

Abstract
Antibiotics have, indeed, altered the course of human history as is evidenced by the increase in human life expectancy since the 1940s. Many of these natural compounds are produced by bacteria that, by necessity, must have efficient self-resistance mechanisms. The methymycin/pikromycin producing species Streptomyces venezuelae, for example, utilizes β-glucosylation of its macrolide products to neutralize their effects within the confines of the cell. Once released into the environment, these compounds are activated by the removal of the glucose moiety. In S. venezuelae, the enzyme responsible for removal of the sugar from the parent compound is encoded by the desR gene and referred to as DesR. It is a secreted enzyme containing 828 amino acid residues, and it is known to be a retaining glycosidase. Here, we describe the structure of the DesR/d-glucose complex determined to 1.4-Å resolution. The overall architecture of the enzyme can be envisioned in terms of three regions: a catalytic core and two auxiliary domains. The catalytic core harbors the binding platform for the glucose ligand. The first auxiliary domain adopts a “PA14 fold,” whereas the second auxiliary domain contains an immunoglobulin-like fold. Asp 273 and Glu 578 are in the proper orientation to function as the catalytic base and proton donor, respectively, required for catalysis. The overall fold of the core region places DesR into the GH3 glycoside hydrolase family of enzymes. Comparison of the DesR structure with the β-glucosidase from Kluyveromyces marxianus shows that their PA14 domains assume remarkably different orientations.
Keywords: β-glucosidase, self-resistance, macrolide antibiotics, enzyme mechanism, family 3 glycoside hydrolases, X-ray structure
Introduction
Streptomycetes are Gram-positive, spore-forming bacteria typically observed in soil habitats. Due to their stunning ability to produce complex compounds of medical relevance, they have been and continue to be extensively studied. More than 500 distinct antibiotics have been shown to be produced by these organisms, 60 of which are used in human and veterinary medicine.1,2 Indeed, a multibillion dollar industry revolves around these organisms and their ability to synthesize therapeutics.
Of particular interest is Streptomyces venezuelae, a soil dwelling bacterium isolated from a field near Caracas, Venezuela. This bacterium produces chloramphenicol (chloromycetin), which served as the first broad-spectrum antibiotic used in clinical settings.3 Due to its significant side effects, however, it has since been replaced with newer less toxic drugs in developed countries. In addition to chloramphenicol, S. venezuelae also produces methymycin, pikromycin, and other related macrolide-based antibacterial agents.4,5
Antibiotic-producing bacteria, such as S. venezuelae, have evolved various self-resistance mechanisms to protect themselves from the bactericidal effects of the compounds they produce. One of these mechanisms involves the addition of a glucose moiety to the macrolide antibiotic, which renders the compound inactive within the confines of the cell.6 Once secreted, however, these antimicrobials must be reactivated, and some bacteria, such as Streptomyces antibioticus and S. venezuelae, use extracellular β-glucosidases for this purpose.7–11
The focus of this investigation is on the product of the desR gene from S. venezuelae, hereafter referred to as DesR.11 It is a β-glucosidase that catalyzes the removal of glucosyl moieties from glycosylated versions of methymycin or neomethymycin as indicated in Scheme 1.11 DesR contains 828 amino acids with a signal peptide sequence located within the first 50 amino acid residues. It has been reported to function as a monomer.11 Based on amino acid sequence alignments, DesR clearly belongs to the family 3 glycoside hydrolases (GH3). Members of this family are involved in a wide range of biological processes ranging from the assimilation of glycosides to the recycling of cellular components.12 Their active sites contain a conserved aspartate that functions as a nucleophile and a glutamate that serves as a proton donor.13 Their catalytic mechanisms proceed through retention of configuration about the anomeric carbon.13 In the first step of the reaction, the conserved glutamate donates a proton to the leaving group whereas the conserved aspartate attacks the sugar C-1 carbon to form an enzyme intermediate with an α-linkage. Subsequently, the glutamate, now deprotonated, activates a water molecule, which in turn attacks the enzyme intermediate to release the sugar moiety.
Figure 1.
