

Abstract
Apoptosis of macrophages has been reported as an effective host strategy to control the growth of intracellular pathogens, including pathogenic mycobacteria. Tumour necrosis factor-α (TNF-α) plays an important role in the modulation of apoptosis of infected macrophages. It exerts its biological activities via two distinct cell surface receptors, TNFR1 and TNFR2, whose extracellular domain can be released by proteolysis forming soluble TNF receptors (sTNFR1 and sTNFR2). The signalling through TNFR1 initiates the majority of the biological functions of TNF-α, leading to either cell death or survival whereas TNFR2 mediates primarily survival signals. Here, the expression of TNF-α receptors and the apoptosis of alveolar macrophages were investigated during the early phase of infection with attenuated and virulent mycobacteria in mice. A significant increase of apoptosis and high expression of TNFR1 were observed in alveolar macrophages at 3 and 7 days after infection with attenuated Mycobacterium bovis but only on day 7 in infection with the virulent M. bovis. Low surface expression of TNFR1 and increased levels of sTNFR1 on day 3 after infection by the virulent strain were associated with reduced rates of apoptotic macrophages. In addition, a significant reduction in apoptosis of alveolar macrophages was observed in TNFR1−/− mice at day 3 after bacillus Calmette–Guérin infection. These results suggest a potential role for TNFR1 in mycobacteria-induced alveolar macrophage apoptosis in vivo. In this scenario, shedding of TNFR1 seems to contribute to the modulation of macrophage apoptosis in a strain-dependent manner.
Keywords: apoptosis, macrophage, Mycobacterium bovis, tumour necrosis factor receptor 1, tumour necrosis factor receptor 2
Introduction
Tuberculosis (TB) is one of the oldest infectious diseases affecting humanity, and although it has been a target of battle for more than a 100 years, it still represents a serious health problem, mainly in less developed countries where high rates of disease are reported annually.1,2 The bacilli that cause TB are grouped in the Mycobacterium tuberculosis complex, which is a group of closely related mycobacteria including M. tuberculosis and Mycobacterium bovis. Mycobacterium tuberculosis is the main causative agent of TB in humans.3 However, some studies of TB have shown that M. bovis is considerably more virulent than M. tuberculosis for mice because of its ability to cause progressive pathology in the lungs and the time taken for it to kill its host.4,5 This higher virulence of M. bovis, characterized by increased intracellular survival and growth, may be related to the bacterial capacity to modulate macrophage activity.6
Pathogenic mycobacteria are highly adapted pathogens that have developed several strategies to ensure their permanence and replication within macrophages that constitute their main host cell.3,7 Apoptosis of infected macrophages has been seen as an alternative strategy of the host to eliminate the environment of protection and replication of mycobacteria. Besides depriving the pathogen of its preferred growth niche, this type of cell death prevents the spread of infection by sequestering the mycobacteria within apoptotic bodies, and also stimulates the adaptive immune response by mechanisms of cross-presentation.8–11 However, in vitro and in vivo studies indicate that virulent strains of mycobacteria, in contrast to attenuated strains, may actively modulate the apoptosis of macrophages depending on the load of bacilli inside the cell.12,13 At low loads, virulent strains induce less apoptosis compared with attenuated strains, hence preserving a protective intracellular environment for bacterial growth. But, when the burden of the bacilli within the cell becomes high, which can happen after a period of replication, virulent strains also induce apoptosis in macrophages and this may rapidly progress to necrosis, allowing the bacillus to escape from the macrophage.13,14 This suggests that the inhibition or induction of macrophage apoptosis promoted by virulent strains of mycobacteria may reflect their need for growth and exit from the host cell during the initial stage of replication and spread in the host.
Modulation of macrophage apoptosis during infection with mycobacteria is a complex process that appears to involve multiple factors, among which, tumour necrosis factor-α (TNF-α) plays a critical role.15,16 Tumour necrosis factor-α is a pleiotropic cytokine that exerts its actions through two distinct cell surface receptors, the 55 000 molecular weight receptor TNFR1 (or TNFR p55) and the 75 000 molecular weight receptor TNFR2 (or TNFR p75), which are expressed in a variety of cells. Although the two receptors share significant homology in their extracellular domain, their cytoplasmic regions show considerable differences. TNFR1 contains a death domain in its cytoplasmic region whereas TNFR2 has lost this domain, hence TNFR1 can induce cell survival signals as well as cell death signals, whereas TNFR2 primarily induces survival signals.17,18 However, some studies suggest that TNFR2 might potentiate the death signal mediated by TNFR1.19,20 TNFR1 and TNFR2 are initially synthesized as membrane proteins whose extracellular domain can be proteolytically cleaved in a process termed ectodomain shedding. TNFR shedding results in the release of soluble TNF receptors (sTNFR1 and sTNFR2), which can compete with the membrane TNFRs for binding to TNF-α, so inhibiting its activity. In addition, the decrease in the number of membrane receptor molecules as a result of receptor shedding may transiently desensitize cells to TNF-α action.21–23
Tumour necrosis factor-α participates in several stages of the anti-mycobacterial immune response. It is involved in macrophage activation, enhances cytokine and chemokine production favouring recruitment of inflammatory cells to the site of infection, and mediates apoptosis in macrophages.24–26 The TNF-α signalling by TNFR1 seems to be essential for host resistance to infection by mycobacteria. Mice genetically deficient for TNFR1 (TNFR1−/−) are extremely susceptible to infection with pathogenic mycobacteria and die within 4–5 weeks.27,28 Although some studies have demonstrated the importance of TNFR1 during mycobacterial infection, the role of TNFR2 in antimycobacterial immunity is still unclear.29 However, it has been suggested that virulent mycobacteria evade macrophage apoptosis by inducing the release of soluble TNFR2, which forms complexes with TNF-α and decreases its activity.30
Signalling via TNFRs plays an important role in determining the outcome of infections caused by several pathogens, including mycobacteria.31 It has been shown that some intracellular pathogens have developed mechanisms to regulate the interaction between TNF-α and its receptors, thereby inhibiting antimicrobial functions of TNF-α.32,33 We therefore hypothesized that virulent mycobacteria could modulate macrophage apoptosis by interfering in the expression of TNFRs. In the present study, the relationship between the expression of TNFRs and apoptosis of alveolar macrophages during early infection with virulent and attenuated mycobacteria in mice was investigated.
