

Abstract
Alzheimer's disease (AD) is the most common form of dementia, with prevalence progressively increasing with aging. Pathological hallmarks of the disease include accumulation of amyloid β-protein (Aβ) peptides and neurofibrillary tangles in the brain associated with glial activation and synaptotoxicity. In addition, AD involves peripheral and brain endogenous inflammatory processes that appear to enhance disease progression. More than a decade ago a new therapeutic paradigm emerged for AD, namely the activation of the adaptive immune system directly against the self-peptide Aβ, aimed at lowering its accumulation in the brain. This was the first time that a brain peptide was used to vaccinate human subjects in a manner similar to classic viral or bacterial vaccines. The vaccination approach has taken several forms, from initially active to passive and then back to modified active vaccines. As the first two approaches to date failed to show sufficient efficacy, the last is presently being evaluated in ongoing clinical trials. The present review summarizes the immunogenic characteristics of Aβ in humans and mice and discusses past, present and future Aβ-based immunotherapeutic approaches for AD. We emphasize potential pathogenic and beneficial roles of CD4 T cells in light of the pathogenesis and the general decline in T-cell responsiveness evident in the disease.
Keywords: Alzheimer's disease, amyloid β-protein (Aβ), CD4 T cells, Aβ antibodies, immunotherapy
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly, characterized by progressive memory loss and cognitive decline. One of the primary pathological features of the disease, in addition to neurofibrillary tangles, dystrophic neurites and significant neuronal loss in affected brain regions, is the extracellular aggregation of the amyloid β-protein (Aβ) peptide in the brain.1–4 Amyloid-β is produced from the amyloid precursor protein (APP) following proteolytic cleavage by β- and γ-secretases. Mutations in the preseneline-1 gene (PS1), which encodes for a transmembrane protein that functions as part of the γ-secretase complex, are associated with increased production of Aβ42 over the less aggregative form Aβ40, and are considered among the primary causes of early-onset familial AD.5,6
A growing body of evidence demonstrates that Aβ plaques induce an inflammatory reaction in the brain,7–9 whereas oligomeric forms of Aβ exert synaptotoxicity.3,4,10 In addition, in recent years information has accumulated demonstrating the marked pathological effects of Aβ on brain vasculature, a phenomenon termed cerebral amyloid angiopathy, that causes vascular inflammation, brain haemorrhages, compromised perivascular drainage and altered blood flow.11–13 Inflammatory processes such as microgliosis, astrocytosis, dystrophic microglia, complement activation, cytokine elevation and acute-phase protein changes are thought to represent, at least in part, a response to the accumulation of Aβ in the vasculature and parenchyma of the brain. A compromised immune system associated with aging may substantially impact on these processes and lead to compromised brain function and neuronal repair processes, which enhance the progression of AD. The current review summarizes the existing knowledge regarding the characteristics of Aβ-reactive CD4 T cells in animal models and in humans, and discusses Aβ-based immunotherapeutic approaches for AD in the context of disease pathogenesis and immunosenescence.
Main body
Aβ autoimmunity in humans and mice
More than a decade ago a new concept emerged in the study of AD, namely eliciting adaptive immune responses to attenuate the accumulation of Aβ in the brain. This was the first time that a self peptide was introduced to the body as a vaccine, similar to classic vaccination approaches used against various pathogens. As this approach may have brought about the most promising therapeutic approach for AD, it also challenged our previous knowledge of autoimmunity, immune tolerance and brain–immune interactions.
Amyloid-β-specific immunotherapy can considerably reduce amyloid burden and improve cognitive functions in animal models of AD.14–21 Although pre-clinical studies have proved successful, an initial clinical trial of active Aβ vaccine (AN-1792 trial performed by Elan) was halted because of the development of severe inflammatory reactions in the brains of some vaccinated AD patients.22–24 The severe side-effects were attributed to the use of the full length of the Aβ peptide together with QS21, a very strong adjuvant, the combination of which presumably led to the occurrence of pathogenic T cells at the brain vasculature and parenchyma.22,23,25 Nevertheless, the study of Aβ-reactive T cells is key to unravelling their occurrence and characteristics in healthy humans as well as in patients with AD, and hence a key to designing safer immune-based approaches for AD therapy.
Although the general dogma would not anticipate the occurrence of effector Aβ-reactive CD4 T cells in the circulation of human subjects, not only were they detected in almost all individuals tested but significantly more elderly subjects and AD patients showed strong Aβ-reactive T-cell responses compared with middle-aged subjects.26 The Aβ T-cell responses were primarily HLA-DR-dependent, and the presented T-cell epitopes derived primarily from residues 15–42 of Aβ (see Table 1). About 20% of all the subjects were found to bear HLA-DR alleles, which either did not stimulate Aβ-reactive T-cell lines or induced only a mild response. The great variability of HLA-DR alleles in humans, which is associated with multiple Aβ T-cell epitopes,26,27 presumably reflects a great variability in the magnitude of T-cell activation in humans and therefore the variations in specific Aβ-antibody titres evoked in AD patients following Aβ42 vaccination.28,29
Table 1.