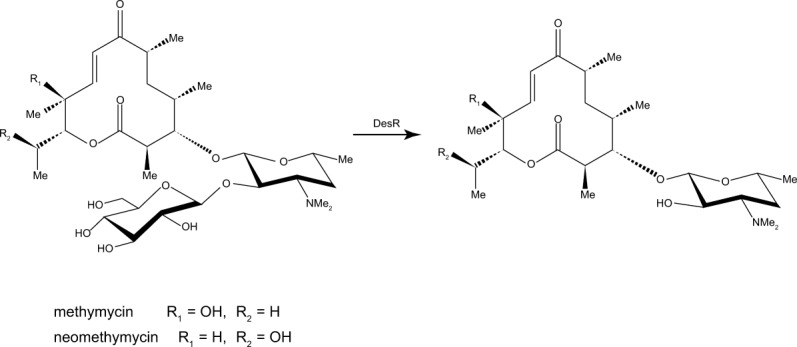
DesR functions as a glucosidase.
In this article, we describe the structure of DesR in complex with d-glucose solved to 1.4-Å resolution. This investigation represents the first glimpse of a β-glucosidase that functions on macrolide antibiotics and thus serves as a paradigm for other GH3 family members involved in bacterial self-resistance.
Results and Discussion
Overall structure of DesR
The DesR crystals used in this investigation contained two monomers in the asymmetric unit that buried a total subunit:subunit surface area of approximately 1000 Å2. These monomers were related by an approximate twofold rotational axis (179.9°). In light of this symmetrical arrangement, ultracentrifugation experiments were subsequently conducted to establish the correct oligomerization state of the enzyme. As previously suggested by gel filtration,11 DesR behaved as a monomer in these experiments, and the dimeric appearance of the protein in the asymmetric unit is apparently a function of the crystallization conditions. The two polypeptide chains in the asymmetric unit correspond with a root-mean-square deviation of 0.20 Å for 616 structurally equivalent α-carbons.
To fulfill its biological role, DesR is secreted from the bacterium via a protein leader sequence of unknown length.11 In the structure presented here, the first monomer in the X-ray coordinate file extends from Ser 51 to Trp 828, whereas the second monomer stretches from Tyr 49 to Trp 828. There were no breaks in the electron density map for either of the two polypeptide chain backbones. Thus, for the sake of simplicity, and because the electron density was somewhat stronger for a few surface loops, the following discussion refers only to the second molecule in the X-ray coordinate file.
Shown in Figure 2(a) is a ribbon representation of DesR. The protein, with overall dimensions of ∼76 × 67 × 67 Å3, consists of 15 α-helices and 33 β-strands that form a decidedly trilobal structure. The core domain, shown in light blue and wheat, is composed of Tyr 49 to Thr 443 and Pro 548 to Tyr 711. It can be envisioned as two subdomains. The N-terminal subdomain contains a distorted seven-stranded β-barrel with two of the strands running antiparallel. The C-terminal end of the barrel provides the binding platform for the glucose moiety. The second subdomain of the core is composed of a six-stranded mixed β-sheet flanked on either side by two α-helices.
Figure 2.
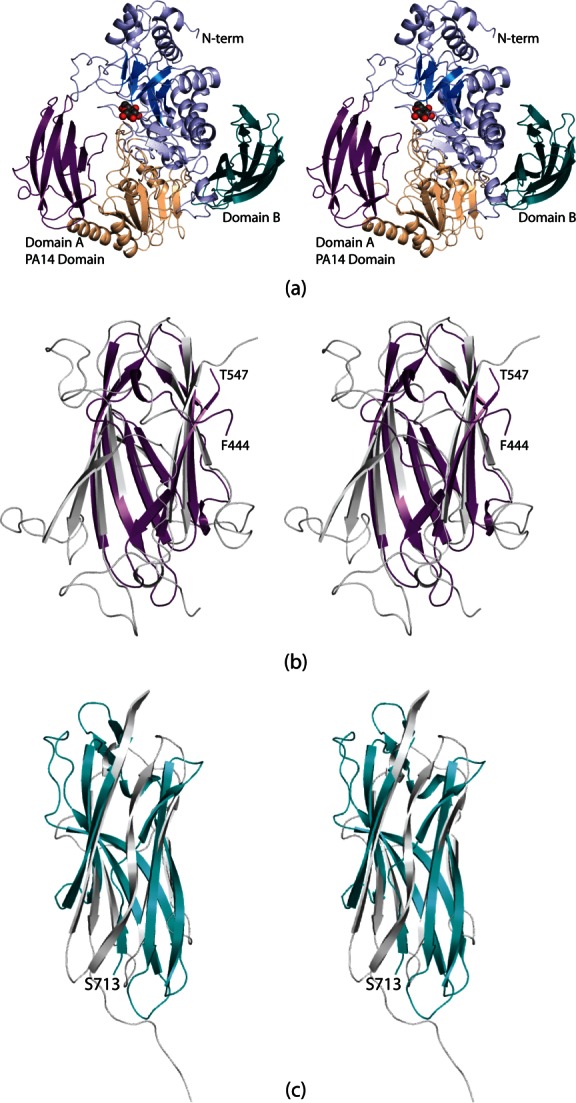
Structure of DesR. A ribbon representation of DesR is displayed in (a). The architecture of the enzyme can be envisioned in terms of three regions: a large core domain and two smaller β-barrels referred to as Domains A and B and shown in violet and teal, respectively. The core is highlighted in blue and wheat to emphasize the subdomains. The glucose ligand, depicted in a sphere representation, sits at the C-terminal end of the distorted β-barrel in the core. Domain A has a similar fold to the PA14 domain observed in the β-glucosidase from K. marxianus. A superposition of these domains from DesR (in violet) and the K. marxianus β-glucosidase (light gray) is shown in (b). The B domain adopts an immunoglobulin-type architecture. A superposition of Domain B onto the structure of telokin, which has an immunoglobulin fold, is presented in (c). Domain B and telokin are color-coded in deep teal and light gray, respectively. All figures were prepared with PyMOL.14