Materials and methods
Animals
C57BL/6 male mice at 8–10 weeks old were obtained from Federal University of Minas Gerais, Belo Horizonte. The TNFR1-deficient mice (TNFR1−/−) were kindly provided by Dr João Santana da Silva (University of São Paulo, Ribeirão Preto) and were maintained in pathogen-free facilities. Mice were housed under barrier conditions in microisolator cages and were allowed free access to sterile chow and water. All procedures were in accordance with the principles of the Brazilian Code for the Use of Laboratory Animals and were approved by the Ethics Committee on the use of laboratory animals of the Federal University of Juiz de Fora.
Mycobacteria and infection
Mycobacterium bovis bacillus Calmette–Guérin (BCG; Moreau substrain) was obtained from Ataulpho de Paiva Foundation, Rio de Janeiro. Wild-type M. bovis (ATCC 19274) was kindly provided by the National Institute of Quality Control in Health – Oswaldo Cruz Foundation, Rio de Janeiro. The attenuated and virulent strains of M. bovis were cultured in Lowenstein–Jensen (L–J) medium for 21 days. Colonies were harvested at mid-log phase and stirred vigorously with sterile glass beads for 5 min and resuspended in PBS. To further disrupt clumps, the bacterial suspensions were sonicated (20 watts for 5 seconds). The viable counts of bacteria were determined by serial dilutions and plating into six-well plates containing L–J medium.
To achieve intratracheal infection, mice were anaesthetized intraperitoneally with 0.2 ml of an anaesthetic solution containing 0.9% NaCl, 2% xylazine and 5% ketamine. The trachea was exposed via a small midline incision, and 106 viable bacteria in 50 μl PBS were injected using a microsyringe. Control mice were injected with 50 μl PBS. The incision was then sutured with sterile silk, and the mice were kept in a vertical position until the effect of the anaesthestic had passed.
Bronchoalveolar lavage
On day 3 and day 7 post-infection, mice (n = 6/group) were killed by overdose of anaesthesia. Bronchoalveolar lavage fluid was collected from control and infected mice. Briefly, a small incision was made in the trachea and the flexible part of an 18-gauge catheter attached to a 1-ml syringe was inserted into the trachea and 1 ml PBS was slowly injected into the lungs and then withdrawn. This procedure was repeated five to seven times. Cells recovered by bronchoalveolar lavage (BAL) were counted and viabilities were ascertained by Trypan blue dye exclusion.
Flow cytometric analysis of cell surface markers and apoptosis
For multicolour FACS analysis, cells obtained in BAL were incubated with specific monoclonal antibodies labelled with FITC, phycoerythrin or allophycocyanin at 4° for 30 min in the dark. After washing with PBS containing 0.1% sodium azide (Sigma-Aldrich, St. Louis, MO), cells were incubated with anti-CD11b (M1/70) and anti-CD11c (HL3) monoclonal antibodies purchased from BD Pharmingen (San Diego, CA), and anti-TNFR1 (3H3104) and anti-TNFR2 (TR75-89) from Santa Cruz Biotechnology (Santa Cruz, CA). Alveolar macrophages were gated as described by Gonzalez-Juarrero et al.34 according to their expression of CD11b and CD11c. Surface expression of TNFR1 and TNFR2 in CD11b− CD11c+/high alveolar macrophages was analysed. Relative fluorescence intensities were recorded from a total of 10 000 events. For analysis of apoptotic alveolar macrophages, stained cells were washed in annexin buffer (10 × buffer of 0.1 m HEPES, 1.4 m NaCl, and 25 mm CaCl2 diluted to 1 × in dH2O) and resuspended in 100 μl annexin buffer with 5 μl/well annexin V-allophycocyanin (BD Pharmingen). After 10-min incubation at room temperature in the dark, 5 μl 7-aminoactinomycin D (7-AAD; BD Pharmingen) was added and incubated for an additional 5 min. Cells were washed and resuspended in annexin buffer and the percentages of apoptotic alveolar macrophages (annexinV+ 7-AAD−) were determined. Cell acquisition was performed with a dual-laser flow cytometer, FACSCalibur (Becton Dickinson, San Jose, CA). Ten thousand events were acquired per sample and analysed using cell quest software (Becton Dickinson).
Soluble TNFR1 and TNFR2 ELISA
Bronchoalveolar lavage fluid obtained from infected and uninfected control mice at different time-points was centrifuged and the supernatant was tested for the presence of sTNFR1 and sTNFR2 by ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
TNF-α measurement
A 100-mg sample of the right lung from infected and uninfected control mice was homogenized using 1 ml of 0.05% Tween 20-PBS containing anti-proteases (0.1 mm PMSF, 0.1 mm benzethonium chloride, 10 mm EDTA and 20 kallikrein inhibitor (KI) units aprotinin A). The samples were then centrifuged for 10 min at 3000 g and the supernatants were collected and frozen at −70° until further use. The levels of TNF-α in lung homogenate supernatants were measured by ELISA using commercially available antibodies, following the instructions supplied by the manufacturer (BD Biosciences Pharmingen, San Diego, CA). The reading was made in a microplate reader (Spectramax 190; Molecular Devices, Sunnyvale, CA) at 450 nm. The amount of cytokine was calculated from the standard curve, for the different concentrations of the recombinant cytokine.
Determination of bacterial growth
The cytospin slides of pulmonary cells obtained in BAL were stained for acid-fast bacilli using the Ziehl–Neelsen stain. The number of macrophages containing M. bovis in a total of 100 macrophages per sample was recorded, and the number of bacilli per infected macrophage was scored at 1–10 bacilli per macrophage or > 10 bacilli per macrophage. The percentage of alveolar macrophages infected and the percentage of macrophages infected with the scored number of bacteria per cell were determined.