Amyloid-β (Aβ) T-cell epitopes and responsiveness in several MHC II and HLA genetic backgrounds
| Strain/MHC II or HLA | Aβ1–42 T-cell responsiveness | T-cell epitope within Aβ residues | References | |
|---|---|---|---|---|
| Mice | C57BL/6 / I-Ab | + | 16–30 | 30–32,113 |
| SJL / I-As | +++ | 10–24 | 30,31 | |
| BALB/c / I-Ad | ++ | 1–28 | 113 | |
| NOD / I-Ag7 | +++ | 10–24 | 30 | |
| Congenic mice | C57BL/6 / I-As | + | 10–24 | 31 |
| NOD / I-Ab | + | 16–30 | 30 | |
| Humanized mice | DR15 | +++ | 25–42 | 27 |
| DR4 | + ++ | 16–33 1–16, 1–28 | 27,32 | |
| DRB1*0101 | ++ | 1–28 | 113 | |
| DR3 | + | 1–16 | 32 | |
| DQ8 | + | 1–42 | 32 | |
| Human subjects | DRB1*0101/1301/1001 | ND | 15–35 | 27 |
| DRB1*0401/0404 | ND | 18–32 | 27 | |
| DRB1*1501 | ND | 25–42 | 27 |
ND, not determined.
In mice, Aβ-reactive T cells were analysed following Aβ1–42 immunization and re-stimulation with Aβ1–42 or with shorter Aβ peptides in vitro. In human subjects, Aβ T-cell epitopes were analysed in isolated peripheral blood mononuclear cells stimulated initially with Aβ1–42 and thereafter with 15-residue-long overlapping peptides between 1 and 42 residues of Aβ. T-cell responsiveness was measured by the magnitude of antigen-driven T-cell proliferation and cytokine production following immunization. In humanized mice bearing the DRB1 1501 and 0401 alleles, peptides between residues 25 and 42 and between residues 18 and 32 served as the dominant T-cell epitopes, as observed also for T-cell lines derived from human subjects with these HLA genetic backgrounds.26 Aβ42 immunization of humanized HLA-DR4 and HLA-DR3/DQ8 transgenic mice evoked Aβ-reactive T-cell responses which could be partially stimulated by Aβ1–16,32 and DRB1 0101 humanized mice elicited T-cell responses to an epitope between residues 1–28.113 Since overlapping peptides between residues 15 and 42 of Aβ were not used in these studies, it is unclear whether additional weak T-cell epitopes are located at the N-terminus of Aβ or whether a truncated portion of the epitope was presented to the T cells
Animal models allow one to more accurately investigate the contribution of an MHC class II genetic background to Aβ immunogenicity associated with the dominant epitope presented to T cells. They also allow a more efficient examination of the effect of various vaccination paradigms (i.e. route of administration and choice of adjuvant) on the dynamics and characteristics of the immune response elicited (i.e. antibody isotype and titres, and the profile of T-cell cytokines). In mice, Aβ immunogenicity markedly differs between strains; for example, Aβ is highly immunogenic in NOD and SJL mice, which have a dominant T-cell epitope between residues 10 and 24 of Aβ, whereas the peptide evokes only weak T-cell responses in C57BL/6 mice in which the epitope is between residues 16 and 30.30–32 NOD congenic mice bearing the I-Ab class II allele also fail to elicit a strong T-cell response, suggesting that the low immunogenicity of Aβ 16–30 in C57BL/6 mice is primarily a result of a low-affinity epitope selected by the I-Ab MHC class II. However, both C57BL/6 and B6.H-2s congenic mice, but not SJL mice, exhibit enhanced Aβ-specific T-cell responses upon the depletion of regulatory T (Treg) cells, suggesting that under certain genetic backgrounds, Treg cells can significantly affect Aβ immunogenicity.31 As no differences in thymic expression of APP are observed between C57BL/6 and SJL mice, the mechanism behind the effect of Treg cells on Aβ immunogenicity in C57BL/6 mice and the reason it is not effective in the more Aβ-immunogenic SJL mice are yet to be revealed.
Overall, T-cell epitopes markedly vary between mice and humans, with multiple epitopes located primarily between Aβ residues 10 and 30 and between 15 and 42, respectively. Both MHC class II alleles and Treg cells are crucial for determining the strength and phenotype of the adaptive immune response to Aβ following immunization. The fact that almost all human subjects possess Aβ-reactive T cells in their circulation and that these tend to expand with age and with the progression of AD raises a number of questions that are yet to be answered. (i) Are these Aβ-specific T cells positively selected in the thymus or do they simply ‘escape’ negative selection? (ii) Do Aβ-reactive T cells play a role in the progression of AD and, if so, how? (iii) Can they be externally stimulated to beneficially halt the progression of AD? Clearly, Aβ-reactive T cells are activated upon immunization and induce Aβ antibody production, however, one should consider the great variability in T-cell responses that can be anticipated in humans; in fact, this variability may perhaps be translated to personalized medicine.
Aβ-based vaccines
Since Aβ T-cell epitopes are located primarily between residues 10 and 42 of Aβ in mice and in humans, N-terminal portions of Aβ, namely fragments within residues 1–15 comprising dominant Aβ B-cell epitopes, have been used to generate active Aβ vaccines (Fig. 1).17,18,33–35 These peptides were conjugated to carriers such as albumin34,36 or the promiscuous foreign T-cell epitope PADRE37 and were shown to elicit effective Aβ antibody responses without stimulating an Aβ-specific T-cell response. These vaccination studies have led to pre-clinical studies using the N-terminal portion of Aβ presented on the surface of virus particles38 or liposomes,39 or administered as Aβ-coding DNA plasmids or viral vectors,35,40–42 and current clinical trials using such N-terminus Aβ peptides conjugated to diphtheria toxin or tetanus toxin are being carried out. The non-self carriers in these vaccines, although they avoid the T-cell response against Aβ, presumably evoke a strong T-cell response against the foreign epitopes and high titres of Aβ-specific antibodies (Fig. 1). In contrast to non-self carriers, our group generated a conjugate of Aβ1–15 and heat-shock protein 458 (hsp 458), a 17-amino-acid residue peptide derived from hsp 60.43 Compared with Aβ1–42, Aβ–hsp 60 immunization of humanized mice carrying the HLA-DR allele DRB1*DR1501 evoked a very mild T-cell response, evident by a significantly lower production of interferon-γ (IFN-γ) and interleukin-17 (IL-17) by draining lymph node-derived T cells.44 Notably, the mild T-cell response induced by Aβ–hsp 60 induced a gradual increase in specific Aβ antibody titres, which were sufficient for effective clearance of Aβ plaques from the brain of aged APP-transgenic mice. In addition to its function as a T-cell epitope,43,45,46 hsp 458 also activates the Toll-like receptor 4 pathway47 and so T-cell-independent antibody production evoked by Aβ–hsp 60 immunization is plausible.
