

Abstract
Intrahepatic cholangiocarcinoma (ICC) is an aggressive malignancy with very poor prognosis. Genome-wide, high-throughput technologies have made major advances in understanding the molecular basis of this disease, although important mechanisms are still unclear. Recent data have revealed specific genetic mutations (for example, KRAS, IDH1 and IDH2), epigenetic silencing, aberrant signaling pathway activation (for example, interleukin (IL)-6/signal transducer and activator of transcription 3 (STAT3), tyrosine kinase receptor-related pathways) and molecular subclasses with unique alterations (for example, proliferation and inflammation subclasses). In addition, some ICCs share common genomic traits with hepatocellular carcinoma. All this information provides the basis to explore novel targeted therapies. Currently, surgery at early stage is the only effective therapy. At more advanced stages, chemotherapy regimens are emerging (that is, cisplatin plus gemcitabine), along with molecular targeted agents tested in several ongoing clinical trials. Nonetheless, a first-line conclusive treatment remains an unmet need. Similarly, there are no studies assessing tumor response related with genetic alterations. This review explores the recent advancements in the knowledge of the molecular alterations underlying ICC and the future prospects in terms of therapeutic strategies leading towards a more personalized treatment of this neoplasm.
Keywords: cholangiocarcinoma, molecular pathogenesis, targeted therapies
INTRODUCTION
Cholangiocarcinoma (CC) is a relative rare hepatobiliary cancer that primarily arises from the transformation of cholangiocytes of the epithelial bile ducts.1,2 It is a heterogeneous malignancy that comprises two different pathological entities, intrahepatic cholangiocarcinoma (ICC), which arises from the small bile ducts in the liver, and extrahepatic CC (ECC), which involves large hilar bile ducts and the extrahepatic biliary tree. In the past two decades, the incidence of ICC, as well as its mortality rate, has been increasing worldwide, reflecting the poor survival associated with this neoplasia.3,4 By contrast, the rate of ECC is stable or even decreasing. Both entities have distinct risk factors, histological features and clinical outcomes along with different pattern of genetic mutations, expression profiling and epigenetic changes indicating different biological tumor types.5–7 These clinical and biological differences make difficult the interpretation of data derived from both clinical and experimental studies where both entities are included indistinctly. In this review, we will focus specifically on ICC, trying to trace the line from the basic knowledge of its pathogenesis to the rationale for putative targeted therapies.
EPIDEMIOLOGY AND RISK FACTORS
Globally, ICC accounts for around 10% of all primary hepatic cancers, being the second most common after hepatocellular carcinoma (HCC)4 with an annual age-standardized incidence rate <1.5 cases per 100 000 population in western countries.8 Epidemiological data has associated the development of ICC with cirrhosis, hepatolithiasis and hepatitis virus infection.6,9,10 The highest incidence of ICC is found in Thailand and other areas in Eastern Asia because of chronic inflammation of bile ducts after liver fluke infections.11,12 However, in developed countries, ICC often arises not only in cirrhotic livers because of chronic hepatitis or metabolic syndrome6,13 but also in non-cirrhotic livers because of the absence of a clear etiological risk factor. To date, there are no specific available markers for ICC diagnosis. Distinguishing ICC from other entities, such as metastatic carcinoma from gallbladder or pancreatic cancer and HCC, can be made by clinical history, radiological explorations and pathology.7 Although a number of potential molecular biomarkers (for example, mucin 4, metalloproteinases 7 and 9) have been proposed, none of them has reached standard clinical application.14,15
Only recently the 7th American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (UICC) have provided for the first time a tumor node metastasis staging system for ICC, mirroring the growing medical importance of this malignancy.16 According to this new staging system, previous T2 and T3 subgroups are now combined into a simplified T2 group and the previous T4 group has been redefined as T3. Hence, the 7th staging system includes: stage I—T1N0M0; stage II—T2N0M0; stage III—T3N0M0; stage IVa—T4N0M0 or N1M0 (any T), and stage IVb M1 (any T, any N). This staging system posits the presence of multiple tumors, vascular invasion and metastatic disease as powerful predictors of adverse outcome,17 whereas the tumor size is not considered a significant prognostic factor.18 Overall, the understanding of prognostic factors in ICC still remains incomplete.
MOLECULAR PATHOGENESIS
Our knowledge of the molecular alterations underlying the development of ICC is still far from complete, but recent research efforts have improved the understanding of ICC pathogenesis. Herein, we overview the molecular pathogenesis along with the latest findings in terms of genetic and epigenetic alterations, chromosomal aberrations, microRNAs (miRNAs) and molecular pathways disturbances (Figure 1 and Table 1).
Figure 1.
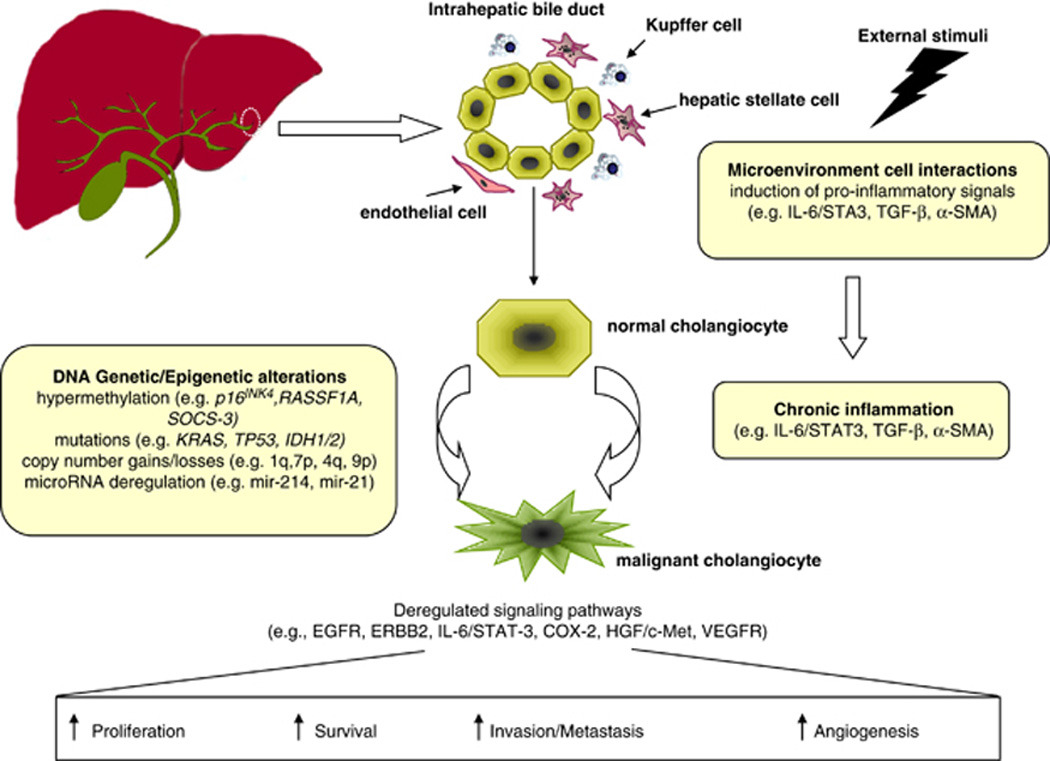
Summary of key molecular alterations involved in ICC carcinogenesis. Despite the absence of clear etiological risk factors or underlying disease, external stimuli (for example, liver fluke or hepatitis viral infection) favor the induction of proinflammatory signals mediated by several cellular types lying in the microenvironment. The release of growth-promoting factors and cytokines during chronic inflammation (for example, IL-6, tumor growth factor β) promotes cholangiocytes’ proliferation. This phenomenon along with the accumulation of genetic and epigenetic alterations in oncogenes and oncosuppressors leads to the malignant transformation of normal cholangiocytes and to the deregulation of main signaling pathways (for example, EGFR, ERBB2, HGF/MET, VEGFR) involved in the hallmarks of cancer, such as proliferation, survival, invasion and enhanced angiogenesis.
Table 1.
