from 100 to 1.5625 ng/ml, were analyzed using a sandwich ELISA specific for Siglec-14 as described in Materials and Methods. Values are means ± SEM. b Correlations between serum Siglec-14 and SIGLEC14 genotype. Siglec-14 serum concentrations on average reflect the number of wildtype (SIGLEC14-positive) alleles. c The Siglec-14 serum concentrations in COPD patient sera before and during exacerbation. The lines connect Siglec-14 concentrations in samples collected before and during exacerbations (stable and exacerbation, respectively) from the same individuals (n = 21; homozygous wildtype and heterozygous patients only). d Correlations between Siglec-14 serum concentrations and frequency of exacerbations (using the definition of Rodriguez-Roisin). e Correlation between Siglec-14 serum concentrations and post-bronchodilator FEV1 %
Thus, measurement of Siglec-14 serum concentration may be used as a surrogate for SIGLEC14 genotyping to evaluate susceptibility to COPD exacerbations. Moreover, there was a positive correlation between Siglec-14 serum concentrations and exacerbation frequency (using the definition of Rodriguez-Roisin) among homozygous wildtype and heterozygous individuals (analyzed by Poisson regression using a generalized linear model), even after SIGLEC14 gene dosage (n = 1 or 2) was included as a covariate (p = 0.0002). In addition, there was a negative correlation between serum Siglec-14 concentrations and post-bronchodilator FEV1 % (p = 0.0402; Fig. 4e). These results suggest that Siglec-14 serum concentration may be a useful predictor of COPD exacerbation susceptibility and consequential decline in pulmonary function.
Discussion
We demonstrated that Siglec-14 interacts with NTHi and enhances proinflammatory responses of immune cells in a tissue culture model, and that COPD patients with wildtype (SIGLEC14-positive) alleles suffer more frequent exacerbations than homozygous SIGLEC14-null patients. A possible mechanism behind this genotype–phenotype correlation can be summarized as follows (Fig. 5): myeloid cells of COPD patients expressing Siglec-14 respond to airway infection by NTHi more strongly and produce larger amounts of proinflammatory cytokines, which recruit inflammatory cells, eventually leading to COPD exacerbation. An additional factor may be the expression of SIGLEC14/5 fusion gene product (identical to Siglec-5 in amino acid sequence) on monocytes of SIGLEC14-null allele carriers, attenuating a proinflammatory response. The expression of sialic acids by NTHi likely represents an evolutionary process with the advantage of mimicking sialic acid-based “self-associated molecular patterns” (SAMPs) [48], which are normally recognized by inhibitory Siglecs such as Siglec-5. The cross-recognition by Siglec-14 (activating counterpart) probably represents an evolutionary response of the host to this evolutionary development in the bacteria. However, in the setting of a chronic disease such as COPD, such a host response may also generate unwanted inflammation, to the detriment of the host.
Fig. 5.
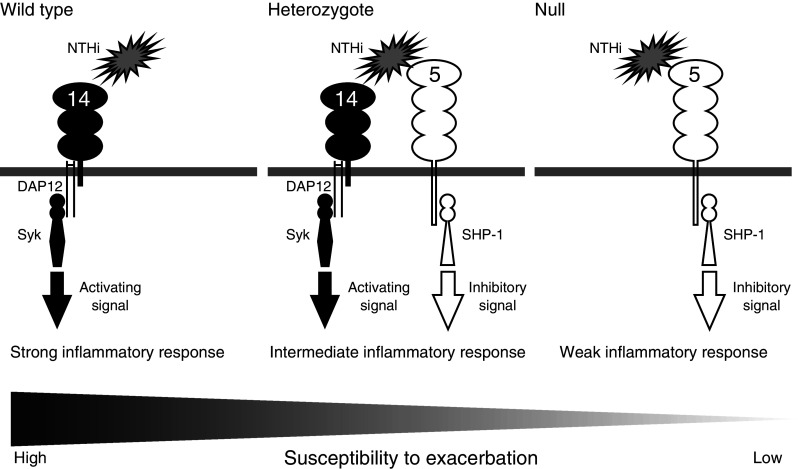
Possible mechanism behind the correlation between SIGLEC14 genotype and COPD exacerbation-prone phenotype. The wildtype (SIGLEC14-positive) allele confers expression of Siglec-14 on monocytes, while the SIGLEC14-null (SIGLEC14/5 fusion) allele confers expression of Siglec-5. Monocytes of homozygous wildtype individuals express Siglec-14 only, those of heterozygous individuals both Siglec-14 and Siglec-5, and those of homozygous SIGLEC14-null individuals Siglec-5 only. Siglec-14 associates with adapter protein DAP12 and the tyrosine kinase Syk to transduce cell-activating signal upon engagement. Siglec-5, on the other hand, associates with tyrosine phosphatase SHP-1 to transduce a cell-inhibitory signal. The myeloid cells of those who carry the wildtype allele respond strongly to infectious cues, while those of homozygous SIGLEC14-null individuals respond weakly. Excessive proinflammatory responses in COPD patients lead to exacerbation