Evaluation of TNFR1 and TNFR2 in J774A.1 cells
J774A.1 cells, a mouse macrophage cell line, were seeded on to 24-well tissue culture plates, in RPMI-1640 supplemented with l-glutamine (2 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), non-essential amino acids (1% v/v × 100) and 10% heat-inactivated fetal calf serum, at a density of 5 × 104 cells per well and were incubated at 37° and 5% CO2 for 2 days until 80% confluency. Cells were infected with M. bovis BCG at a multiplicity of infection (MOI) of 10 : 1 bacteria per macrophage. After 2 hr of incubation at 37° and 5% CO2, the medium was removed and the plates were washed three times with pre-warmed PBS to remove non-associated bacteria, and fresh antibiotic-free complete medium was added. After 24, 48 and 72 hr of incubation, J774A.1 cells were scraped from the bottom of the wells and washed three times in PBS containing 0.05% BSA (blocking buffer) at 300 g for 5 min. Cells were stained with FITC-conjugated or phycoerythrin-conjugated monoclonal antibodies specific for TNFR1 (3H3104) and TNFR2 (TR75-89) (Santa Cruz Biotechnology), or with the corresponding isotype control antibody, for 30 min at 4° in the dark, followed by a washing step in blocking buffer. Cells were then resuspended in PBS and analysed by flow cytometry using FACSCalibur (Becton Dickinson). The mean fluorescence intensity for each sample was acquired and analysed using cell quest software.
Statistical analysis
Results are expressed as means ± standard error (SE). Differences between groups were analysed by Mann–Whitney U-test using the graphpad Prism 5.00 for windows (GraphPad Software, San Diego, CA). Differences were considered statistically significant when P < 0.05. All experiments were performed at least twice with comparable results.
Results
Mycobacterial infection alters surface expression of TNF receptors in J774A.1 macrophages
Interactions between TNF and TNFR initiate multiple signalling pathways that affect cell proliferation, survival, differentiation and apoptosis depending on the activation state of the cells and the expression levels of the TNFR molecules.35 Initially, in vitro studies were performed to assess whether infection with mycobacteria alters the expression of TNFR1 and TNFR2 receptors on the surface of macrophages. J774A.1 macrophages were infected with M. bovis BCG (MOI 10 : 1) and surface expression of TNFR1 and TNFR2 was analysed by cytometry. Mycobacterium bovis BCG infection induced a significant and gradual increase of TNFR1 in the macrophage cell surface (Fig. 1a). In contrast, a significant reduction of TNFR2 expression was found in the infected cells (Fig. 1b).
Figure 1.
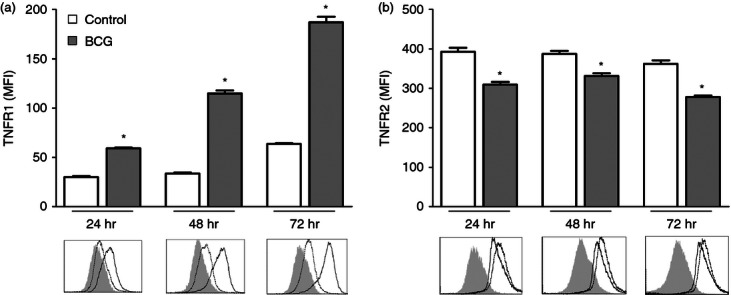
Surface tumour necrosis factor receptor (TNFR) expression in J774A.1 cells after Mycobacterium bovis bacillus Calmette–Guérin (BCG) infection. J774A.1 macrophages were either mock-infected or infected with M. bovis BCG at a multiplicity of infection (MOI) of 10 : 1. After 24, 48 and 72 hr, cells were stained with FITC-conjugated anti-mouse TNFR1 (a), or phycoerythrin-conjugated anti-mouse TNFR2 (b) and analysed by flow cytometry. The intensity of TNFRs was expressed as the mean fluorescence intensity (MFI) of at least 10 000 cells per sample. Representative histograms show TNFR1 or TNFR2 surface expression on mock-infected cells (dotted line) and BCG-infected cells (black line) or isotype control (light grey-filled area). The means and standard errors from one representative experiment of at least three is shown. *P < 0.001 versus mock-infected control.
TNFR1 and TNFR2 expression in alveolar macrophages after infection with virulent and attenuated M. bovis
To determine in vivo whether attenuated and virulent strains of M. bovis induce the same profile of TNFR expression in alveolar macrophages, C57BL/6 mice were intratracheally infected either with an attenuated M. bovis strain (BCG) or with a virulent M. bovis strain (ATCC 19274). After 3 and 7 days of infection, alveolar macrophages were gated into CD11b− CD11c+/high (Fig. 2a) and surface expression of TNFR1 and TNFR2 was evaluated by flow cytometry (Fig. 2b).
Figure 2.