Molecular alterations in intrahepatic cholangiocarcinoma
| Gene or molecule |
Type of alteration |
Total samples, n |
Frequency in ICCa (range) (%) |
References |
|---|---|---|---|---|
| Genetic mutations | ||||
| KRAS | Activating mutations | 470 | 22 (5–57) | 22,24,31–35 |
| IDH1/2 | Activating mutations | 433 | 14 (10–28) | 43–45 |
| TP53 | Inactivating mutations | 277 | 15 (0.7–37) | 24,32–34,40 |
| BRAF | Activating mutations | 279 | 7 (1–22) | 22,24,38 |
| EGFR | Activating mutations | 226 | 2 (0–20) | 22,24,37 |
| Epigenetic changes—promoter hypermethylation | ||||
| p16INK4a/CDKN2 | 207 | 47 (11–83) | 34,35,53,56 | |
| p14ARF | 166 | 18 (9–76) | 35,53,56 | |
| APC | 115 | 29 (21–46) | 53,56 | |
| GSTP | 110 | 25 (21–31) | 53,56 | |
| RASSF1A | 64 | 56 (47–64) | 49,55,56 | |
| RUNX3 | 53 | 42 | 58 | |
| SOCS-3 | 26 | 27 | 57 | |
| Chromosomal aberrations (>20% prevalence)b | ||||
| 1q | Gains | 149 | 32 | 24 |
| 7p | Gains | 211 | 24 (25–32) | 24,47,48,50 |
| 4q | Losses | 211 | 22 (18–46) | 24,48–50 |
| 3p | Losses | 171 | 44 (41–68) | 24,50 |
| 6q | Losses | 149 | 52 | 24 |
| 9p | losses | 211 | 40 (26–55) | 24,48–50 |
| 9q | Losses | 171 | 43 (36–45) | 24,50 |
| 13q | Losses | 149 | 38 | 24 |
| 17p | Losses | 192 | 24 (21–55) | 24,47–49 |
Abbreviations: EGFR, epidermal growth factor receptor; ICC, intrahepatic cholangiocarcinoma.
The frequency in ICC has been calculated by considering the number of samples presenting the molecular alteration over the total number of samples evaluated in different studies.
Chromosomal aberrations observed in more than 20% of the total samples analyzed in different studies.
Overview of molecular pathogenesis
Activation of Notch and Wnt signaling governs intrahepatic bile duct development and proliferation and progenitor cell activation, but the direct implication of these cascades in cholangiocarcino-genesis is not established.19 Although important mechanisms underlying the pathogenesis of ICC are still unclear, recent genome-wide technologies have provided novel insights into the molecular understanding of this disease. In the classical model of ICC pathogenesis, promotion of tumor development follows chronic biliary inflammation (with the release of inflammatory cytokines inducing inducible nitric oxide synthase in cholangiocytes, favoring mutagenesis, impaired DNA repair and cyclooxygenase-2 (COX-2) upregulation) and cholestasis (where bile acid signaling promotes cholangiocyte growth via activation of growth factors).2,20,21 Once clonal proliferation led by epidermal growth factor receptor (EGFR), RAS/mitogen-activated protein kinase (MAPK), interleukin (IL)-6 and MET is established, additional alterations regulated by genetic or epigenetic mechanisms in cholangiocytes or stromal cells induce limitless replicative potential (telomerase reverse transcriptase (TERT) activation), evasion of apoptosis (mediated by COX-2, BCL-2), neoangiogenesis (vascular endothelial growth factor (VEGF) and angiopoietin-2), and invasion and metastasis (matrix metalloproteinases overexpression and E-cadherin downregulation).1,2,20–22
ICC results from malignant transformation of cholangiocytes, and in a subset of cases it also arises from progenitor cells. Recent data indicate common genomic traits between ICC and HCC, supporting the hypothesis of common cell ancestors in specific molecular subclasses.23 Transcriptome analysis suggests that the poor prognostic subclass of ICC share genomic traits and signatures of poor-prognosis HCC,22,24 which are associated with stem-like molecular signatures.25–27 Current evidence suggests that 20–25% of HCCs derive from stem cells, whereas the rest derive from adult hepatocytes.28 In a recent meta-analysis exploring molecular subclasses of HCC in more than 600 patients,29 the S2 subclass was characterized by stem cell phenotype (high EpCAM and AFP expression), AKT/mechanistic target of rapamycin (mTOR) and MYC activation and poor prognosis. Moreover, both ICC and HCC share common copy number variations, including chromosomal gains (1q, 8q and 17q) and losses (4q, 8p, 13q and 17p) together with high-level amplifications of 11q–13.24,30 Finally, ICC shares dominant risk factors associated with HCC development, mostly cirrhosis, hepatitis B virus and hepatitis C virus infections, and metabolic syndrome due to diabetes and/or overweight.6,13
Genetic mutations in ICC
Several studies have evaluated the role of mutations in ICC as well as their potential impact in prognosis and utility for diagnosis. Conclusive data in this regard is limited by the small number of samples analyzed in most of the studies and the mixed nature of CC specimens. Activating mutations of KRAS are frequent (22%, range 5–57%),22,24,31–35 particularly in hotspots located at codon 12, and have been pointed as independent predictors of worse survival rate after hepatectomy.31,36 These data, however, need further validation in independent cohorts of samples. BRAF and EGFR mutations have been reported in 7% (1–22%) and 2% (0–20%), respectively.22,24,37,38 On the other hand, NRAS or phosphatidylinositol 3-kinase (PI3K) mutations seem to be rare events in ICC.39 The tumor suppressor gene TP53 appears mutated in more than 50% of human malignancies. A large number of TP53 loss-of-function mutations have been reported in ICC at different prevalence (0.7–37%) with an overall frequency of 15%.24,32–34,40 The contribution of the cell cycle regulator to the development of ICC has been proven in experimental animal models.41,42 Lately, there has been a growing interest in assessing the role of mutations in isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2). Overall, mutations in these genes were identified in 14% of 433 ICCs.43–45 Mutations in IDH1 and IDH2 co-occurred with increased protein levels of TP53 and were associated with DNA hypermethylation. The functional relevance of IDH mutations in the biliary tract remains to be determined.
There is no reported study assessing whole-genome sequencing in ICC, and the sole data available include eight cases of liver fluke-related CCs.46 In this study, 206 somatic mutations affecting 187 genes were identified in known cancer genes (KRAS, TP53) and in 10 novel mutated genes involved in histone modification, genomic instability and G-protein signaling (for example, MLL3, ROBO2, PEG3 and GNAS).
Chromosomal aberrations in ICC
There are only a few studies reporting chromosomal imbalances in ICC. Four of them investigated copy number variations by applying comparative genomic hybridization to small series of ICC patients from Eastern countries47–49 and Europe.50 Even though the studies from the Eastern countries revealed common pattern of alterations, including gains at 8q, 17q and 20q and losses at 4q, 17p and 18q, the European study revealed only partial overlap and a higher karyotypic complexity. The common alterations were restricted to gains at 7p and 8q and losses at 1p, 4q and 9p. These discrepancies might reflect differences in ethnicity as well as etiological backgrounds. Moreover, the small number of cases analyzed in each study further limits the interpretation of data. Recently, Sia et al.24 first applied a single-nucleotide polymorphism array to analyze copy number variation in more than 149 formalin-fixed ICC tissues collected in Europe and the United States. The analysis identified a variety of chromosomal alterations, including gains at 1q and 7p and several losses for 3p, 4q, 6q, 9pq, 13q, 14q, 8p, 17p and 21q, some of them confirming results previously reported. Notably, significant overlap was found between the two European series, further suggesting that the different ethnicity could explain discrepancies observed among eastern and western studies.
Epigenetic and miRNA changes in ICC
Human cancer exhibits aberrant epigenetic regulation through promoter hypermethylation51 along with miRNAs deregulation.52 The methylation profile of several tumor suppressor genes has been investigated in ICC, including p16INK4a/CDKN2 (47%, range 11–83%), RASSF1A (56%, range 47–64%) and APC (29%, range 21–46%).34,35,49,53–56 Other relevant aberrantly methylated genes include SOCS-3, implicated in IL-6/ signal transducer and activator of transcription 3 (STAT3) activation, which promoter is hypermethylated in 27% of CC tumors,57 p14ARF, which prevents TP53 degradation and hence cell cycle arrest in 18% (range 9–76%) of tumors,35,53,56 and the transcription factor RUNX3 in 42% of ICC tumors.58
At the same time, recent evidence suggests that miRNAs’ expression pattern has an important role in the development and progression of ICC. Studies evaluating the function of single oncogenic miRNAs have been reported, such as mir-21459 and mir-21.60,61 Furthermore, a unique 38-miRNA profile has been identified in a cohort of 27 ICCs,62 and some of them are associated with aberrant signaling pathways (for example, hepatocyte growth factor (HGF)/MET, IL-6/STAT3, and so on). More recently, a link between miR-200c, stem cell traits and poor prognosis has been proposed.27 Nevertheless, data should be interpreted with caution, and the exact role of miRNAs either as oncoMIRs or as prognostic markers remains to be elucidated.