Figure 1.
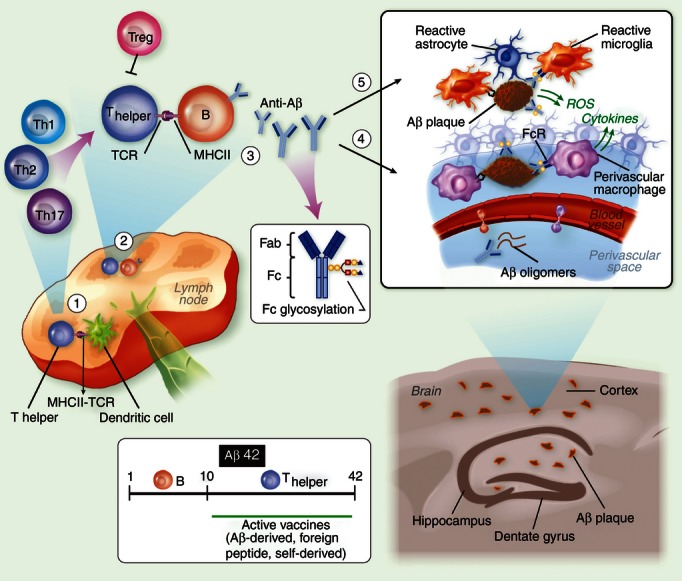
Amyloid β antibody-based immunotherapy of Alzheimer's disease (AD). (1) The immune response elicited by Aβ immunization begins in draining lymph nodes where dendritic cells present the MHC II-bound antigen to naive CD4 T cells. The antigen may be Aβ42 or a shorter peptide conjugated to a carrier of choice, such as heat-shock protein p458 or PADRE. (2) T cells then migrate to B-cell follicles, where they promote Aβ-specific B-cell proliferation and differentiation to plasma and memory B cells. Depending on the adjuvant and the carrier used, T cells polarize to either a pro-inflammatory or anti-inflammatory phenotype. In the case of Aβ or other self-derived carriers, T-cell responsiveness may be attenuated because of anergy or the presence of specific regulatory T cells. (3) Secreted antibodies may be of various isotypes and specificities to Aβ42 and with various glycoforms at the Fc portion, processes regulated by the cytokine milieu. (4) Antibodies target Aβ at the brain vasculature and enhance Aβ clearance from the brain. Clearance of soluble Aβ is accomplished through perivascular drainage or Fc receptor (FcR) -mediated uptake. Clearance of compact Aβ plaques is less effective, although the activation of perivascular macrophages via activating FcR may enhance their phagocytic function. Such a reaction, however, can facilitate an inflammatory reaction at the brain vasculature influenced by microglial/macrophage scavenger receptors114,115 and also by the Aβ B-cell epitope, the Fc glycosylation pattern and/or the type of Fc receptors (i.e. activating or inhibitory FcRs). The inflammatory reaction at the vasculature may also be influenced by the adjuvant and carrier of choice, a process that may enhance clearance on the one hand but promote brain inflammation and microhaemorrhages on the other. (5) Similar processes occur within the brain parenchyma once antibodies target Aβ plaques. As some monocytes infiltrate the brain and target Aβ plaques, the capacity of antibody-mediated clearance may increase.
Clinical trials using either Aβ42 active vaccination or anti-Aβ passive vaccination have so far failed to show treatment efficacy, so eliciting a beneficial adaptive immune response to Aβ appears to be more complicated than was originally thought. Indeed, clearance of Aβ plaques in mouse models of AD may be partially misleading because it may not accurately represent key pathological features of the disease. This could have several explanations. (i) Most animal models of AD are treated prophylactically (i.e. in a prevention mode) or following the initial Aβ deposition in the brain. They are rarely, if at all, conducted in ages and disease stages equivalent to human AD patients, in which immunity declines and brain inflammation is markedly enhanced. (ii) The increase in Aβ42/40 ratio in some mouse models of AD induces a more condensed form of plaques where the capacity of Aβ clearance in the brain is considerably reduced.48,49 This may represent a shift towards a fast-progressing form of AD where Aβ antibodies, either naturally occurring or generated following vaccination, are insufficient to promote a therapeutic effect. (iii) The inflammatory reaction at the vasculature and parenchyma in AD patients may be facilitated by the Aβ-specific antibodies depending on their titers, epitope specificity, the Fc glycosylation pattern or the type of Fc receptor.50–52 In addition, a robust expansion of Aβ-specific B cells occurs, which may lead to ectopically enhanced activation of pathogenic Aβ-specific T cells (Fig. 1). (iv) The loss of synapses and neurons, which leads to progressive cognitive decline throughout the course of AD, is moderate in most mouse models of the disease, so the impact of Aβ clearance on this key aspect of the disease is unclear. Stimulating an immune response that promotes Aβ clearance as well as neuronal repair (e.g. via cytokines and neurotrophic factors53–55), which may be administered in a prevention mode, may therefore be considered a future goal for AD immunotherapy. Factors such as the vaccine carriers (either derived from self or non-self proteins), the routes and timing of vaccine administration and the choice of adjuvants may substantially decrease some of the risks described above and therefore improve treatment efficacy.