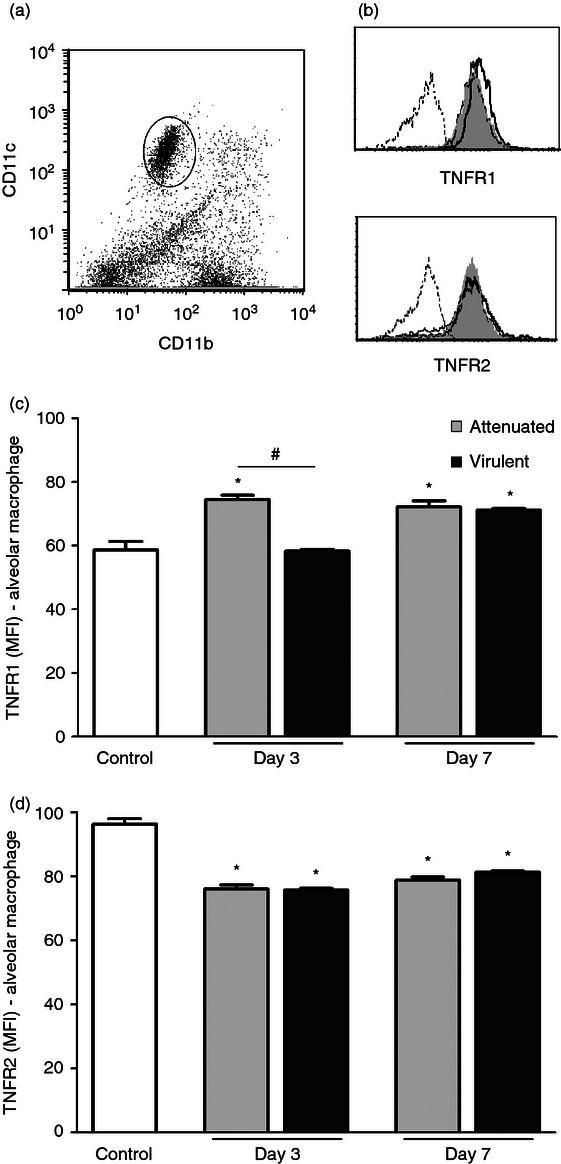
Surface expression of tumour necrosis factor receptor 1 (TNFR1) and TNFR2 on alveolar macrophages after infection with attenuated and virulent Mycobacterium bovis. C57BL/6 mice were intratracheally infected with attenuated (bacillus Calmette–Guérin Moreau) or virulent (ATCC19274) M. bovis. Control mice were inoculated with PBS. After 3 and 7 days, cells were obtained by bronchoalveolar lavage. Alveolar macrophages were identified as CD11b− CD11c+/high (a) and cell surface expression of TNFR1 and TNFR2 was assessed by FACS, as shown by representative histograms (b). Results of specific staining for TNFR1 (c) and TNFR2 (d) were expressed as the mean channel fluorescence intensity from 10 000 events per sample. Each bar represents mean ± SE of one representative experiment of three with similar results. *P < 0.05 versus control, #P < 0.05.
A significant increase in surface expression of TNFR1 in alveolar macrophages was observed on day 3 and day 7 after infection with the attenuated M. bovis (Fig. 2c). In contrast, in the infection with the virulent strain, increased expression of TNFR1 only occurred on day 7 after infection (Fig. 2c). On day 3, the expression of TNFR1 induced by the virulent M. bovis strain was similar to that detected in uninfected control mice and was significantly lower in comparison to the levels observed using the attenuated strain. On day 7, surface expression of TNFR1 was not significantly different between the attenuated and virulent strains of M. bovis. TNFR2 expression on alveolar macrophages was lower after infection with both strains of M. bovis (Fig. 2d).
The observed reduction of TNFR2 cell surface expression after infection with both strains of M. bovis, as well as the lower expression of TNFR1 on day 3 post-infection with the virulent strain did not seem to result from transcriptional down-regulation of TNFR gene expression as evaluated by real-time RT-PCR (data not shown). This suggests that regulation of surface expression of the TNFR1 and TNFR2 in macrophages during infection with mycobacteria occurs at the post-transcriptional level.
Changes in surface expression of TNFRs in macrophages may be because of shedding of the receptors
Levels of sTNFR1 and sTNFR2 in BAL fluid were measured by ELISA to assess whether the ectodomain shedding of TNFR1 and TNFR2 could explain the reduction in the surface expression of these receptors. The amount of sTNFR1 increased significantly only on day 3 after infection with the virulent strain (Fig. 3a), and suggests that shedding of TNFR1 may be implicated in the lower expression of this receptor on day 3. Similarly, reduction of TNFR2 surface expression could be explained by the shedding of the receptor, since a significant increase of sTNFR2 in BALF was detected on day 3 and day 7 after infection with both M. bovis strains (Fig. 3b).
Figure 3.
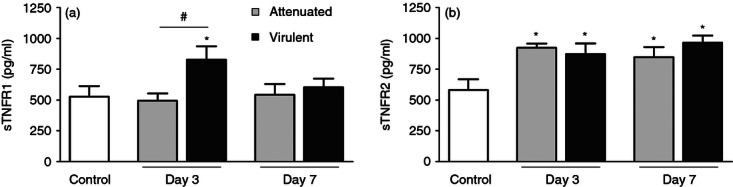
Detection of soluble tumour necrosis factor receptor 1 (sTNFR1) and sTNFR2. Levels of sTNFR1 (a) and sTNFR2 (b) in bronchoalveolar lavage fluid from mice infected with attenuated or virulent Mycobacterium bovis and uninfected control were assessed by ELISA. Each bar represents mean ± SE of one representative experiment of three with similar results. *P < 0.05 versus control, #P < 0.05.
Increased TNF-α production after infection with attenuated and virulent M. bovis
Signalling of TNF-α can be affected at several levels, including not only the regulation of surface TNFR expression and release of their soluble forms, but also the regulation of ligand expression. Mycobacteria have developed specific mechanisms that modulate TNF production by macrophages.18,36 Levels of TNF-α were quantified by ELISA on day 3 and day 7 after infection with the two M. bovis strains. Lung TNF-α levels increased in both infections. On day 3 TNF-α levels were not significantly different between the attenuated and the virulent M. bovis infections. However, after 7 days of infection, the virulent strain induced more TNF-α than the attenuated strain (Fig. 4).
Figure 4.
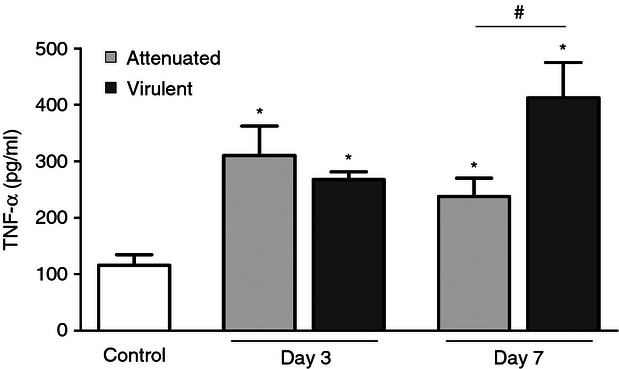
Quantification of tumour necrosis factor-α (TNF-α). Levels of TNF-α in the lung from mice infected with attenuated and virulent Mycobacterium bovis strains were measured by ELISA. Each bar represents mean ± SE. Results from one representative experiment of three with similar results is shown *P < 0.05 versus control, #P < 0.05.