The role of stroma in ICC
ICCs are desmoplastic cancers frequently surrounded by a dense stroma with marked cellular admixture. Only recently the significance of this cancer microenvironment has been elucidated and increasing evidence suggests its crucial role in cancer progression and in the promotion of resistance to therapy. Interestingly, the genomic profiling of the epithelium and the stroma from 23 microdissected CCs identified a total of 1442 differentially expressed genes.22 Notably, IL-6 and TGFB3 were found upregulated in the stroma along with chemokine receptors and ligands, cytokines receptors and interleukins. The stromal signature was found associated with poor prognosis. Hence, it seems that targeting the ICC-associated stroma could represent a new valid therapeutic strategy. The ICC-associated stroma is often enriched with mesenchymal cells, including activated macrophages and cancer-associated fibroblasts. To date, many studies have suggested that tumor-associated macrophages may contribute to tumor growth, development and prognosis in several cancers.63 In ICC, recent evidence suggests that patients with higher levels of CD163-positive macrophages show poor disease-free survival.64 On the other side, α-smooth muscle actin-positive cancer-associated fibroblasts are able to induce cell proliferation, migration, invasion and epithelial–mesenchymal transition in an organotypic model of ICC in vitro.21,65
Biological differences between intra- and extrahepatic CC
CCs include a group of tumors largely heterogeneous and can be classified as ECC and ICC. These two entities differ in terms of incidence, risk factors, clinical presentation and molecular biology.4,7 ECC is the most common form of CC accounting for 80% of cases, but its incidence has remained stable or even slightly declined during the past four decades. Conversely, the incidence of ICC has increased worldwide.3,4,66,67 In terms of risk factors, ECCs have been associated with chronic inflammation of the biliary tract including primary sclerosing cholangitis in the western countries and hepatolithiasis in Asian countries,1,2,7 whereas ICC share risk factors with HCC, such as cirrhosis, chronic hepatitis B and C infection, Type 2 diabetes mellitus, obesity and alcohol.6,68,69 Hepatic infections by liver flukes are associated with higher incidence of both ICC and ECC in Asian countries.1,7,70 ICC arises from intrahepatic bile ducts and its typical morphological presentation is of an incidental hepatic mass lesion with a well-demarcated nodule.1,7 These tumors can grow to a large size as they remain asymptomatic for a long period of time.71–73 In contrast, ECC arises from epithelia of large ducts and can be often detected at an early stage owing to signs of biliary obstruction and cholangitis.2,7
More novel findings at molecular level have pointed to genetic differences between both tumor types.5,56,74–76 A unique altered expression of 1633 and 80 genes has been identified in ICC and ECC, respectively, when compared with normal biliary epithelium.75 Aberrant methylation of RASSF1A is more common in ECC (83% vs 47% in ICC), whereas methylation of GSTP occurred more frequently in ICC (31% vs 6% in ECC).56 In addition, somatic mutations in the metabolic enzymes IDH1 and IDH2 have shown to be more prevalent in ICC (22–28%) than in ECC (0–7%).43,44 Finally, BRAF mutations (7%) have been only reported in ICCs (Table 1).5 Genome-wide high-throughput sequencing and methylome analysis comparing both entities will refine the understanding of their differentiated molecular traits.
SIGNALING PATHWAYS
The development of targeted therapies in cancer has been increasingly guided by the tumor’s genetic profile. An example is the identification of oncogene addiction loops that has led to the use of antibodies blocking ERBB2 (HER2/neu) in breast cancer and EGFR and ALK inhibitors in lung cancer. As a consequence of the above-described genetic and epigenetic alterations, several pathways have been found deregulated in ICC, including inflammatory pathways, cell cycle and growth factors signaling. Although they contain potential drivers of carcinogenesis, to date no oncogenic addition loop has been documented.
Some pathways have been found to be deregulated, including the most common IL-6/STAT signaling, growth factors (for example, EGF, HGF/MET, VEGF) and KRAS/MAPKs. Other emerging pathways, including Hedgehog,77 WNT/catenin78,79 and Hippo,80 have been only occasionally described in ICC. Below we review key pathways of importance in the disease in terms of candidate targeted therapies.
IL-6/STAT signaling
Inflammation has been closely linked to an increased risk of ICC. Overall, JAK/STAT signaling activation accounts for 50% of ICC, and may affect more than 70% of the ICC inflammation subclass.24 In particular, IL-6 is an important oncogenic player in the growth of malignant cholangiocytes57,81 and its overexpression may be a consequence of the epigenetic silencing of SOCS-3, the suppressor of cytokine signaling.57,82 IL-6 is secreted by CC cells in response to inflammatory stimuli in an autocrine or paracrine manner and acts upstream or downstream of potent oncogenes. Binding of IL-6 to the gp130 receptor triggers receptor dimerization, leading to the activation of gp130-associated JAK kinases (JAK1, JAK2 and TYK2) and subsequent activation of STAT3, which induces the transcription of target genes essential for cell growth, differentiation and proliferation. The silencing of SOCS-3 might explain, in part, the IL-6-mediated activation of STAT3. Treatment of CC cells with demethylating agents restored SOCS-3 expression, downregulating MCL1 and sensitizing CC cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis.57,82 Furthermore, IL-6 increases the telomerase activity facilitating malignant cholangiocytes to evade senescence,83 and it is also involved in the altered methylation pattern of relevant growth factor receptors, including EGFR,84–86 and in the expression of miRNA belonging to the let-7 family.87
EGFR signaling
Members of the EGFR family, most notably EGFR and ERBB2 (HER-2/ neu), have been implicated in the ICC pathogenesis. Overexpression of these receptors (10–32%) has been reported in ICC patients,88–90 but mutations are infrequent (Table 1).37 On the other side, the oncogenic role of ERBB2 has been shown in a tissue-specific transgenic model that developed intrahepatic biliary tumors in 30% of cases.91 Aberrant phosphorylation of EGFR family receptors activates MAPK/ERK and p38, which in turn increases COX-2 and induces inhibition of apoptosis and promotion of tumor growth.92–93 Several preclinical studies with anti-EGFR targeted drugs, such as erlotinib and cetuximab, have demonstrated in vitro a decrease in cell proliferation of ICC cell lines,94 although in vivo tumor growth inhibition requires blocking both ERBB1 and ERBB2 receptors by lapatinib.22,95 Recently, it has been reported that vandetanib, an antagonist of EGFR, VEGFR2 and RET kinases, caused significant ICC growth inhibition in vivo.96 However, the clinical experience with anti- EGFR therapies showed questionable benefits, suggesting that further investigations will be required to delineate the relevance of these targets (Table 2).
Table 2.