The core region of DesR is interrupted by the insertion of an auxiliary motif, designated Domain A, in Figure 2(a). This domain, delineated by Phe 444 to Thr 547, contains eight antiparallel β-strands. These β-strands adopt the PA14 domain topology, which was first observed in the protective antigen of anthrax toxin.15,16 In most cases PA14 domains are thought to be involved in carbohydrate binding. A superposition of the PA14 domains found in DesR and the β-glucosidase from Kluyveromyces marxianus is displayed in Figure 2(b). The two domains superimpose with a root-mean-square deviation of 2.0 Å for 95 structurally equivalent α-carbons.
Following the core motif is Domain B, which extends from Ser 713 to the C-terminus. It is composed of nine β-strands, seven of which form an immunoglobulin-like motif. Shown in Figure 2(c) is a superposition of Domain B onto the structure of telokin, an abundant protein found in smooth muscle and known to exhibit an immunoglobulin fold.17 These two domains superimpose with a root-mean-square deviation of 3.5 Å for 79 target α-carbons. The biological function of Domain B is unknown, but immunoglobulin folds have been implicated in protein:protein interactions and cellular communications.
The crystals of DesR were grown in the presence of d-glucose, which was observed binding in both monomers of the asymmetric unit. Shown in Figure 3(a) is the electron density corresponding to the glucosyl moiety in monomer 2. The sugar binds to the protein as the β-anomer, and it adopts the4C1 conformation. A close-up view of the region surrounding the glucose ligand is presented in Figure 3(b). The sugar is tightly bound in the active site by numerous hydrogen bonding interactions. With the exception of one water molecule, all of the hydrogen bonding interactions occur through protein side chains. Specifically, the C-1 hydroxyl lies within 2.4 Å of Glu 578 (Subdomain 2 of the core). It is this glutamate, which is conserved amongst the GH3 hydrolase family that is thought to serve as the proton donor during the hydrolysis reaction. The guanidinium groups of Arg 162 and Arg 206, and the carboxylate group of Asp 273 are situated within hydrogen bonding distance of the C-2 hydroxyl (Subdomain 1 of the core). Asp 273 is the conserved nucleophile in the GH3 family, and one of its carboxylate oxygens resides within 2.9 Å of the C-1 carbon of the glucose molecule. The C-3 hydroxyl group is tetrahedrally surrounded by Arg 162, Lys 195, and His 196 (Subdomain 1 of the core). Lys 195 serves to bridge the C-3 and C-4 hydroxyls whereas the side chain of Asp 98 interacts with both the C-4 and the C-6 hydroxyl groups. There is one water molecule that is positioned at 2.8 Å from the C-1 hydroxyl group. This water, in turn, is hydrogen-bonded to a MOPS buffer located at the opening to the active site pocket. DesR contains seven cis-peptide bonds (Gly 99, Pro 150, His 196, Ala 198, Pro 384, Pro 455, and Pro 675). Of these, Gly 99, Pro 150, His 196, and Ala 198 reside near the active site [Fig. 3(b)]. In addition to these cis-peptides, a disulfide bridge formed between Cys 239 and Cys 250 abuts one side of the active site pocket [Fig. 3(c)]. This bridge adopts a right-handed hook conformation with the α-carbons separated by 5.3 Å. There is a decided cleft between the core region of DesR and Domain A (Fig. 4). This area may provide the binding pocket for the aglycone ring of the antibiotic. Experiments to test this are presently underway.