Apoptosis of alveolar macrophages induced by M. bovis and the participation of TNFR1
Binding of TNF-α to TNFR1 and TNFR2 can lead to cell survival or apoptosis.37 A significant increase in apoptosis of alveolar macrophages was observed on days 3 and 7 of infection with the attenuated strain and only on day 7 of infection with the virulent strain. On day 3, the virulent strain induced significantly less apoptosis of alveolar macrophages than the attenuated strain (Fig. 5). Interestingly, the surface expression of TNFR1 was lower on day 3 of infection with the virulent strain compared with the attenuated strain of M. bovis (Fig. 2c), suggesting a potential role for TNFR1 in modulation of alveolar macrophage apoptosis induced by mycobacterium. To further evaluate the contribution of TNFR1, the frequency of alveolar macrophage apoptosis in TNFR1-deficient mice (TNFR1−/−) during M. bovis BCG infection was investigated. The infected TNFR1−/− mice exhibited lower macrophage apoptosis compared with the wild-type mice, indicating that TNFR1 plays an important role in macrophage apoptosis induced by mycobacteria (Fig. 6).
Figure 5.
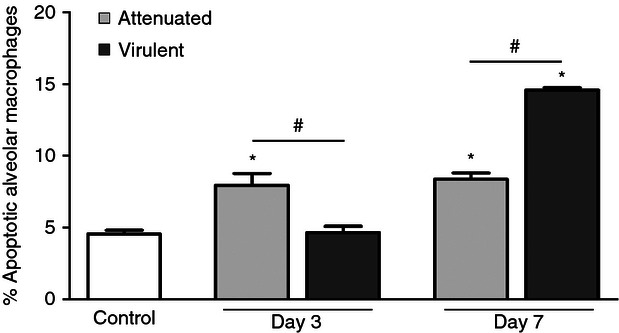
Apoptosis of alveolar macrophages after infection with attenuated or virulent Mycobacterium bovis. C57BL/6 mice were intratracheally infected with attenuated or virulent M. bovis. After 3 and 7 days of infection, alveolar macrophages were stained with annexin V and 7-aminoactinomycin D for analysis of apoptosis by flow cytometry. Each bar represents mean ± SE. Results from one representative experiment of three with similar results is shown. *P < 0.05 versus control, #P < 0.05.
Figure 6.
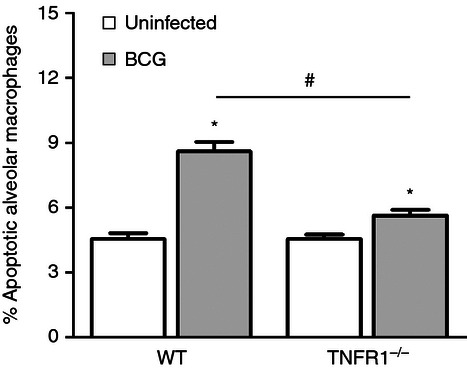
Apoptosis of alveolar macrophages in tumour necrosis factor receptor 1-deficient (TNFR1−/−) mice. Wild-type C57BL/6 mice or TNFR1−/− mice were intratracheally infected with Mycobacterium bovis BCG. After 3 days of infection, alveolar macrophages were stained with annexin V and 7-aminoactinomycin D for analysis of apoptosis by flow cytometry. Each bar represents mean ± SE. One representative experiment of two with similar results is shown. *P < 0.05 versus control, #P < 0.05.
Counts of bacilli in alveolar macrophages
In a previous study, we found that modulation of macrophage apoptosis during mycobacterial infection appears to be influenced by the intracellular bacterial load.13 The number of bacilli in macrophages was evaluated in this study using acid-fast staining to investigate a possible relationship between bacterial load and expression of TNFR1. In infection with the attenuated strain, the percentage of infected macrophages decreased from 23% on day 3 to 16% on day 7 post-infection. On both days, most infected macrophages contained from 1 to 10 bacilli (Fig. 7a), with high occurrence of macrophages containing an individual bacillus (Fig. 7c) mainly on day 3 of infection. In contrast, in infection with the virulent strain, the percentage of infected macrophages increased from 29% to 41% between the third and seventh days of infection. On day 3, most infected macrophages (88%) contained < 10 bacilli (Fig. 7b,d), whereas on day 7 most macrophages (82%) presented > 10 bacilli (Fig. 7b,e).
Figure 7.
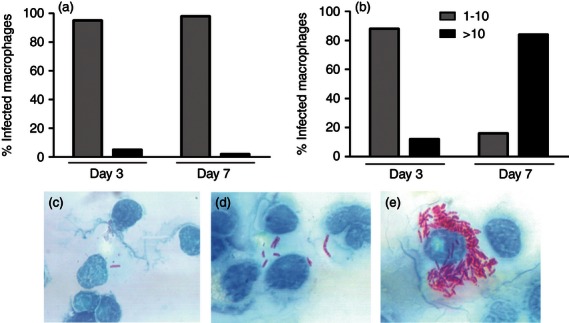
Number of bacilli in alveolar macrophages infected with attenuated (a) or virulent (b) Mycobacterium bovis at day 3 and day 7 post-infection. Cytocentrifuge slides were stained for acid-fast bacilli using the Ziehl–Neelsen stain and the number of bacilli per macrophage was scored as 1–10 bacilli per macrophage (c,d) and > 10 bacilli per macrophage (e). Results are representative of two different experiments. (c, d, e × 1000).