Completed clinical trials with targeted therapies
| Treatment | Targets | Clinical phase |
Patients, n | Patients with ICC (%) |
Results |
References | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CR (%) | PR (%) | ORR (%) | SD (%) | Median OS (months) |
||||||
| Sorafenib | VEGFR, PDGFR, BRAF | II | 46 | 60 | 0 | 2.2 | 2.2 | 30 | 4.4 | 119 |
| Sorafenib | VEGFR, PDGFR, BRAF | II | 31 | ND | 0 | 0 | 0 | 39 | 9 | 120 |
| Erlotinib | EGFR | II | 42 | 32 | 0 | 8 | 8 | 43 | 7.5 | 135 |
| Sunitinib | VEGFR, PDGFR, KIT | II | 56 | 63 | 0 | 8.9 | 8.9 | 32 | 4.8 | 136 |
| Selumetinib | MEK1/2 | II | 28 | 60 | 0 | 12 | 12 | 68 | 9.8 | 137 |
| Erlotonib/bevacizumab | EGFR, VEGFA | II | 53 | 66 | 0 | 12 | 12 | 51 | 9.9 | 138 |
| GEMOX+bevacizumab | VEGFA | II | 35 | 63 | 0 | 40 | 40 | 29 | 12.7 | 122 |
| GEMOX±erlotonib | EGFR | III | 135 vs 133 | ND | 0 vs 2 | 30 vs 14 | 30 vs 16 | 36 vs 51 | 9.5 vs 9.5 | 124 |
| GEMOX+cetuximab | EGFR | II | 30 | 60 | 10 | 53 | 63 | 17 | 15.2 | 123 |
| GEMOX/capecitabine +panitumumab | EGFR | II | 42 | 24 | 2.4 | 31 | 33 | 50 | 9.8 | 139 |
Abbreviations: CP, complete response; EGFR, epidermal growth factor receptor; GEMOX, Gemicitabine and Oxaplatin; ICC, intrahepatic cholangiocarcinoma; KIT, c-kit proto-oncogene receptor tyrosine kinase; ND, not determined; ORR, overall response rate; OS, overall survival; PDGFR, platelet-derived growth factor receptor; PR, partial response; SD, stable disease. VEGFA, vascular endothelial growth factor A; VEGFR, vascular endothelial growth factor receptor.
HGF/MET signaling
MET is considered a key regulator of invasive growth. The interaction of HGF with its receptor MET triggers the activation of major signaling cascades, including MAPK, PI3K/AKT and STAT.97 MET is overexpressed in ICC (12–58%).98,99 Several experimental models have linked overexpression of MET with overexpression of members of the EGFR family98,100 and have shown the capacity of HGF to stimulate migration and invasion in CC cells.101 Nonetheless, MET inhibitors have not yet entered clinical trials.
Angiogenesis
VEGF has an important role in tumor-associated neoangiogenesis. Activation of the VEGF receptors leads to survival, proliferation and migration of endothelial cells.102 VEGF has been found expressed in 51% of 106 ICCs89 and its expression level correlates with poor prognosis.103 Moreover, a recent study showed that sorafenib, a multikinase inhibitor acting predominantly against BRAF and VEGFR, presents potent antitumor activity in both in vitro and in vivo preclinical models of human ICC.104 However, the role of VEGF in ICC needs to be further explored.
Emerging pathways in ICC: Wnt and Hedgehog signaling
The Wnt/β-catenin pathway is an evolutionarily conserved pathway essential for normal cellular processes (that is, development, growth and survival) and its dysregulation has been found associated with numerous malignancies. So far, few studies in ICC reported aberrant nuclear localization (15%) and reduced membranous expression of β-catenin.78,79,105 However, the mechanism beyond Wnt activation in ICC has not been elucidated. In fact, genetic mutations in β-catenin, Axin 1 and APC are rare events.78,79 Hedgehog pathway has an important role in survival, proliferation, development and self-renewal. The role of this pathway in ICC pathogenesis has not been explored thoroughly, and just some preclinical studies demonstrated an indirect role of this cascade in promoting tumorigenesis.77,106
Animal models of ICC
Only a few animal models have been able to recapitulate key molecular and clinical features of human ICC progression.107 Among them, a unique ‘patient-like’ rat model of ICC that closely mimics the disease has been proposed. It consists of an orthotopic model based on the inoculation in the bile duct of isogenic rats of the highly tumorigenic BDEneu rat epithelial cell line,108 which results in rapid ICC tumor growth, accompanied by bile duct obstruction and peritoneal metastases. Another model consists of mutant mice harboring albumin-Cre-mediated somatic activation of KRASG12D and deletion of TP53 in the hepatic parenchyma.42 This is based on the observation that tissue-specific activation of KRASG12D alone results in the development of invasive ICC with long latency that is strongly accelerated by combining with heterozygous or homozygous deletion of TP53 (mean survival of 56 vs 19 weeks, respectively). Clearly, these models might represent a valuable platform for the understanding of the progression of ICC and for the preclinical testing of promising novel therapies.
Molecular classification of ICC
Molecular classification of cancer should aid in understanding the biological subclasses and drivers of the disease and optimize benefits from molecular therapies and enrich trial populations. Molecular stratification can be based on biomarkers as predictors of response to targeted drugs or biomarkers as prognostic factors. Few molecular subclasses have been adopted by guidelines of management, and they are particularly based on biomarker predictors of treatment response. This is the case of amplification of ERBB2 and responders to trastuzumab in breast cancer,109 EGFR mutational status or ALK status and response to erlotinib and crizotinib, respectively, in non-small-cell lung cancer,110,111 and BRAF mutations to identify responders to BRAF inhibitors in melanoma.112 No such case has been described in ICC.
Recent advancements have been made defining molecular subclasses in ICC based on whole-transcriptome analysis and other biological parameters.22,24,113,114 The first comprehensive study included 104 CC cases—both ICC and ECC—and described two molecular subclasses, one of which with poor prognosis and activation of receptor tyrosine kinases, including EGFR, ERBB2 and MET.22 Exploring the microenvironment of ICC in 23 cases, they identified two subclasses, including one with a stromal signature, including chemokines (CXCR4), cytokines and IL-6, pointing to an alternative therapeutic strategy. More recently, an integrative genomic study of 149 ICC identified two molecular subgroups— inflammation and proliferation—with distinct genomic profiling and clinical outcome.24 The inflammation subclass (40%) showed an enrichment of inflammation and cytokine pathway signatures, overexpression of IL-6, IL-10 and IL-17, and constitutive activation of STAT3. The Proliferation subclass (60%) was characterized by enrichment of activated oncogenic pathways as RAS/MAPK and MET, high-level DNA amplifications at 11q13 and deletions at 14q22.1 and signatures of poor outcome. Further independent validation of ICC subclasses is needed in order to be adopted as stratification factors by ICC guidelines.
CLINICAL MANAGEMENT
Overview and unmet needs
Surgical treatment is the only curative treatment option for ICC. Life expectancy for patients with unresectable ICC is <5% at 5 years,4 whereas it increases to 20–44% at 5 years for patients undergoing resection at early T1–T2 stages. Tumor recurrence is observed frequently.115,116 Adjuvant treatments, including chemotherapy, radiation therapy and photodynamic therapy, have not shown to significantly improve survival or time to recurrence, albeit no large randomized trials have been published.2,7 The development of molecular targeted therapies for the treatment of advanced ICCs has encountered many problems.117 Firstly, the vast majority of clinical trials conducted until now are directed towards biliary tract cancers, including both ICC and ECC, as well as gallbladder carcinomas. Secondly, most studies are small, non-randomized and single-centered trials, resulting in statistically underpowered or biased data. Finally, the development of targeted agents in cancer is increasingly guided by the tumor’s genetic profile and until recently little effort had been dedicated to fully understand the molecular basis of ICC. Despite that, in recent years there has been a renewed interest in developing molecularly targeted therapies in this arena (Table 2), especially in combination with conventional chemotherapy. The completion of the landmark phase III trial (ABC-02 trial), which demonstrated improved overall survival of patients treated with gemcitabine plus cisplatin vs gemcitabine alone (11.7 vs 8 months) defined a novel paradigm for the management of biliary tract cancers.118 The subgroup analysis including around 80 ICC confirms a positive signal of efficacy for the combination therapy, but this result needs to be confirmed within a specific well-powered RCT-only targeting ICC patients. This type of evidence will certainly be required to accept the combination chemotherapy as the standard of care for management of patients in the setting of guidelines and to be the control arm in advanced ICC trials testing novel compounds.
Molecular targeted therapies
In the past years, a growing number of clinical trials have been conducted using few classes of targeted therapies as first-line treatment in advanced biliary tract cancers (Table 2 and Figure 2). These trials were carried out as single agents (for example, sorafenib, erlotinib, sunitinib, selumetinib), combined targeted agents or in combination with conventional chemotherapy (for example, gemcitabine, cisplatin and oxaliplatin). So far, discouraging results have been obtained in several phase II studies where these agents have been used as monotherapy (Table 2), such is the case of sorafenib119,120 and lapatinib.121 Small single-arm phase II studies have reported acceptable results combining gemcitabine plus oxaliplatin with bevacizumab (median survival of 12.7 months; objective responses of 40%)122 or cetuximab (median survival of 15.2 months; objective responses of 63%).123 At the same time, a multicenter, open-label randomized phase III of GEMOX in combination with erlotinib (a tyrosine kinase inhibitor against EGFR) or placebo suggested a marginal benefit of erlotinib in the subgroup analysis of CC.124 Few randomized phase II trials are currently ongoing, but no phase III pivotal trial is active so far (Table 3).