COPD is a multifactorial disease with a complex etiology that is not fully understood. While tobacco smoking plays a fundamental role in COPD pathogenesis in the developed world, confounding factors, some genetic (e.g., deficiency of α1 antitrypsin [11]) and some nongenetic (e.g., latent adenoviral infection [49]), are also involved. While many studies on host genetic factors influencing COPD pathogenesis have been conducted, those focusing on COPD exacerbations are relatively scarce. Before our study, genetic polymorphisms in four human genes (i.e., SNPs in MBL2, SFTPB, CCL1 and HHIP) were associated with the risk of COPD exacerbations [16–20]. Our study adds a new entry, the first non-SNP-type polymorphism, to this very short list. It is the second mammalian lectin (the other being mannose-binding lectin encoded by the MBL2 gene) found to be associated with the risk of COPD exacerbations, pointing to the importance of glycan recognition in the interactions between infectious agents and the immune system.
Excessive inflammatory responses to infectious agents are a putative cause of the sharp decline in pulmonary function and increased mortality during and after COPD exacerbations. At present, corticosteroids are widely used to control the inflammatory response during episodes of COPD exacerbation, but severe side effects limit their utility in long-term therapy. Our findings suggest that the use of Syk inhibitors may be beneficial in mitigating the infection-induced inflammatory response. Although one of the compounds we used (BAY61-3606) is not in clinical development, the other Syk inhibitor (R406; Rigel/AstraZeneca) has entered clinical trials as a candidate drug to treat rheumatoid arthritis [50]. Treatment with a Syk inhibitor might also be relevant in the treatment of nonhemolytic transfusion reactions (including transfusion-related acute lung injury), some of which may be caused by anti-Siglec-14 alloantibody present in transfused blood plasma [51].
Our finding that patients with wildtype alleles have more severe airflow limitation implies that individuals with a propensity for frequent exacerbations are more prone to a rapid decline in respiratory function. This logic may extend to previous findings regarding the genetic and nongenetic factors involved in COPD pathogenesis. For example, latent adenoviral infection of the respiratory system reportedly contributes to COPD pathogenesis by enhancing the inflammatory response elicited by tobacco smoke [49, 52]. Latent adenoviral infection may increase the frequency and/or intensity of exacerbations elicited by viral or bacterial infection as well, which may play a role not only in COPD pathogenesis but also in its progression.
Although we initially suspected a link between SIGLEC14 genotype and COPD exacerbations based on the interaction between Siglec-14 and NTHi, Siglec-14 may influence inflammatory responses to other pathogens as well, and its effect on COPD exacerbations may not be limited to that caused by NTHi. For example, Siglec-14 expression on myeloid cells may generally influence the inflammatory response to acute viral infection of the airway and lung epithelia by altering cell activation thresholds, leading to COPD exacerbation [53].
The correlation between soluble Siglec-14 serum concentration and exacerbation frequency, even after correcting for wildtype allele number, was unexpected. Although it was beyond the scope of this initial work, it would be interesting to pursue a prospective study enrolling more COPD patients to examine potential correlations between Siglec-14 serum concentration and exacerbation frequency, possibly in combination with patient stratification (based on the cause of exacerbations). Such work may provide information regarding the clinical utility of Siglec-14 serum concentration for COPD exacerbation prognosis. It is also important for the mechanism(s) by which myeloid cells release soluble Siglec-14 to be elucidated.