Aβ-reactive CD4 T cells in brain inflammation and repair
Given the immunogenicity of Aβ as demonstrated in humans and mice, it is clear that Aβ-reactive T cells can be boosted to promote pathogenic autoimmunity. In the following section we discuss the molecular and cellular setting that drives the homing of Aβ T cells to the brain and whether such a process can be used to enhance neuronal repair mechanisms in the aging and diseased brain.
The role of T cells in the brain has been widely studied in recent years. Trafficking T cells to the brains of APP-transgenic mice over-expressing transforming growth factor-β or IL-1β did not cause cellular or behavioural abnormalities56,57 and brain-specific T cells have been shown to play beneficial roles in murine models of brain injury,58,59 amyotrophic lateral sclerosis60 and stroke.61 Such specific T cells, or the cytokines they produce, participate in numerous activities such as increasing the uptake of Aβ plaques,30,62,63 releasing regulatory cytokines,55 increasing the expression of neurotrophic factors,54,64,65 increasing the buffering capacity of glutamate66 and enhancing neurogenesis.53,54,67,68 Our group has recently demonstrated that Aβ-reactive T cells are able to effectively target Aβ plaques in the brains of APP-transgenic mice and enhance the phagocytic activity of adjacent microglia (see Fig. 2 and refs 30 and 62), at least partially via IFN-γ-induced TREM2 and SIRPβ1 expression, which were recently suggested as DAP12-associated phagocytic receptors on microglia.69,70 Amyloid-β may be presented to T cells via co-localized MHCIIhigh antigen-presenting cells, which either differentiate from brain-endogenous microglia or are recruited from the blood as a result of increased CCL2 expression. Interferon-γ emerges as a unique cytokine, which on one hand facilitates T-cell migration into and within the brain parenchyma,71–73 and on the other hand promotes immunoregulatory processes74–76 and neuronal repair in the brain.53,66,74,77–82 Provided that IFN-γ signals to all neural populations, further research is required to determine how IFN-γ orchestrates its various effects in the brain. Clearly, the overall amounts of IFN-γ in the brain are crucial to shift its function from devastating at high levels83 to beneficial at low levels.53,74,78,80 Additional cytokines such as IL-10 and transforming growth factor-β, together with a profile of chemokines and neurotrophic factors secreted by the T cells, may prove therapeutic for the AD brain.
Figure 2.
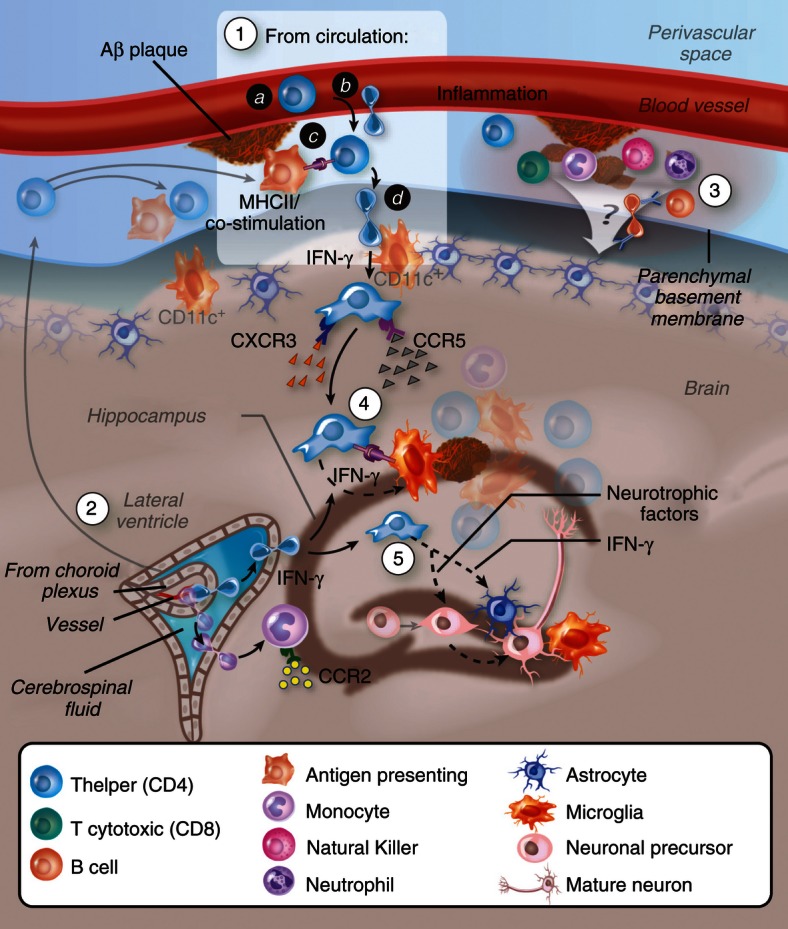
Migration of Aβ-specific T cells towards Aβ plaques in the brain parenchyma. (1) T cells may undergo activation following Aβ immunization or following drainage of Aβ or antigen-presenting cells (APCs) that carry Aβ to peripheral lymph nodes. Aβ-reactive helper T (Th) cells adhere and transmigrate into the perivascular space of Aβ-deposited vasculature in the brain (a, b). To cross the glia limitans, Th cells need to be re-stimulated by dendritic cells or, possibly, by other competent APCs located at the perivascular space and/or juxtavascular with processes sent into the perivascular space.116 Similar T-cell infiltration processes may occur at the choroid plexus (2), and/or the leptomeninges85,117,118 followed by their dissemination in the central nervous system subarachnoid space. Low levels of interferon-γ (IFN-γ) promote the infiltration process. Adhesion molecules such as P-selectin, vascular cell adhesion molecule-1 or intercellular adhesion molecule 1 (interacting with P-selectin glycoprotein ligand-1, integrin α4 and lymphocyte function-associated antigen-1, respectively, on the T cells) and chemokine signalling (such as via CCR5 and CXCR3) play a key role in mediating the extravasation of the T cells through the blood–brain barrier (BBB) or the blood–cerebrospinal fluid barrier. (3) Leucocytes accumulate at the subarachnoid and perivascular spaces and may impact on the overall inflammatory reaction at both the vasculature and parenchyma. (4) Once Aβ Th cells cross the glia limitans they migrate and accumulate around Aβ plaques, possibly interacting with APCs (i.e. microglia, or peripheral monocytes or dendritic cells recruited towards CCL2) that present Aβ T-cell epitopes. Cytokines such as IFN-γ are secreted by the T cells and facilitate Aβ clearance either by brain endogenous microglia or by infiltrating microglia-like cells. (5) T cells secreting IFN-γ and/or neurotrophic factors stimulate neural precursor cell proliferation and differentiation.