Figure 2.
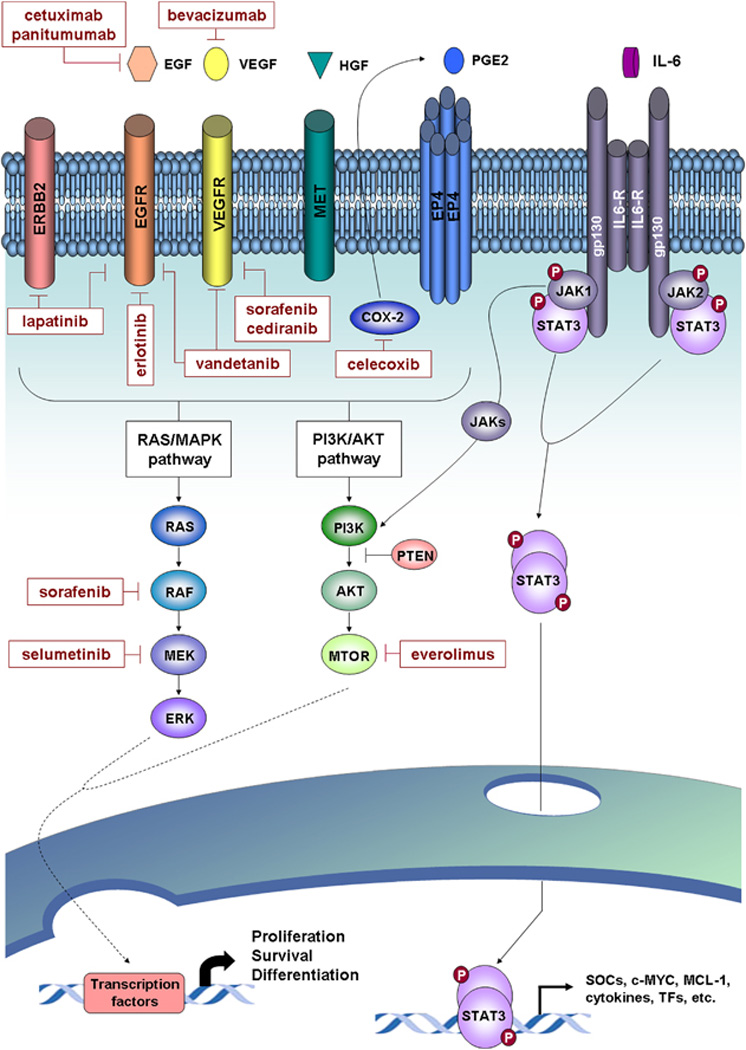
Signaling pathways and molecular therapies in ICC. Major deregulated oncogenic signaling pathways identified for ICC and targeted molecular drugs evaluated in preclinical and clinical studies are represented. Activation of tyrosine kinase receptors (for example, EGFR, VEGFR, MET, and so on) and also others, such prostaglandin receptor 4 (EP4), triggers the activation of two major signaling pathways including the RAS/MAPK and PI3K/AKT pathways. These pathways implicate sequential activation of downstream tyrosine kinases, which lead to the regulation of gene expression through the activation of specific transcription factors. COX-2 is a key enzyme implicated in inflammation and cell growth through the biosynthesis of prostaglandin E2 (PGE2). In IL-6/STAT3 signaling, the binding of IL-6 to the receptor leads to gp130 receptor dimerization and associated JAK phosphorylation. These then provide a docking place for the transcription factor STAT3, which also is phosphorylated and dimerized. Activated STAT3 is translocated to the nucleus and induces the transcription of targeted genes implicated in cell processes, such as proliferation, cell growth and differentiation.
Table 3.
Ongoing clinical trials using targeted therapya
| Treatment | Targets | Clinical phase | Number of trials | Trial type |
|---|---|---|---|---|
| Everolimus | mTOR | II | 2 | NRCT |
| Chemotherapyb±cetuximab | EGFR | II | 2 | NRCT/RCT |
| Chemotherpy±panitumumab | EGFR | II | 5 | NRCTc/RCT |
| Chemotherpy+bevacizumab | VEGFA | II | 2 | NRCT |
| Chemotherpy±cediranib | VEGFR | II, II/III | 2 | NRCT/RCT |
| Chemotherpy±vandetanib | VEGFR, EGFR | I, II | 2 | NRCT/RCT |
| Chemotherpy+sorafenib | BRAF, VEGFR, PDGFR | I/II | 1 | NRCT |
| Chemotherpy+selumetinib | MEK1/2 | I/II | 1 | NRCT |
Abbreviations: EGFR, epidermal growth factor receptor; KIT, c-kit proto-oncogene receptor tyrosine kinase; mTOR, mechanistic target of rapamycin; NRCT, nonrandomized clinical trial; PDGFR, platelet-derived growth factor receptor; RCT, randomized clinical trial; VEGFA, vascular endothelial growth factor A; VEGFR, vascular endothelial growth factor receptor.
Information retrieved from clinicaltrials.gov.
Chemotherapy (for example, gemcitabine, cisplatin and mFOLFOX6).
Two of the NRCT include patients with wild-type KRAS and BRAF.
A more comprehensive understanding of ICC pathogenesis may lead to uncover new candidate therapeutic targets. For example, herein we emphasized the importance of MET signaling, IL-6/JAK/ STAT3 pathway and COX-2 in ICC (Figure 2). Currently, there are multiple MET inhibitors in clinical development for solid tumors but none for testing patients with ICC.125 Also, the use of recently developed and clinically evaluated novel JAK1 and JAK2 inhibitors126–130 along with some novel STAT3 inhibitors131 and antibodies against IL-6 receptor may be considered an appealing therapeutic strategy. Finally, considering that selective targeting of COX-2 with celecoxib reduces CC proliferation in vitro,93,132–134 COX-2-mediated pathway may represent a promising target.
CONCLUSIONS
There is still a limited understanding of the molecular abnormalities involved in ICC pathogenesis. Only recently, there has been a growing effort dedicated to clarifying the involvement of several signaling pathways and key drivers. However, an approach aimed at identifying oncogenic loops and at linking these discoveries with the design of therapeutic algorithms is still missing. In this regard, the identification of two molecular subclasses with specific molecular traits needs further validation to guide a more stratified treatment approach. As more we understand the molecular basis of CC, novel candidate targets, such as MET, EGFR and JAK/STAT, may become attractive, and clinical trials testing drugs blocking these pathways are encouraged. So far, advanced ICC is considered an orphan cancer with no established first-line treatment option, hence representing an unmet medical need. Although combined chemotherapy might provide survival advantages, results need to be confirmed in specific trails for this tumor type. Hopefully, the latest technological advancements (for example, next-generation sequencing technology) will significantly improve our understanding of the main drivers of this neoplasm. These advancements should lead to a more adequate trial design and stratified medicine.