Discussion
Pathogenic mycobacteria employ several strategies to escape the immune response and settle into macrophages, the preferred site for intracellular persistence and growth of mycobacteria.38 Studies comparing the host response to attenuated and virulent strains of mycobacteria contribute to the elucidation of such strategies. Recognition that manipulation of macrophage death pathways is one of the complex methods used by mycobacteria to evade the host's defences39,40 has motivated many groups to investigate the mechanisms involved in this process, and some reports have established a central role for TNF-α in the modulation of apoptosis induced by mycobacteria.8,15,16 Tumour necrosis factor signals through two transmembrane receptors, TNFR1 and TNFR2.17,18 Signalling initiated by TNFR1 can result in cell survival, apoptosis or necroptosis depending on the cellular environment, whereas TNFR2 primarily induces survival signals.37,41,42 Although most activities of TNF require TNFR1 signalling, different cell types co-express TNFR1 and TNFR2 and require cooperation between these two receptors to generate TNF responses, including some cases of TNF-mediated apoptosis.43,44
In this study, the relationship between the expression of TNFRs and apoptosis of alveolar macrophages was investigated in mice during the early stage of infection with attenuated and virulent strains of M. bovis. A significant increase of macrophage apoptosis and high expression of TNFR1 were observed in alveolar macrophages on days 3 and 7 after infection with the attenuated mycobacteria but only on day 7 after infection with the virulent M. bovis. In contrast, low surface expression of TNFR1 observed on day 3 after infection with the virulent strain was associated with reduced rates of apoptotic macrophages. In addition, a significant reduction in apoptosis of alveolar macrophages was observed in TNFR1−/− mice on day 3 after infection with M. bovis BCG, which suggests a potential role of TNFR1 in modulation of macrophage apoptosis in the early phase of mycobacterial infection.
Different pathogens can modulate the expression of TNFRs to inhibit TNF-induced antimicrobial functions. It has been shown that Epstein–Barr virus decreases the expression of TNFR1 by down-regulation of the promoter, and so prevents TNFR1-induced cell death signalling.32 Chlamydia trachomatis, a pathogen that actively inhibits apoptosis of the host cell, decreases surface TNFR1 expression in infected cells. This decrease was suggested to be the result of receptor shedding or of the sequestering of the receptor within chlamydial inclusions.33 In the present work, increased levels of sTNFR1 in BAL were observed on day 3 post-infection with the virulent M. bovis, and was associated with lower expression of TNFR1 on the alveolar macrophage plasma membrane. Hence, the reduction of the TNFR1 surface expression affected by the shedding process may represent an immune escape mechanism whereby mycobacterium subverts apoptosis induced by TNF-α.
The regulation of TNFR shedding has been previously proposed as one possible mechanism by which mycobacteria inhibit macrophage apoptosis. Balcewicz-Sablinska et al.30 suggested that virulent mycobacteria evade apoptosis of host macrophages by shedding of TNFR2. The authors observed that virulent M. tuberculosis H37Rv induced greater release of sTNFR2 in comparison with the attenuated H37Ra, resulting in neutralization of TNF-α bioactivity by soluble receptor–ligand complex formation (sTNFR2–TNF). In the present study, an increase of sTNFR2 in BAL was correlated to a lower surface expression of TNFR2 after infection with either the attenuated or virulent strain of M. bovis. Here, shedding of TNFR2 does not appear to be influenced by the M. bovis virulence and does not seem to have any modulatory effect on macrophage apoptosis. Nevertheless, the importance of this process during mycobacterial infection should be taken into account, because cross-talk between TNFR2 and TNFR1 occurs on multiple levels and may be influenced by the amounts of both the membrane-bound and the soluble forms of the receptor.45
Soluble TNFRs bind to TNF-α. Although this binding can stabilize and preserve the bioactive trimeric form of TNF-α, it appears to act mainly as an antagonist of the TNF-α activity, competing for the ligand with the cell surface receptors.46,47 Hence, as suggested, release of sTNFR2 with formation of inactive TNF-α–sTNFR2 complexes, would contribute to prevent the TNF–TNFR1 binding, and to reduce macrophage apoptosis.30 On the other hand, it has been proposed that TNFR2 can also increase the apoptotic response mediated by TNFR1.48,49 Some studies have suggested that TNFR2 might enhance TNFR1-dependent responses by recruiting TNF-α to the cell surface and by delivering this cytokine to TNFR1 in a ligand-passing model.50,51 However, other studies suggest that TNFR2 can increase the apoptosis induced by TNFR1 via its own signalling activity, by means of TNFR2-mediated negative regulation of TNFR-associated factor 2 (TRAF2) function.44,52 The TRAF2 is recruited to both TNFR1 and TNFR2 complexes during the initial cell signalling process, and forms a heterodimeric complex with TRAF1. This complex interacts with the cellular inhibitor of apoptosis proteins cIAP-1 and cIAP-2 and delivers an anti-apoptotic signal that is essential for cellular survival.53–55 According to this model, stimulation by TNFR2 could strongly enhance TNFR1-induced cell death because of TNFR2-mediated depletion of TRAF2, which leads to abrogation of TRAF2-dependent TNFR1-induced anti-apoptotic signalling.43,52 Therefore, the complex cross-talk between TNFR1 and TNFR2 may result in both stimulatory as well as inhibitory effects that influence the balance between cell survival and apoptosis.45
Based on the above discussion, both the increased levels of sTNFR2 and the low surface expression of TNFR2 could contribute in different ways to inhibition of macrophage apoptosis. However, reduced macrophage apoptosis was only detected when TNFR1 surface expression was lower and levels of sTNFR1 were higher. The stability of individual ligand–receptor complexes may provide a possible explanation for this finding. Krippner-Heidenreich et al.56 demonstrated that the interaction of TNF-α with TNFR1 and TNFR2 differs strongly. Whereas TNF-α produces stable complexes bound with TNFR1 (half-life = 33 min), its binding with TNFR2 (half-life = 1.1 min) is transient. For this reason we can speculate that similarly, variation in binding stability of TNF–sTNFR1 and TNF–sTNFR2 could influence the inhibitory effect of these complexes.