Our study had limitations. Although the number of patients recruited was substantial (n = 135), the patients at our institution are uniform in ethnicity. As the frequency of the SIGLEC14-null allele is higher in Asia than in other regions [28], our results may be primarily relevant to Asian patients. Second, the follow-up period was limited to 1 year. However, a previous study on COPD pathophysiology showed that patients should be followed for at least 1 year to allow for seasonal effects and accurate estimation of exacerbation frequency [54]. It has also been reported that the frequency with which patients have exacerbations remains relatively stable from year to year [55].
In summary, we demonstrated that COPD patients who express Siglec-14 suffer more frequent exacerbations than those who do not. This knowledge may be utilized to stratify patient populations for personalized management, such as more intensive monitoring or earlier medical intervention against exacerbations in patients who express Siglec-14. Furthermore, Siglec-14 and its downstream signaling pathway may be promising targets for therapeutic intervention to treat COPD exacerbations and limit disease progression.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Leela Davies, Xiaoxia Wang and Mina Fujishiro for experimental help, Andrea Verhagen and Sandra Diaz for general laboratory help, and Syed Raza Ali for general discussion. We also thank Dr. Michael A. Apicella (University of Iowa) for the generous gift of NTHi strain 2019. This work was supported by Global COE program “Frontier Biomedical Science Underlying Organelle Network” from the Ministry of Education, Culture, Sports, Science and Technology of Japan to Osaka University (to T.A. and N.T.); the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), Japan (to K.K. and N.T.); and the National Institutes of Health/National Heart, Lung and Blood Institute (NIH/NHLBI) Programs of Excellence in Glycosciences (P01HL107150 to A.V. and V.N.).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
T. Angata and T. Ishii contributed equally to this work.
Contributor Information
Takashi Angata, Email: angata@riken.jp.
Kozui Kida, Email: kkida@nms.ac.jp.
Naoyuki Taniguchi, Email: tani52@wd5.so-net.ne.jp.
References
- 1.Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization . World Health Statistics 2008. Geneva: WHO; 2008. [Google Scholar]
- 3.Chenna PR, Mannino DM. Outcomes of severe COPD exacerbations requiring hospitalization. Semin Respir Crit Care Med. 2010;31:286–294. doi: 10.1055/s-0030-1254069. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Aymerich J, Serra Pons I, Mannino DM, Maas AK, Miller DP, Davis KJ. Lung function impairment, COPD hospitalisations and subsequent mortality. Thorax. 2011;66:585–590. doi: 10.1136/thx.2010.152876. [DOI] [PubMed] [Google Scholar]
- 5.Decramer M, Nici L, Nardini S, Reardon J, Rochester CL, Sanguinetti CM, Troosters T. Targeting the COPD exacerbation. Respir Med. 2008;102(Suppl 1):S3–S15. doi: 10.1016/S0954-6111(08)70003-9. [DOI] [PubMed] [Google Scholar]
- 6.Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 7.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 8.Barnes PJ. New treatments for COPD. Nat Rev Drug Discov. 2002;1:437–446. doi: 10.1038/nrd820. [DOI] [PubMed] [Google Scholar]
- 9.Brinkman RR, Dube MP, Rouleau GA, Orr AC, Samuels ME. Human monogenic disorders—a source of novel drug targets. Nat Rev Genet. 2006;7:249–260. doi: 10.1038/nrg1828. [DOI] [PubMed] [Google Scholar]
- 10.Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 11.Ganrot PO, Laurell CB, Eriksson S. Obstructive lung disease and trypsin inhibitors in alpha-1-antitrypsin deficiency. Scand J Clin Lab Invest. 1967;19:205–208. doi: 10.3109/00365516709090627. [DOI] [PubMed] [Google Scholar]
- 12.Ishii T, Matsuse T, Teramoto S, Matsui H, Miyao M, Hosoi T, Takahashi H, Fukuchi Y, Ouchi Y. Glutathione S-transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax. 1999;54:693–696. doi: 10.1136/thx.54.8.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molfino NA. Genetics of COPD. Chest. 2004;125:1929–1940. doi: 10.1378/chest.125.5.1929. [DOI] [PubMed] [Google Scholar]