ACKNOWLEDGEMENTS
Josep M Llovet is supported by grants from the US National Institutes of Diabetes and Digestive and Kidney Diseases (1R01DK076986), the European Commission’s Framework Programme 7 (HEPTROMIC; 259744), the Asociación Española Contra el Cáncer, the Spanish National Health Institute (SAF-2010-16055) and the Samuel Waxman Cancer Research Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303–1314. doi: 10.1016/S0140-6736(05)67530-7. [DOI] [PubMed] [Google Scholar]
- 2.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353–1357. doi: 10.1053/jhep.2001.25087. [DOI] [PubMed] [Google Scholar]
- 4.Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115–125. doi: 10.1055/s-2004-828889. [DOI] [PubMed] [Google Scholar]
- 5.Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol. 2010;28:3531–3540. doi: 10.1200/JCO.2009.27.4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol. 2012;57:69–76. doi: 10.1016/j.jhep.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sempoux C, Jibara G, Ward SC, Fan C, Qin L, Roayaie S, et al. Intrahepatic cholangiocarcinoma: new insights in pathology. Semin Liver Dis. 2011;31:49–60. doi: 10.1055/s-0031-1272839. [DOI] [PubMed] [Google Scholar]
- 8.Parkin DM, Whelan SL, Ferlay J, Teppo L, Thomas DB. Cancer Incidence in Five Continents Vol VIII: IARC Scientific Publication No. 155I. Lyon: ARC Press; 2002. [Google Scholar]
- 9.El-Serag HB, Engels EA, Landgren O, Chiao E, Henderson L, Amaratunge HC, et al. Risk of hepatobiliary and pancreatic cancers after hepatitis C virus infection: a population-based study of US veterans. Hepatology. 2009;49:116–123. doi: 10.1002/hep.22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaib YH, El-Serag HB, Davila JA, Morgan R, McGlynn KA. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case-control study. Gastroenterology. 2005;128:620–626. doi: 10.1053/j.gastro.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 11.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 12.Shin HR, Oh JK, Masuyer E, Curado MP, Bouvard V, Fang YY, et al. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 2010;101:579–585. doi: 10.1111/j.1349-7006.2009.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54:463–471. doi: 10.1002/hep.24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leelawat K, Sakchinabut S, Narong S, Wannaprasert J. Detection of serum MMP-7 and MMP-9 in cholangiocarcinoma patients: evaluation of diagnostic accuracy. BMC Gastroenterol. 2009;9:30. doi: 10.1186/1471-230X-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibahara H, Tamada S, Higashi M, Goto M, Batra SK, Hollingsworth MA, et al. MUC4 is a novel prognostic factor of intrahepatic cholangiocarcinoma-mass forming type. Hepatology. 2004;39:220–229. doi: 10.1002/hep.20031. [DOI] [PubMed] [Google Scholar]
- 16.Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. 7th edn. New York, NY: Springer; 2010. [Google Scholar]
- 17.de Jong MC, Nathan H, Sotiropoulos GC, Paul A, Alexandrescu S, Marques H, et al. Intrahepatic cholangiocarcinoma: an international multi-institutional analysis of prognostic factors and lymph node assessment. J Clin Oncol. 2011;29:3140–3145. doi: 10.1200/JCO.2011.35.6519. [DOI] [PubMed] [Google Scholar]
- 18.Nathan H, Pawlik TM. Staging of intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2010;26:269–273. doi: 10.1097/MOG.0b013e328337c899. [DOI] [PubMed] [Google Scholar]
- 19.Sirica AE, Nathanson MH, Gores GJ, Larusso NF. Pathobiology of biliary epithelia and cholangiocarcinoma: proceedings of the Henry M. and Lillian Stratton Basic Research Single-Topic Conference. Hepatology. 2008;48:2040–2046. doi: 10.1002/hep.22623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berthiaume EP, Wands J. The molecular pathogenesis of cholangiocarcinoma. Semin Liver Dis. 2004;24:127–137. doi: 10.1055/s-2004-828890. [DOI] [PubMed] [Google Scholar]
- 21.Sirica AE, Campbell DJ, Dumur CI. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2011;27:276–284. doi: 10.1097/MOG.0b013e32834405c3. [DOI] [PubMed] [Google Scholar]
- 22.Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 24.Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals two subclasses with distinct clinical outcome. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.01.001. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS, Jang JJ, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010;70:3034–3041. doi: 10.1158/0008-5472.CAN-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oishi N, Kumar MR, Roessler S, Ji J, Forgues M, Budhu A, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of mir-200c and EMT in intrahepatic cholangiocarcinoma. Hepatology. 2012;56:1792–1803. doi: 10.1002/hep.25890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 29.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen TC, Jan YY, Yeh TS. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann Surg Oncol. 2012;19(Suppl 3):S675–S681. doi: 10.1245/s10434-012-2224-7. [DOI] [PubMed] [Google Scholar]
- 32.Furubo S, Harada K, Shimonishi T, Katayanagi K, Tsui W, Nakanuma Y. Protein expression and genetic alterations of p53 and ras in intrahepatic cholangiocarcinoma. Histopathology. 1999;35:230–240. doi: 10.1046/j.1365-2559.1999.00705.x. [DOI] [PubMed] [Google Scholar]
- 33.Momoi H, Itoh T, Nozaki Y, Arima Y, Okabe H, Satoh S, et al. Microsatellite instability and alternative genetic pathway in intrahepatic cholangiocarcinoma. J Hepatol. 2001;35:235–244. doi: 10.1016/s0168-8278(01)00106-4. [DOI] [PubMed] [Google Scholar]
- 34.Tannapfel A, Benicke M, Katalinic A, Uhlmann D, Kockerling F, Hauss J, et al. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47:721–727. doi: 10.1136/gut.47.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tannapfel A, Sommerer F, Benicke M, Weinans L, Katalinic A, Geissler F, et al. Genetic and epigenetic alterations of the INK4a-ARF pathway in cholangiocarcinoma. J Pathol. 2002;197:624–631. doi: 10.1002/path.1139. [DOI] [PubMed] [Google Scholar]
- 36.Isa T, Tomita S, Nakachi A, Miyazato H, Shimoji H, Kusano T, et al. Analysis of microsatellite instability, K-ras gene mutation and p53 protein overexpression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2002;49:604–608. [PubMed] [Google Scholar]
- 37.Leone F, Cavalloni G, Pignochino Y, Sarotto I, Ferraris R, Piacibello W, et al. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin Cancer Res. 2006;12:1680–1685. doi: 10.1158/1078-0432.CCR-05-1692. [DOI] [PubMed] [Google Scholar]
- 38.Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52:706–712. doi: 10.1136/gut.52.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deshpande V, Nduaguba A, Zimmerman SM, Kehoe SM, Macconaill LE, Lauwers GY, et al. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer. 2011;11:60. doi: 10.1186/1471-2407-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannapfel A, Weinans L, Geissler F, Schutz A, Katalinic A, Kockerling F, et al. Mutations of p53 tumor suppressor gene, apoptosis, and proliferation in intrahepatic cholangiocellular carcinoma of the liver. Dig Dis Sci. 2000;45:317–324. doi: 10.1023/a:1005412626515. [DOI] [PubMed] [Google Scholar]
- 41.Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006;66:6622–6627. doi: 10.1158/0008-5472.CAN-05-4609. [DOI] [PubMed] [Google Scholar]
- 42.O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–1567. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kipp BR, Voss JS, Kerr SE, Barr Fritcher EG, Graham RP, Zhang L, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43:1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Wang P, Dong QZ, Zhang C, Kuan PF, Liu Y, Jeck WR, et al. Mutations in isocitrate dehydrogenase 1 and 2 are associated with DNA hypermethylation in intrahepatic cholangiocarcinomas. Cancer Res. 2012 e-pub ahead of print. [Google Scholar]
- 46.Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 47.Koo SH, Ihm CH, Kwon KC, Park JW, Kim JM, Kong G. Genetic alterations in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Genet Cytogenet. 2001;130:22–28. doi: 10.1016/s0165-4608(01)00460-5. [DOI] [PubMed] [Google Scholar]
- 48.Uhm KO, Park YN, Lee JY, Yoon DS, Park SH. Chromosomal imbalances in Korean intrahepatic cholangiocarcinoma by comparative genomic hybridization. Cancer Genet Cytogenet. 2005;157:37–41. doi: 10.1016/j.cancergencyto.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Wong N, Li L, Tsang K, Lai PB, To KF, Johnson PJ. Frequent loss of chromosome 3p and hypermethylation of RASSF1A in cholangiocarcinoma. J Hepatol. 2002;37:633–639. doi: 10.1016/s0168-8278(02)00269-6. [DOI] [PubMed] [Google Scholar]