The specific mechanisms underlying the migration, activation and survival of the T cells within the brain parenchyma are yet to be identified. The model illustrated in Fig. 2 demonstrates that following Aβ immunization, Aβ-specific T cells target the brain vasculature in which Aβ is deposited. Expression of IFN-γ in the brain of a mouse AD model in limited amounts, which cause no spontaneous infiltration of bone marrow-derived cells, abnormal glial activation or neurological deficits,53,84 is required for the migration of T cells within the brain parenchyma.30,85 Three conditions can therefore promote Aβ-specific T-cell entry to the brain parenchyma: (i) deposition of Aβ at the brain vasculature, compromising the blood–brain barrier and inducing a local inflammatory reaction86–88 (ii) the stimulation of Aβ-specific T cells initially in the lymph node and then by perivascular and leptomeningial dendritic cells in the brain89–92–94 and (iii) low brain levels of IFN-γ, which is perhaps a master regulator of T-cell adhesion, antigen presentation, chemokine expression and signalling and T-cell migration.
Previous findings have primarily implicated brain-specific CD4 T cells in the pathology of experimental autoimmune encephalomyelitis, a model for multiple sclerosis in which myelin-specific T cells penetrate the central nervous system and promote axonal demyelination.91,95–98 Although not inducing an autoimmune disease, the lymphocytic reaction of both B and T cells to Aβ is potentially pathogenic because of the risks of meningoencephalitis,23 entry of cytotoxic CD8 T cells into the brain, strong pro-inflammatory cytokine profile of the CD4 T cells99 and brain haemorrhages caused by Aβ antibodies.100–102 However, if the pathogenic capacity of Aβ-specific T cells can be neutralized (for instance by stimulating a non-pathogenic cytokine profile) these T cells may play a beneficial role in promoting both Aβ clearance and neuronal repair with minimal risk of adverse side-effects.
In summary, it is commonly accepted that effector and regulatory functions of lymphocytes are altered with aging103,104 and that further immune manifestations accompany the progression of AD.7–9,105,106 Such alterations presumably increase the severity of infectious diseases and chronic inflammation and are reflected in the brain as increased levels of pro-inflammatory cytokines such as IL-1β, tumour necrosis factor-α and IL-6, which enhance neurotoxicity and may impair key functions of microglia in neuronal function and repair.107–109 Most immune intervention approaches, although performed in mouse models of AD, do not fully address the aforementioned immune alterations occurring with aging and with the progression of AD, which may significantly impact the outcome of treatment. Future studies of immunotherapy may therefore consider the following approaches, separately or in combination with the Aβ antibody treatment: (i) improving immunity through direct immune interventions (e.g. balancing key cytokines and chemokines)110,111 and through indirect approaches such as exercise, appropriate nutrition and stress management, which may significantly contribute to higher immune potency and regulation; (ii) effectively blocking prominent inflammatory cascades underlying auto-inflammation, such as those mediated by tumour necrosis factor-α or IL-1; and (iii) cell therapy (either with monocytes, dendritic cells or CD4 T cells directed at Aβ or at other brain antigens) to facilitate the function of immune cells within the brain. Such immune interventions may reduce immune-mediated neuronal damage overall and enhance neuronal repair,110,112 aspects that may be crucial to achieve treatment efficacy in patients with AD.
Disclosures
The authors declare that they have no conflict of interests.