The importance of TNFR1 shedding in mycobacterial infection was demonstrated in a study using transgenic mice that produce different amounts of sTNFR1. Transgenic mice expressing low levels of sTNFR1 were protected against BCG infection by means of increased bactericidal mechanisms. In contrast, mice expressing high levels of sTNFR1 failed to develop an effective immune response and succumbed to BCG infection, which was attributed to inhibition of circulating TNF-α and to reduced signalling via both TNFR1 and TNFR2.31,57 Hence, although a significant increase of TNF-α was observed on day 3 of infection with both strains of M. bovis, in infection with the virulent strain the lower expression of TNFR1 and increased levels of sTNFR1 may have contributed to inhibition of TNF-induced macrophage apoptosis, favouring permanence and replication of virulent mycobacteria within the host cell at this point when few bacilli per macrophage were observed. In contrast, on day 7, the expression of TNFR1 in macrophages did not differ between the two strains; however, the virulent strain induced significantly more TNF-α than the attenuated strain, which could possibly be related to the large increase in macrophage apoptosis observed by the infection with the virulent strain. Apoptosis of a large number of macrophages at this point, when most of these infected cells contained > 10 bacilli, could facilitate the exit of mycobacterium from the host cell to infect new cells, as apoptotic cells lyse and become necrotic, a process called ‘secondary necrosis’, when the load of dying cells exceeds the local capacity for phagocyte-mediated clearance.58,59 Taken together, these results support the hypothesis that virulent M. bovis modulates macrophage apoptosis according to the intracellular bacterial load. However, a direct relationship between the number of bacilli and TNFR1 expression cannot be concluded in general for all strains, because, although the number of bacilli may influence the surface expression of TNFR1, the modulation of this process seems to be dependent on the virulence of mycobacteria.
The manipulation of macrophage cell death by virulent mycobacteria appears to involve both inhibition of apoptosis and induction of secondary necrosis, which can favour the intracellular replication of bacillus and its exit from the host cell, respectively.36 However, it is still unclear whether during the initial phase of infection these cell death programmes represent independent processes or alternative decisions of a common pathway.60 In this context, regulation of TNFR1 expression could favour both cell death processes. Recent studies have demonstrated that in the absence of caspase 8 activity, activation of TNFR1 does not result in apoptosis, but instead it initiates a programmed necrosis pathway that is referred to as necroptosis.41,42 Cell morphology of necroptosis is similar to that observed after necrosis or secondary necrosis following apoptosis, and includes early loss of plasma membrane integrity,61 which may favour the spread of the bacillus. Indeed, it was demonstrated that at high intracellular load, virulent M. tuberculosis rapidly induces cell death in a caspase-independent manner14 of which the morphological changes were distinct from typical apoptosis or necrosis but showed some features of these death modes, including plasma membrane disruptions62 that may be compatible with necroptosis induced by TNFR1. However, additional studies are needed to evaluate the occurrence of necroptosis during mycobacterial infection and to determine whether signalling mediated by TNFR1 may have different consequences for the cell death pathway induced by attenuated and virulent M. bovis strains.
In conclusion, this study suggests that TNFR1 plays an important role in inducing apoptosis of the macrophage during the early stage of mycobacterial infection and that the reduction of surface expression of TNFR1 via shedding can be a mechanism used by virulent mycobacteria to inhibit macrophage apoptosis. Additional studies are needed to evaluate the mechanistic details involved in this process.
Acknowledgments
The support from the Brazilian Funding Agencies CNPq, CAPES and FAPEMIG is gratefully acknowledged. We are also grateful to Dr João Santana da Silva (University of São Paulo, Ribeirão Preto, Brazil) for providing TNFR1−/− mice.
Disclosures
The authors indicated no potential conflicts of interest.
References
- 1.Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. 2003;16:463–96. doi: 10.1128/CMR.16.3.463-496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. 2011;240:252–68. doi: 10.1111/j.1600-065X.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–76. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- 4.Dunn PL, North RJ. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect Immun. 1995;63:3428–37. doi: 10.1128/iai.63.9.3428-3437.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medina E, Ryan L, LaCourse R, North RJ. Superior virulence of Mycobacterium bovis over Mycobacterium tuberculosis (Mtb) for Mtb-resistant and Mtb-susceptible mice is manifest as an ability to cause extrapulmonary disease. Tuberculosis. 2006;86:20–7. doi: 10.1016/j.tube.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Andrade MR, Amaral EP, Ribeiro SC, Almeida FM, Peres TV, Lanes V, D'Império-Lima MR, Lasunskaia EB. Pathogenic Mycobacterium bovis strains differ in their ability to modulate the proinflammatory activation phenotype of macrophages. BMC Microbiol. 2012;12:166. doi: 10.1186/1471-2180-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe. 2008;3:399–407. doi: 10.1016/j.chom.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fratazzi C, Arbeit RD, Carini C, Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Macrophage apoptosis in mycobacterial infections. J Leukoc Biol. 1999;66:763–4. doi: 10.1002/jlb.66.5.763. [DOI] [PubMed] [Google Scholar]
- 10.Schaible UE, Winau F, Sieling PA, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–46. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 11.Winau F, Weber S, Sad S, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–17. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–20. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues MF, Barsante MM, Alves CCS, Souza MA, Ferreira AP, Amarante-Mendes GP, Teixeira HC. Apoptosis of macrophages during pulmonary Mycobacterium bovis infection: correlation with intracellular bacillary load and cytokine levels. Immunology. 2009;128:e691–9. doi: 10.1111/j.1365-2567.2009.03062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Remold HG, Ieong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol. 2006;176:4267–74. doi: 10.4049/jimmunol.176.7.4267. [DOI] [PubMed] [Google Scholar]
- 15.Rojas M, Olivier M, Gros P, Barrera LF, García LF. TNF-α and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J Immunol. 1999;162:6122–31. [PubMed] [Google Scholar]
- 16.Spira A, Carroll JD, Liu G, Aziz Z, Shah V, Kornfeld H, Keane J. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol. 2003;29:545–51. doi: 10.1165/rcmb.2002-0310OC. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–3. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 18.Hehlgans T, Pfeffer K. The intriguing biology of the tumor necrosis factor/tumor necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss T, Grell M, Hessabi B, Bourteele S, Müller G, Scheurich P, Wajant H. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: requirement of the TNF receptor-associated factor-2 binding site. J Immunol. 1997;158:2398–404. [PubMed] [Google Scholar]