- 50.Homayounfar K, Gunawan B, Cameron S, Haller F, Baumhoer D, Uecker S, et al. Pattern of chromosomal aberrations in primary liver cancers identified by comparative genomic hybridization. Hum Pathol. 2009;40:834–842. doi: 10.1016/j.humpath.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 52.Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, Kim WH, Jung HY, Yang MH, Kang GH. Aberrant CpG island methylation of multiple genes in intrahepatic cholangiocarcinoma. Am J Pathol. 2002;161:1015–1022. doi: 10.1016/S0002-9440(10)64262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandhu DS, Shire AM, Roberts LR. Epigenetic DNA hypermethylation in cholangiocarcinoma: potential roles in pathogenesis, diagnosis and identification of treatment targets. Liver Int. 2008;28:12–27. doi: 10.1111/j.1478-3231.2007.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tischoff I, Markwarth A, Witzigmann H, Uhlmann D, Hauss J, Mirmohammadsadegh A, et al. Allele loss and epigenetic inactivation of 3p21.3 in malignant liver tumors. Int J Cancer. 2005;115:684–689. doi: 10.1002/ijc.20944. [DOI] [PubMed] [Google Scholar]
- 56.Yang B, House MG, Guo M, Herman JG, Clark DP. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod Pathol. 2005;18:412–420. doi: 10.1038/modpathol.3800287. [DOI] [PubMed] [Google Scholar]
- 57.Isomoto H, Mott JL, Kobayashi S, Werneburg NW, Bronk SF, Haan S, et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–396. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dachrut S, Banthaisong S, Sripa M, Paeyao A, Ho C, Lee SA, et al. DNA copy-number loss on 1p36.1 harboring RUNX3 with promoter hypermethylation and associated loss of RUNX3 expression in liver fluke-associated intrahepatic cholangiocarcinoma. Asian Pac J Cancer Prev. 2009;10:575–582. [PubMed] [Google Scholar]
- 59.Li B, Han Q, Zhu Y, Yu Y, Wang J, Jiang X. Down-regulation of miR-214 contributes to intrahepatic cholangiocarcinoma metastasis by targeting Twist. FEBS J. 2012;279:2393–2398. doi: 10.1111/j.1742-4658.2012.08618.x. [DOI] [PubMed] [Google Scholar]
- 60.Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 61.Selaru FM, Olaru AV, Kan T, David S, Cheng Y, Mori Y, et al. MicroRNA-21 is overexpressed in human cholangiocarcinoma and regulates programmed cell death 4 and tissue inhibitor of metalloproteinase 3. Hepatology. 2009;49:1595–1601. doi: 10.1002/hep.22838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Yan HX, Yang W, Hu L, Yu LX, Liu Q, et al. The role of microRNA expression pattern in human intrahepatic cholangiocarcinoma. J Hepatol. 2009;50:358–369. doi: 10.1016/j.jhep.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 64.Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101:1913–1919. doi: 10.1111/j.1349-7006.2010.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okabe H, Beppu T, Hayashi H, Horino K, Masuda T, Komori H, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2009;16:2555–2564. doi: 10.1245/s10434-009-0568-4. [DOI] [PubMed] [Google Scholar]
- 66.Hammill CW, Wong LL. Intrahepatic cholangiocarcinoma: a malignancy of increasing importance. J Am Coll Surg. 2008;207:594–603. doi: 10.1016/j.jamcollsurg.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 67.Shaib YH, Davila JA, McGlynn K, El-Serag HB. Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol. 2004;40:472–477. doi: 10.1016/j.jhep.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi M, Ikeda K, Saitoh S, Suzuki F, Tsubota A, Suzuki Y, et al. Incidence of primary cholangiocellular carcinoma of the liver in Japanese patients with hepatitis C virus-related cirrhosis. Cancer. 2000;88:2471–2477. doi: 10.1002/1097-0142(20000601)88:11<2471::aid-cncr7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 69.Yamamoto S, Kubo S, Hai S, Uenishi T, Yamamoto T, Shuto T, et al. Hepatitis C virus infection as a likely etiology of intrahepatic cholangiocarcinoma. Cancer Sci. 2004;95:592–595. doi: 10.1111/j.1349-7006.2004.tb02492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bragazzi MC, Cardinale V, Carpino G, Venere R, Semeraro R, Gentile R, et al. Cholangiocarcinoma: epidemiology and risk factors. Transl Gastrointest Cancer. 2012;1:21–32. [Google Scholar]
- 71.Aishima S, Kuroda Y, Nishihara Y, Iguchi T, Taguchi K, Taketomi A, et al. Proposal of progression model for intrahepatic cholangiocarcinoma: clinicopathologic differences between hilar type and peripheral type. Am J Surg Pathol. 2007;31:1059–1067. doi: 10.1097/PAS.0b013e31802b34b6. [DOI] [PubMed] [Google Scholar]
- 72.Malhi H, Gores GJ. Cholangiocarcinoma: modern advances in understanding a deadly old disease. J Hepatol. 2006;45:856–867. doi: 10.1016/j.jhep.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okuda K, Nakanuma Y, Miyazaki M. Cholangiocarcinoma: recent progress. Part 1: epidemiology and etiology. J Gastroenterol Hepatol. 2002;17:1049–1055. doi: 10.1046/j.1440-1746.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 74.Karamitopoulou E, Tornillo L, Zlobec I, Cioccari L, Carafa V, Borner M, et al. Clinical significance of cell cycle-and apoptosis-related markers in biliary tract cancer: a tissue microarray-based approach revealing a distinctive immunophenotype for intrahepatic and extrahepatic cholangiocarcinomas. Am J Clin Pathol. 2008;130:780–786. doi: 10.1309/AJCP35FDCAVANWMM. [DOI] [PubMed] [Google Scholar]
- 75.Miller G, Socci ND, Dhall D, D’Angelica M, DeMatteo RP, Allen PJ, et al. Genome wide analysis and clinical correlation of chromosomal and transcriptional mutations in cancers of the biliary tract. J Exp Clin Cancer Res. 2009;28:62. doi: 10.1186/1756-9966-28-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guedj N, Zhan Q, Perigny M, Rautou PE, Degos F, Belghiti J, et al. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. J Hepatol. 2009;51:93–101. doi: 10.1016/j.jhep.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 77.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–2088. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sugimachi K, Taguchi K, Aishima S, Tanaka S, Shimada M, Kajiyama K, et al. Altered expression of beta-catenin without genetic mutation in intrahepatic cholangiocarcinoma. Mod Pathol. 2001;14:900–905. doi: 10.1038/modpathol.3880409. [DOI] [PubMed] [Google Scholar]
- 79.Tokumoto N, Ikeda S, Ishizaki Y, Kurihara T, Ozaki S, Iseki M, et al. Immunohistochemical and mutational analyses of Wnt signaling components and target genes in intrahepatic cholangiocarcinomas. Int J Oncol. 2005;27:973–980. [PubMed] [Google Scholar]
- 80.Li H, Wolfe A, Septer S, Edwards G, Zhong X, Abdulkarim AB, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32:38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogenactivated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30:1128–1133. doi: 10.1002/hep.510300522. [DOI] [PubMed] [Google Scholar]
- 82.Kobayashi S, Werneburg NW, Bronk SF, Kaufmann SH, Gores GJ. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054–2065. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 83.Yamagiwa Y, Meng F, Patel T. Interleukin-6 decreases senescence and increases telomerase activity in malignant human cholangiocytes. Life Sci. 2006;78:2494–2502. doi: 10.1016/j.lfs.2005.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem. 2001;276:39508–39511. doi: 10.1074/jbc.C100343200. [DOI] [PubMed] [Google Scholar]
- 85.Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–1065. doi: 10.1016/j.jhep.2005.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006;66:10517–10524. doi: 10.1158/0008-5472.CAN-06-2130. [DOI] [PubMed] [Google Scholar]
- 87.Meng F, Wehbe-Janek H, Henson R, Smith H, Patel T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene. 2008;27:378–386. doi: 10.1038/sj.onc.1210648. [DOI] [PubMed] [Google Scholar]
- 88.Nakazawa K, Dobashi Y, Suzuki S, Fujii H, Takeda Y, Ooi A. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol. 2005;206:356–365. doi: 10.1002/path.1779. [DOI] [PubMed] [Google Scholar]
- 89.Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98:418–425. doi: 10.1038/sj.bjc.6604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sirica AE. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J Gastroenterol. 2008;14:7033–7058. doi: 10.3748/wjg.14.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kiguchi K, Carbajal S, Chan K, Beltran L, Ruffino L, Shen J, et al. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001;61:6971–6976. [PubMed] [Google Scholar]