References
- 1.Rosenmann H, Grigoriadis N, Eldar-Levy H, et al. A novel transgenic mouse expressing double mutant tau driven by its natural promoter exhibits tauopathy characteristics. Exp Neurol. 2008;212:71–84. doi: 10.1016/j.expneurol.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:11039–41. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Walsh DM, Selkoe DJ. A β oligomers – a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 5.Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 6.Moehlmann T, Winkler E, Xia X, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ 42 production. Proc Natl Acad Sci USA. 2002;99:8025–30. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGeer EG, Klegeris A, McGeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(Suppl. 1):94–7. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Vom Berg J, Prokop S, Miller KR, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–9. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 10.Shankar GM, Li S, Mehta TH, et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herzig MC, Winkler DT, Burgermeister P, et al. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–60. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 12.Meyer EP, Ulmann-Schuler A, Staufenbiel M, Krucker T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc Natl Acad Sci USA. 2008;105:3587–92. doi: 10.1073/pnas.0709788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 14.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–12. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer β-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452–5. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 17.Lemere CA, Maron R, Spooner ET, et al. Nasal A β treatment induces anti-A β antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann N Y Acad Sci. 2000;920:328–31. doi: 10.1111/j.1749-6632.2000.tb06943.x. [DOI] [PubMed] [Google Scholar]
- 18.Weiner HL, Lemere CA, Maron R, et al. Nasal administration of amyloid-β peptide decreases cerebral amyloid burden in a mouse model of Alzheimer's disease. Ann Neurol. 2000;48:567–79. [PubMed] [Google Scholar]
- 19.Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Aβ1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging. 2001;22:721–7. doi: 10.1016/s0197-4580(01)00245-7. [DOI] [PubMed] [Google Scholar]
- 20.Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001;159:439–47. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 23.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 24.Schenk D. Amyloid-β immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–8. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 25.Gilman S, Koller M, Black RS, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 26.Monsonego A, Zota V, Karni A, et al. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zota V, Nemirovsky A, Baron R, Fisher Y, Selkoe DJ, Altmann DM, Weiner HL, Monsonego A. HLA-DR alleles in amyloid β-peptide autoimmunity: a highly immunogenic role for the DRB1*1501 allele. J Immunol. 2009;183:3522–30. doi: 10.4049/jimmunol.0900620. [DOI] [PubMed] [Google Scholar]
- 28.Hock C, Konietzko U, Papassotiropoulos A, et al. Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–5. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 29.Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 30.Monsonego A, Imitola J, Petrovic S, et al. Aβ-induced meningoencephalitis is IFN-γ-dependent and is associated with T cell-dependent clearance of Aβ in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:5048–53. doi: 10.1073/pnas.0506209103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toly-Ndour C, Lui G, Nunes MM, Bruley-Rosset M, Aucouturier P, Dorothee G. MHC-independent genetic factors control the magnitude of CD4+ T cell responses to amyloid-β peptide in mice through regulatory T cell-mediated inhibition. J Immunol. 2011;187:4492–500. doi: 10.4049/jimmunol.1003953. [DOI] [PubMed] [Google Scholar]
- 32.Das P, Chapoval S, Howard V, David CS, Golde TE. Immune responses against Aβ1–42 in HLA class II transgenic mice: implications for Aβ1–42 immune-mediated therapies. Neurobiol Aging. 2003;24:969–76. doi: 10.1016/s0197-4580(03)00036-8. [DOI] [PubMed] [Google Scholar]
- 33.Lemere CA, Maron R, Selkoe DJ, Weiner HL. Nasal vaccination with β-amyloid peptide for the treatment of Alzheimer's disease. DNA Cell Biol. 2001;20:705–11. doi: 10.1089/10445490152717569. [DOI] [PubMed] [Google Scholar]
- 34.Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsiveness to amyloid β-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:10273–8. doi: 10.1073/pnas.191118298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nat Rev Neurol. 2010;6:108–19. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bard F, Barbour R, Cannon C, et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci USA. 2003;100:2023–8. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghochikyan A, Mkrtichyan M, Petrushina I, Movsesyan N, Karapetyan A, Cribbs DH, Agadjanyan MG. Prototype Alzheimer's disease epitope vaccine induced strong Th2-type anti-Aβ antibody response with Alum to Quil A adjuvant switch. Vaccine. 2006;24:2275–82. doi: 10.1016/j.vaccine.2005.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zurbriggen R, Amacker M, Kammer AR, Westerfeld N, Borghgraef P, Van Leuven F, Van der Auwera I, Wera S. Virosome-based active immunization targets soluble amyloid species rather than plaques in a transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 2005;27:157–66. doi: 10.1385/JMN:27:2:157. [DOI] [PubMed] [Google Scholar]
- 39.Muhs A, Hickman DT, Pihlgren M, et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Natl Acad Sci USA. 2007;104:9810–15. doi: 10.1073/pnas.0703137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okura Y, Miyakoshi A, Kohyama K, Park IK, Staufenbiel M, Matsumoto Y. Nonviral Aβ DNA vaccine therapy against Alzheimer's disease: long-term effects and safety. Proc Natl Acad Sci USA. 2006;103:9619–24. doi: 10.1073/pnas.0600966103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Movsesyan N, Ghochikyan A, Mkrtichyan M, et al. Reducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine – a novel immunotherapeutic strategy. PLoS ONE. 2008;3:e2124. doi: 10.1371/journal.pone.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220:95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- 43.Amir-Kroll H, Nussbaum G, Cohen IR. Proteins and their derived peptides as carriers in a conjugate vaccine for Streptococcus pneumoniae: self-heat shock protein 60 and tetanus toxoid. J Immunol. 2003;170:6165–71. doi: 10.4049/jimmunol.170.12.6165. [DOI] [PubMed] [Google Scholar]