- 14.Hersh CP, Demeo DL, Lange C, et al. Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am J Respir Cell Mol Biol. 2005;33:71–78. doi: 10.1165/rcmb.2005-0073OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foreman MG, DeMeo DL, Hersh CP, Carey VJ, Fan VS, Reilly JJ, Shapiro SD, Silverman EK. Polymorphic variation in surfactant protein B is associated with COPD exacerbations. Eur Respir J. 2008;32:938–944. doi: 10.1183/09031936.00040208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin CL, Siu LK, Lin JC, Liu CY, Chian CF, Lee CN, Chang FY. Mannose-binding lectin gene polymorphism contributes to recurrence of infective exacerbation in patients with COPD. Chest. 2011;139:43–51. doi: 10.1378/chest.10-0375. [DOI] [PubMed] [Google Scholar]
- 18.Yang IA, Seeney SL, Wolter JM, et al. Mannose-binding lectin gene polymorphism predicts hospital admissions for COPD infections. Genes Immun. 2003;4:269–274. doi: 10.1038/sj.gene.6363961. [DOI] [PubMed] [Google Scholar]
- 19.Takabatake N, Shibata Y, Abe S, et al. A single nucleotide polymorphism in the CCL1 gene predicts acute exacerbations in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:875–885. doi: 10.1164/rccm.200603-443OC. [DOI] [PubMed] [Google Scholar]
- 20.Pillai SG, Kong X, Edwards LD, Cho MH, Anderson WH, Coxson HO, Lomas DA, Silverman EK. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:1498–1505. doi: 10.1164/rccm.201002-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. doi: 10.1056/NEJMoa0909883. [DOI] [PubMed] [Google Scholar]
- 22.Hood DW, Makepeace K, Deadman ME, Rest RF, Thibault P, Martin A, Richards JC, Moxon ER. Sialic acid in the lipopolysaccharide of Haemophilus influenzae: strain distribution, influence on serum resistance and structural characterization. Mol Microbiol. 1999;33:679–692. doi: 10.1046/j.1365-2958.1999.01509.x. [DOI] [PubMed] [Google Scholar]
- 23.Mandrell RE, McLaughlin R, Aba Kwaik Y, Lesse A, Yamasaki R, Gibson B, Spinola SM, Apicella MA. Lipooligosaccharides (LOS) of some Haemophilus species mimic human glycosphingolipids, and some LOS are sialylated. Infect Immun. 1992;60:1322–1328. doi: 10.1128/iai.60.4.1322-1328.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev. 2002;102:439–470. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 25.Varki A, Angata T. Siglecs – the major subfamily of I-type lectins. Glycobiology. 2006;16:1R–27R. doi: 10.1093/glycob/cwj008. [DOI] [PubMed] [Google Scholar]
- 26.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 27.Angata T, Hayakawa T, Yamanaka M, Varki A, Nakamura M. Discovery of Siglec-14, a novel sialic acid receptor undergoing concerted evolution with Siglec-5 in primates. FASEB J. 2006;20:1964–1973. doi: 10.1096/fj.06-5800com. [DOI] [PubMed] [Google Scholar]
- 28.Yamanaka M, Kato Y, Angata T, Narimatsu H. Deletion polymorphism of SIGLEC14 and its functional implications. Glycobiology. 2009;19:841–846. doi: 10.1093/glycob/cwp052. [DOI] [PubMed] [Google Scholar]
- 29.Greiner LL, Watanabe H, Phillips NJ, Shao J, Morgan A, Zaleski A, Gibson BW, Apicella MA. Nontypeable Haemophilus influenzae strain 2019 produces a biofilm containing N-acetylneuraminic acid that may mimic sialylated O-linked glycans. Infect Immun. 2004;72:4249–4260. doi: 10.1128/IAI.72.7.4249-4260.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor RE, Gregg CJ, Padler-Karavani V, et al. Novel mechanism for the generation of human xeno-autoantibodies against the nonhuman sialic acid N-glycolylneuraminic acid. J Exp Med. 2010;207:1637–1646. doi: 10.1084/jem.20100575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- 32.Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med. 1987;106:196–204. doi: 10.7326/0003-4819-106-2-196. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest. 2000;117:398S–401S. doi: 10.1378/chest.117.5_suppl_2.398S. [DOI] [PubMed] [Google Scholar]