- 92.Endo K, Yoon BI, Pairojkul C, Demetris AJ, Sirica AE. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36:439–450. doi: 10.1053/jhep.2002.34435. [DOI] [PubMed] [Google Scholar]
- 93.Han C, Leng J, Demetris AJ, Wu T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004;64:1369–1376. doi: 10.1158/0008-5472.can-03-1086. [DOI] [PubMed] [Google Scholar]
- 94.Jimeno A, Rubio-Viqueira B, Amador ML, Oppenheimer D, Bouraoud N, Kulesza P, et al. Epidermal growth factor receptor dynamics influences response to epidermal growth factor receptor targeted agents. Cancer Res. 2005;65:3003–3010. doi: 10.1158/0008-5472.CAN-04-3586. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Z, Oyesanya RA, Campbell DJ, Almenara JA, Dewitt JL, Sirica AE. Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy. Hepatology. 2010;52:975–986. doi: 10.1002/hep.23773. [DOI] [PubMed] [Google Scholar]
- 96.Yoshikawa D, Ojima H, Kokubu A, Ochiya T, Kasai S, Hirohashi S, et al. Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br J Cancer. 2009;100:1257–1266. doi: 10.1038/sj.bjc.6604988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 98.Miyamoto M, Ojima H, Iwasaki M, Shimizu H, Kokubu A, Hiraoka N, et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer. 2011;105:131–138. doi: 10.1038/bjc.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Terada T, Nakanuma Y, Sirica AE. Immunohistochemical demonstration of MET overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum Pathol. 1998;29:175–180. doi: 10.1016/s0046-8177(98)90229-5. [DOI] [PubMed] [Google Scholar]
- 100.Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: A link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatology. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 101.Leelawat K, Leelawat S, Tepaksorn P, Rattanasinganchan P, Leungchaweng A, Tohtong R, et al. Involvement of c-Met/hepatocyte growth factor pathway in cholangiocarcinoma cell invasion and its therapeutic inhibition with small interfering RNA specific for c-Met. J Surg Res. 2006;136:78–84. doi: 10.1016/j.jss.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 102.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 103.Park BK, Paik YH, Park JY, Park KH, Bang S, Park SW, et al. The clinicopathologic significance of the expression of vascular endothelial growth factor-C in intrahepatic cholangiocarcinoma. Am J Clin Oncol. 2006;29:138–142. doi: 10.1097/01.coc.0000204402.29830.08. [DOI] [PubMed] [Google Scholar]
- 104.Sugiyama H, Onuki K, Ishige K, Baba N, Ueda T, Matsuda S, et al. Potent in vitro and in vivo antitumor activity of sorafenib against human intrahepatic cholangiocarcinoma cells. J Gastroenterol. 2011;46:779–789. doi: 10.1007/s00535-011-0380-3. [DOI] [PubMed] [Google Scholar]
- 105.Settakorn J, Kaewpila N, Burns GF, Leong AS. FAT, E-cadherin, beta catenin, HER 2/neu, Ki67 immuno-expression, and histological grade in intrahepatic cholangiocarcinoma. J Clin Pathol. 2005;58:1249–1254. doi: 10.1136/jcp.2005.026575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;9:44–54. doi: 10.1038/nrgastro.2011.222. [DOI] [PubMed] [Google Scholar]
- 107.Sirica AE, Dumur CI, Campbell DJ, Almenara JA, Ogunwobi OO, Dewitt JL. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009;7:S68–S78. doi: 10.1016/j.cgh.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel ‘patient-like’ model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 109.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 110.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–144. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 112.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jinawath N, Chamgramol Y, Furukawa Y, Obama K, Tsunoda T, Sripa B, et al. Comparison of gene expression profiles between Opisthorchis viverrini and non-Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology. 2006;44:1025–1038. doi: 10.1002/hep.21330. [DOI] [PubMed] [Google Scholar]
- 114.Obama K, Ura K, Li M, Katagiri T, Tsunoda T, Nomura A, et al. Genome-wide analysis of gene expression in human intrahepatic cholangiocarcinoma. Hepatology. 2005;41:1339–1348. doi: 10.1002/hep.20718. [DOI] [PubMed] [Google Scholar]
- 115.Pascher A, Jonas S, Neuhaus P. Intrahepatic cholangiocarcinoma: indication for transplantation. J Hepatobiliary Pancreat Surg. 2003;10:282–287. doi: 10.1007/s00534-002-0731-9. [DOI] [PubMed] [Google Scholar]
- 116.Yoon JH, Gwak GY, Lee HS, Bronk SF, Werneburg NW, Gores GJ. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J Hepatol. 2004;41:808–814. doi: 10.1016/j.jhep.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 117.Zhu AX, Hezel AF. Development of molecularly targeted therapies in biliary tract cancers: reassessing the challenges and opportunities. Hepatology. 2011;53:695–704. doi: 10.1002/hep.24145. [DOI] [PubMed] [Google Scholar]
- 118.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 119.Bengala C, Bertolini F, Malavasi N, Boni C, Aitini E, Dealis C, et al. Sorafenib in patients with advanced biliary tract carcinoma: a phase II trial. Br J Cancer. 2010;102:68–72. doi: 10.1038/sj.bjc.6605458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.El-Khoueiry AB, Rankin CJ, Ben-Josef E, Lenz HJ, Gold PJ, Hamilton RD, et al. SWOG 0514: a phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest New Drugs. 2011;30:1646–1651. doi: 10.1007/s10637-011-9719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramanathan RK, Belani CP, Singh DA, Tanaka M, Lenz HJ, Yen Y, et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009;64:777–783. doi: 10.1007/s00280-009-0927-7. [DOI] [PubMed] [Google Scholar]
- 122.Zhu AX, Meyerhardt JA, Blaszkowsky LS, Kambadakone AR, Muzikansky A, Zheng H, et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol. 2010;11:48–54. doi: 10.1016/S1470-2045(09)70333-X. [DOI] [PubMed] [Google Scholar]
- 123.Gruenberger B, Schueller J, Heubrandtner U, Wrba F, Tamandl D, Kaczirek K, et al. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol. 2010;11:1142–1148. doi: 10.1016/S1470-2045(10)70247-3. [DOI] [PubMed] [Google Scholar]
- 124.Lee J, Park SH, Chang HM, Kim JS, Choi HJ, Lee MA, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012;13:181–188. doi: 10.1016/S1470-2045(11)70301-1. [DOI] [PubMed] [Google Scholar]
- 125.Borbath C, Porta L, Rimassa B, Daniele S, Salvagni JL, Van Laethem H, et al. Tivantinib in Metþ pretreated hepatocellular carcinoma (HCC): a randomized controlled phase 2 trial (Rct) Sixth ILCA Annual Conference. 2012 (16 Abstract 0-023) [Google Scholar]
- 126.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mesa RA, Yasothan U, Kirkpatrick P. Ruxolitinib. Nat Rev Drug Discov. 2012;11:103–104. doi: 10.1038/nrd3652. [DOI] [PubMed] [Google Scholar]
- 128.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sirica AE, Lai GH, Endo K, Zhang Z, Yoon BI. Cyclooxygenase-2 and ERBB-2 in cholangiocarcinoma: potential therapeutic targets. Semin Liver Dis. 2002;22:303–313. doi: 10.1055/s-2002-34507. [DOI] [PubMed] [Google Scholar]
- 133.Wu GS, Zou SQ, Liu ZR, Tang ZH, Wang JH. Celecoxib inhibits proliferation and induces apoptosis via prostaglandin E2 pathway in human cholangiocarcinoma cell lines. World J Gastroenterol. 2003;9:1302–1306. doi: 10.3748/wjg.v9.i6.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wu T, Leng J, Han C, Demetris AJ. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer Ther. 2004;3:299–307. [PubMed] [Google Scholar]
- 135.Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, et al. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol. 2006;24:3069–3074. doi: 10.1200/JCO.2005.05.3579. [DOI] [PubMed] [Google Scholar]
- 136.Yi JH, Thongprasert S, Lee J, Doval DC, Park SH, Park JO, et al. A phase II study of sunitinib as a second-line treatment in advanced biliary tract carcinoma: a multicentre, multinational study. Eur J Cancer. 2012;48:196–201. doi: 10.1016/j.ejca.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 137.Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29:2357–2363. doi: 10.1200/JCO.2010.33.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lubner SJ, Mahoney MR, Kolesar JL, Loconte NK, Kim GP, Pitot HC, et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J Clin Oncol. 2010;28:3491–3497. doi: 10.1200/JCO.2010.28.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jensen LH, Lindebjerg J, Ploen J, Hansen TF, Jakobsen A. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann Oncol. 2012;23:2341–2346. doi: 10.1093/annonc/mds008. [DOI] [PubMed] [Google Scholar]