- 44.Nemirovsky A, Fisher Y, Baron R, Cohen IR, Monsonego A. Amyloid β-HSP60 peptide conjugate vaccine treats a mouse model of Alzheimer's disease. Vaccine. 2011;29:4043–50. doi: 10.1016/j.vaccine.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 45.Konen-Waisman S, Cohen A, Fridkin M, Cohen IR. Self heat-shock protein (hsp60) peptide serves in a conjugate vaccine against a lethal pneumococcal infection. J Infect Dis. 1999;179:403–13. doi: 10.1086/314590. [DOI] [PubMed] [Google Scholar]
- 46.Quintana FJ, Cohen IR. HSP60 speaks to the immune system in many voices. Novartis Found Symp. 2008;291:101–111. doi: 10.1002/9780470754030.ch8. discussion 11-4, 37-40. [DOI] [PubMed] [Google Scholar]
- 47.Cohen N, Stolarsky-Bennun M, Amir-Kroll H, et al. Pneumococcal capsular polysaccharide is immunogenic when present on the surface of macrophages and dendritic cells: TLR4 signaling induced by a conjugate vaccine or by lipopolysaccharide is conducive. J Immunol. 2008;180:2409–18. doi: 10.4049/jimmunol.180.4.2409. [DOI] [PubMed] [Google Scholar]
- 48.Nemirovsky A, Shapiro J, Baron R, Kompaniets A, Monsonego A. Active Aβ vaccination fails to enhance amyloid clearance in a mouse model of Alzheimer's disease with Aβ42-driven pathology. J Neuroimmunol. 2012;247:95–9. doi: 10.1016/j.jneuroim.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 49.Wang A, Das P, Switzer RC, 3rd, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-β immunotherapy. J Neurosci. 2011;31:4124–36. doi: 10.1523/JNEUROSCI.5077-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adolfsson O, Pihlgren M, Toni N, et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci. 2012;32:9677–89. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edri-Brami M, Rosental B, Hayoun D, et al. Glycans in sera of amyotrophic lateral sclerosis patients and their role in killing neuronal cells. PLoS ONE. 2012;7:e35772. doi: 10.1371/journal.pone.0035772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lunnon K, Teeling JL, Tutt AL, Cragg MS, Glennie MJ, Perry VH. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J Immunol. 2011;186:7215–24. doi: 10.4049/jimmunol.0903833. [DOI] [PubMed] [Google Scholar]
- 53.Baron R, Nemirovsky A, Harpaz I, Cohen H, Owens T, Monsonego A. IFN-γ enhances neurogenesis in wild-type mice and in a mouse model of Alzheimer's disease. FASEB J. 2008;22:2843–52. doi: 10.1096/fj.08-105866. [DOI] [PubMed] [Google Scholar]
- 54.Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci. 2005;233:125–32. doi: 10.1016/j.jns.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 56.Buckwalter MS, Coleman BS, Buttini M, Barbour R, Schenk D, Games D, Seubert P, Wyss-Coray T. Increased T cell recruitment to the CNS after amyloid β 1–42 immunization in Alzheimer's mice overproducing transforming growth factor-β 1. J Neurosci. 2006;26:11437–41. doi: 10.1523/JNEUROSCI.2436-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1β expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood–brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauben E, Butovsky O, Nevo U, et al. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000;20:6421–30. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 60.Holmoy T. T cells in amyotrophic lateral sclerosis. Eur J Neurol. 2008;15:360–6. doi: 10.1111/j.1468-1331.2008.02065.x. [DOI] [PubMed] [Google Scholar]
- 61.Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, Weiner HL. Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol. 2003;171:6549–55. doi: 10.4049/jimmunol.171.12.6549. [DOI] [PubMed] [Google Scholar]
- 62.Fisher Y, Nemirovsky A, Baron R, Monsonego A. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer's disease. PLoS ONE. 2010;5:e10830. doi: 10.1371/journal.pone.0010830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103:11784–9. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aharoni R, Arnon R, Eilam R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J Neurosci. 2005;25:8217–28. doi: 10.1523/JNEUROSCI.1859-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. Neurol Sci. 2006;27(Suppl. 1):S1–S7. doi: 10.1007/s10072-006-0537-7. [DOI] [PubMed] [Google Scholar]
- 66.Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M. Protective autoimmunity: interferon-γ enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- 67.Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T, Kempermann G. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. 2009;182:3979–84. doi: 10.4049/jimmunol.0801218. [DOI] [PubMed] [Google Scholar]
- 68.Mastrangelo MA, Sudol KL, Narrow WC, Bowers WJ. Interferon-γ differentially affects Alzheimer's disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am J Pathol. 2009;175:2076–88. doi: 10.2353/ajpath.2009.090059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaikwad S, Larionov S, Wang Y, Dannenberg H, Matozaki T, Monsonego A, Thal DR, Neumann H. Signal regulatory protein-β1: a microglial modulator of phagocytosis in Alzheimer's disease. Am J Pathol. 2009;175:2528–39. doi: 10.2353/ajpath.2009.090147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tran EH, Prince EN, Owens T. IFN-γ shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–68. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- 72.Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-γ determine lesion localization patterns during EAE pathogenesis. J Exp Med. 2008;205:2633–42. doi: 10.1084/jem.20080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–15. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, Popko B. Interferon-γ–oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–24. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005;19:1347–9. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 76.Wheeler RD, Zehntner SP, Kelly LM, Bourbonniere L, Owens T. Elevated interferon γ expression in the central nervous system of tumour necrosis factor receptor 1-deficient mice with experimental autoimmune encephalomyelitis. Immunology. 2006;118:527–38. doi: 10.1111/j.1365-2567.2006.02395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Butovsky O, Bukshpan S, Kunis G, Jung S, Schwartz M. Microglia can be induced by IFN-γ or IL-4 to express neural or dendritic-like markers. Mol Cell Neurosci. 2007;35:490–500. doi: 10.1016/j.mcn.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 78.Gao X, Gillig TA, Ye P, D'Ercole AJ, Matsushima GK, Popko B. Interferon-γ protects against cuprizone-induced demyelination. Mol Cell Neurosci. 2000;16:338–49. doi: 10.1006/mcne.2000.0883. [DOI] [PubMed] [Google Scholar]
- 79.Kim SJ, Son TG, Kim K, Park HR, Mattson MP, Lee J. Interferon-γ promotes differentiation of neural progenitor cells via the JNK pathway. Neurochem Res. 2007;32:1399–1406. doi: 10.1007/s11064-007-9323-z. [DOI] [PubMed] [Google Scholar]