- 34.American Thoracic Society Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 35.Japanese Respiratory Society The predicted values of spirometry and arterial blood gas analysis in Japanese. J Jpn Respir Soc. 2001;39:1–17. [Google Scholar]
- 36.Nakano Y, Muro S, Sakai H, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Am J Respir Crit Care Med. 2000;162:1102–1108. doi: 10.1164/ajrccm.162.3.9907120. [DOI] [PubMed] [Google Scholar]
- 37.Okazawa M, Muller N, McNamara AE, Child S, Verburgt L, Pare PD. Human airway narrowing measured using high resolution computed tomography. Am J Respir Crit Care Med. 1996;154:1557–1562. doi: 10.1164/ajrccm.154.5.8912780. [DOI] [PubMed] [Google Scholar]
- 38.Orlandi I, Moroni C, Camiciottoli G, Bartolucci M, Pistolesi M, Villari N, Mascalchi M. Chronic obstructive pulmonary disease: thin-section CT measurement of airway wall thickness and lung attenuation. Radiology. 2005;234:604–610. doi: 10.1148/radiol.2342040013. [DOI] [PubMed] [Google Scholar]
- 39.Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27–38. doi: 10.1093/biomet/80.1.27. [DOI] [Google Scholar]
- 40.Jones C, Virji M, Crocker PR. Recognition of sialylated meningococcal lipopolysaccharide by siglecs expressed on myeloid cells leads to enhanced bacterial uptake. Mol Microbiol. 2003;49:1213–1225. doi: 10.1046/j.1365-2958.2003.03634.x. [DOI] [PubMed] [Google Scholar]
- 41.Carlin AF, Lewis AL, Varki A, Nizet V. Group B streptococcal capsular sialic acids interact with siglecs (immunoglobulin-like lectins) on human leukocytes. J Bacteriol. 2007;189:1231–1237. doi: 10.1128/JB.01155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood. 2009;113:3333–3336. doi: 10.1182/blood-2008-11-187302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- 44.Hansel TT, Barnes PJ. New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet. 2009;374:744–755. doi: 10.1016/S0140-6736(09)61342-8. [DOI] [PubMed] [Google Scholar]
- 45.Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, Dales RE. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:349–355. doi: 10.1164/ajrccm.163.2.2003122. [DOI] [PubMed] [Google Scholar]
- 46.Au DH, Bryson CL, Chien JW, Sun H, Udris EM, Evans LE, Bradley KA. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med. 2009;24:457–463. doi: 10.1007/s11606-009-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alsaeedi A, Sin DD, McAlister FA. The effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review of randomized placebo-controlled trials. Am J Med. 2002;113:59–65. doi: 10.1016/S0002-9343(02)01143-9. [DOI] [PubMed] [Google Scholar]
- 48.Varki A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology. 2011;21:1121–1124. doi: 10.1093/glycob/cwr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med. 2001;164:S71–S75. doi: 10.1164/ajrccm.164.supplement_2.2106063. [DOI] [PubMed] [Google Scholar]
- 50.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- 51.Yasui K, Angata T, Matsuyama N, Furuta RA, Kimura T, Okazaki H, Tani Y, Nakano S, Narimatsu H, Hirayama F. Detection of anti-Siglec-14 alloantibodies in blood components implicated in nonhaemolytic transfusion reactions. Br J Haematol. 2011;153(6):794–796. doi: 10.1111/j.1365-2141.2010.08488.x. [DOI] [PubMed] [Google Scholar]
- 52.Hayashi S, Hogg JC. Adenovirus infections and lung disease. Curr Opin Pharmacol. 2007;7:237–243. doi: 10.1016/j.coph.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:115–120. doi: 10.1513/pats.2306030. [DOI] [PubMed] [Google Scholar]
- 54.Donaldson GC, Seemungal T, Jeffries DJ, Wedzicha JA. Effect of temperature on lung function and symptoms in chronic obstructive pulmonary disease. Eur Respir J. 1999;13:844–849. doi: 10.1034/j.1399-3003.1999.13d25.x. [DOI] [PubMed] [Google Scholar]
- 55.Donaldson GC, Seemungal TA, Patel IS, Lloyd-Owen SJ, Wilkinson TM, Wedzicha JA. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J. 2003;22:931–936. doi: 10.1183/09031936.03.00038303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.