- 20.Grell M, Zimmermann G, Gottfried E, et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 1999;18:3034–43. doi: 10.1093/emboj/18.11.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aderka D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996;7:231–40. doi: 10.1016/s1359-6101(96)00026-3. [DOI] [PubMed] [Google Scholar]
- 22.Brockhaus M. Soluble TNF receptor: what is the significance? Intensive Care Med. 1997;23:808–9. doi: 10.1007/s001340050416. [DOI] [PubMed] [Google Scholar]
- 23.Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med. 2004;200:367–76. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J Immunol. 2004;172:6846–57. doi: 10.4049/jimmunol.172.11.6846. [DOI] [PubMed] [Google Scholar]
- 25.Stenger S. Immunological control of tuberculosis: role of tumour necrosis factor and more. Ann Rheum Dis. 2005;64:iv24–8. doi: 10.1136/ard.2005.042531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris J, Hope JC, Keane J. Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. J Infect Dis. 2008;198:1842–50. doi: 10.1086/593174. [DOI] [PubMed] [Google Scholar]
- 27.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 28.Ehlers S, Kutsch S, Ehlers EM, Benini J, Pfeffer K. Lethal granuloma disintegration in mycobacteria-infected TNFRp55–/– mice is dependent on T cells and IL-12. J Immunol. 2000;165:483–92. doi: 10.4049/jimmunol.165.1.483. [DOI] [PubMed] [Google Scholar]
- 29.Jacobs M, Brown N, Allie N, Chetty K, Ryffel B. Tumor necrosis factor receptor 2 plays a minor role for mycobacterial immunity. Pathobiology. 2000;68:68–75. doi: 10.1159/000028116. [DOI] [PubMed] [Google Scholar]
- 30.Balcewicz-Sablinska MK, Keane J, Kornefeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-α. J Immunol. 1998;161:2636–41. [PubMed] [Google Scholar]
- 31.Schlüter D, Deckert M. The divergent role of tumor necrosis factor receptors in infectious diseases. Microbes Infect. 2000;2:1285–92. doi: 10.1016/s1286-4579(00)01282-x. [DOI] [PubMed] [Google Scholar]
- 32.Morrison TE, Mauser A, Klingelhutz A, Kenney SC. Epstein–Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor α-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J Virol. 2004;78:544–9. doi: 10.1128/JVI.78.1.544-549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paland N, Böhme L, Gurumurthy RK, Mäurer A, Szczepek AJ, Rudel T. Reduced display of tumor necrosis factor receptor I at the host cell surface supports infection with Chlamydia trachomatis. J Biol Chem. 2008;283:6438–48. doi: 10.1074/jbc.M708422200. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. 2003;171:3128–35. doi: 10.4049/jimmunol.171.6.3128. [DOI] [PubMed] [Google Scholar]
- 35.Sun M, Fink PJ. A new class of reverse signaling costimulators belongs to the TNF family. J Immunol. 2007;179:4307–12. doi: 10.4049/jimmunol.179.7.4307. [DOI] [PubMed] [Google Scholar]
- 36.Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annu Rev Pathol. 2012;7:353–84. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- 37.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24:1297–305. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 38.de Chastellier C. The many niches and strategies used by pathogenic mycobacteria for survival within host macrophages. Immunobiology. 2009;214:526–42. doi: 10.1016/j.imbio.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Porcelli SA, Jacobs WR., Jr Tuberculosis: unsealing the apoptotic envelope. Nat Immunol. 2008;9:1101–2. doi: 10.1038/ni1008-1101. [DOI] [PubMed] [Google Scholar]
- 40.Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–74. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 42.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2012;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fotin-Mleczek M, Henkler F, Samel D, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115:2757–70. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- 44.Grech AP, Gardam S, Chan T, Quinn R, Gonzales R, Basten A, Brink R. Tumor necrosis factor receptor 2 (TNFR2) signaling is negatively regulated by a novel, carboxyl-terminal TNFR-associated factor 2 (TRAF2)-binding site. J Biol Chem. 2005;280:31572–81. doi: 10.1074/jbc.M504849200. [DOI] [PubMed] [Google Scholar]
- 45.Naudé PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J. 2011;278:888–98. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 46.Peschon JJ, Torrance DS, Stocking KL, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52. [PubMed] [Google Scholar]
- 47.Dai H, Guzman J, Chen B, Costabel U. Production of soluble tumor necrosis factor receptors and tumor necrosis factor-α by alveolar macrophages in sarcoidosis and extrinsic allergic alveolitis. Chest. 2005;127:251–6. doi: 10.1378/chest.127.1.251. [DOI] [PubMed] [Google Scholar]
- 48.Bigda J, Beletsky I, Brakebusch C, Varfolomeev Y, Engelmann H, Bigda J, Holtmann H, Wallach D. Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J Exp Med. 1994;180:445–60. doi: 10.1084/jem.180.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–60. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 50.Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268:18542–8. [PubMed] [Google Scholar]
- 51.Dri P, Haas E, Cramer R, Menegazzi R, Gasparini C, Martinelli R, Scheurich P, Patriarca P. Role of the 75-kDa TNF receptor in TNF-induced activation of neutrophil respiratory burst. J Immunol. 1999;162:460–6. [PubMed] [Google Scholar]
- 52.Weiss T, Grell M, Siemienski K, Mühlenbeck F, Dürkop H, Pfizenmaier K, Scheurich P, Wajant H. TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J Immunol. 1998;161:3136–42. [PubMed] [Google Scholar]
- 53.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–92. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 54.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–52. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 55.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 56.Krippner-Heidenreich A, Tübing F, Bryde S, Willi S, Zimmermann G, Scheurich P. Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J Biol Chem. 2002;277:44155–63. doi: 10.1074/jbc.M207399200. [DOI] [PubMed] [Google Scholar]
- 57.Garcia I, Miyazaki Y, Marchal G, Lesslauer W, Vassalli P. High sensitivity of transgenic mice expressing soluble TNFR1 fusion protein to mycobacterial infections: synergistic action of TNF and IFN-γ in the differentiation of protective granulomas. Eur J Immunol. 1997;27:3182–90. doi: 10.1002/eji.1830271215. [DOI] [PubMed] [Google Scholar]
- 58.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 59.Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–9. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 60.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–66. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 61.Berghe TV, Vanlangenakker N, Parthoens E, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 62.Lee J, Repasy T, Papavinasasundaram K, Sassetti C, Kornfeld H. Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS ONE. 2011;6:e18367. doi: 10.1371/journal.pone.0018367. [DOI] [PMC free article] [PubMed] [Google Scholar]