- 80.Lee J, Kim SJ, Son TG, Chan SL, Mattson MP. Interferon-γ is up-regulated in the hippocampus in response to intermittent fasting and protects hippocampal neurons against excitotoxicity. J Neurosci Res. 2006;83:1552–7. doi: 10.1002/jnr.20831. [DOI] [PubMed] [Google Scholar]
- 81.Song JH, Wang CX, Song DK, Wang P, Shuaib A, Hao C. Interferon γ induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J Biol Chem. 2005;280:12896–901. doi: 10.1074/jbc.M412139200. [DOI] [PubMed] [Google Scholar]
- 82.Wong G, Goldshmit Y, Turnley AM. Interferon-γ but not TNF α promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp Neurol. 2004;187:171–7. doi: 10.1016/j.expneurol.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 83.Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B. Targeted CNS expression of interferon-γ in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci. 1996;7:354–70. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]
- 84.Renno T, Taupin V, Bourbonniere L, et al. Interferon-γ in progression to chronic demyelination and neurological deficit following acute EAE. Mol Cell Neurosci. 1998;12:376–89. doi: 10.1006/mcne.1998.0725. [DOI] [PubMed] [Google Scholar]
- 85.Fisher Y, Nemirovsky A, Baron R, Monsonego A. Dendritic cells regulate amyloid-β-specific T-cell entry into the brain: the role of perivascular amyloid-β. J Alzheimers Dis. 2011;27:99–111. doi: 10.3233/JAD-2011-102034. [DOI] [PubMed] [Google Scholar]
- 86.Boche D, Zotova E, Weller RO, et al. Consequence of Aβ immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- 87.Carare RO, Bernardes-Silva M, Newman TA, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–44. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 88.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18:253–66. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Archambault AS, Sim J, Gimenez MA, Russell JH. Defining antigen-dependent stages of T cell migration from the blood to the central nervous system parenchyma. Eur J Immunol. 2005;35:1076–85. doi: 10.1002/eji.200425864. [DOI] [PubMed] [Google Scholar]
- 91.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–34. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 92.Bartholomaus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462:94–8. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- 93.Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol. 2012;33:579–89. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 94.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–81. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 95.de Rosbo NK, Ben-Nun A. T-cell responses to myelin antigens in multiple sclerosis; relevance of the predominant autoimmune reactivity to myelin oligodendrocyte glycoprotein. J Autoimmun. 1998;11:287–99. doi: 10.1006/jaut.1998.0202. [DOI] [PubMed] [Google Scholar]
- 96.Waldner H, Whitters MJ, Sobel RA, Collins M, Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc Natl Acad Sci USA. 2000;97:3412–7. doi: 10.1073/pnas.97.7.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ercolini AM, Miller SD. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J Immunol. 2006;176:3293–8. doi: 10.4049/jimmunol.176.6.3293. [DOI] [PubMed] [Google Scholar]
- 98.Krishnamoorthy G, Saxena A, Mars LT, et al. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med. 2009;15:626–32. doi: 10.1038/nm.1975. [DOI] [PubMed] [Google Scholar]
- 99.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by TH1 and TH17 cells. Nat Med. 2008;14:337–42. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–34. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 101.Burbach GJ, Vlachos A, Ghebremedhin E, Del Turco D, Coomaraswamy J, Staufenbiel M, Jucker M, Deller T. Vessel ultrastructure in APP23 transgenic mice after passive anti-Aβ immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007;28:202–12. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 102.Racke MM, Boone LI, Hepburn DL, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–36. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maue AC, Yager EJ, Swain SL, Woodland DL, Blackman MA, Haynes L. T-cell immunosenescence: lessons learned from mouse models of aging. Trends Immunol. 2009;30:301–5. doi: 10.1016/j.it.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Panda A, Arjona A, Sapey E, Bai F, Fikrig E, Montgomery RR, Lord JM, Shaw AC. Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol. 2009;30:325–33. doi: 10.1016/j.it.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rocha NP, Teixeira AL, Coelho FM, et al. Peripheral blood mono-nuclear cells derived from Alzheimer's disease patients show elevated baseline levels of secreted cytokines but resist stimulation with β-amyloid peptide. Mol Cell Neurosci. 2012;49:77–84. doi: 10.1016/j.mcn.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 107.Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2012;33:195. doi: 10.1016/j.neurobiolaging.2010.05.008. e1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45:208–12. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 109.Tremblay ME, Zettel ML, Ison JR, Allen PD, Majewska AK. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia. 2012;60:541–58. doi: 10.1002/glia.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Villeda SA. The circulatory systemic environment as a modulator of neurogenesis and brain aging. Autoimmun Rev. 2012 doi: 10.1016/j.autrev.2012.10.014. doi: 10.1016/j.autrev.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 111.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–22. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mirochnic S, Wolf S, Staufenbiel M, Kempermann G. Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus. 2009;19:1008–18. doi: 10.1002/hipo.20560. [DOI] [PubMed] [Google Scholar]
- 113.Kutzler MA, Cao C, Bai Y, et al. Mapping of immune responses following wild-type and mutant AB42 plasmid or peptide vaccination in different mouse haplotypes and HLA Class II transgenic mice. Vaccine. 2006;24:4630–9. doi: 10.1016/j.vaccine.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 114.Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimers Dis. 2012;2012:489456. doi: 10.1155/2012/489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Prodinger C, Bunse J, Kruger M, et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. 2011;121:445–58. doi: 10.1007/s00401-010-0774-y. [DOI] [PubMed] [Google Scholar]
- 117.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–35. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 118.Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. 2013;13:206–18. doi: 10.1038/nri3391. [DOI] [PubMed] [Google Scholar]