Figure 3.
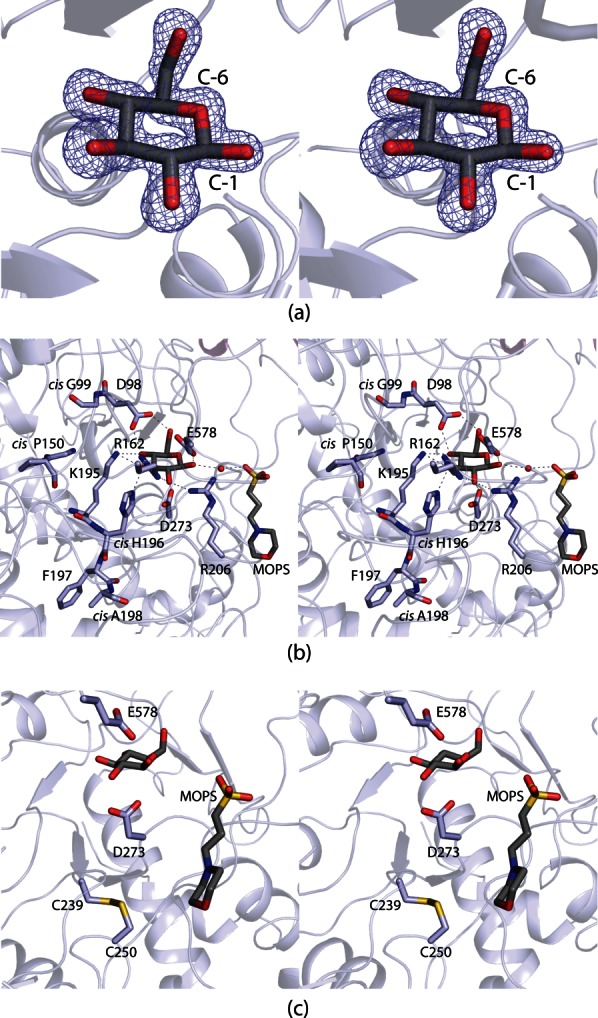
The DesR active site. Electron density corresponding to the bound d-glucose ligand is shown in (a). The map, contoured at 2σ, was calculated with coefficients of the form (Fo − Fc) where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. The DesR model used for the calculation was refined to reduce model bias from the molecular replacement procedure. Those amino acid residues or solvent molecules lying within 3.2 Å of d-glucose are shown in (b). An ordered water molecule is depicted as a red sphere. Possible hydrogen bonding interactions between the carbohydrate and the protein are indicated by the dashed lines. A MOPS buffer was observed binding at the opening of the active site cleft. A close-up view of the disulfide bridge that flanks one side of the active site pocket is presented in (c).
Figure 4.
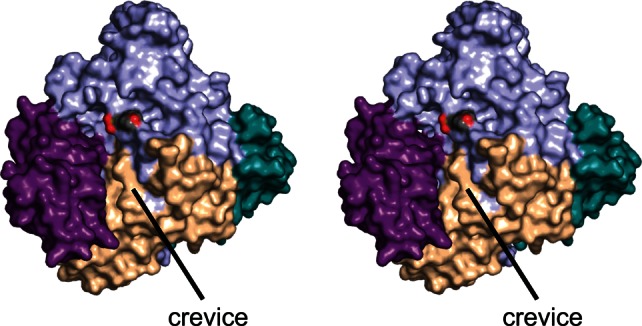
Surface representation of DesR. The color-coding is as described in Figure 2. The oxygens of the MOPS buffer are displayed in red. There is a decided cleft formed at the interface of the core region and Domain A (violet), which leads directly from the active site.
Enzymatic activity of DesR
The natural substrates for DesR, glucosylated methymycin and glucosylated neomethymycin, are not commercially available. Thus, to test the enzymatic activity of the DesR sample utilized in this investigation, we used a chromogenic assay with 4-nitrophenyl β-d-glucopyranoside (pNP-Glc) as the substrate.18 Relevant kinetic parameters are provided in Table I. The catalytic efficiency of the enzyme against pNP-Glc is 1.5 × 103M−1 s−1. When Asp 273, the putative nucleophile, was converted to an asparagine residue via site-directed mutagenesis, the Km increased by a factor of 2 whereas the kcat decreased by approximately four orders of magnitude. These data suggest, as expected from previous research on β-glucosidases, that Asp 273 plays a key role in the catalytic mechanism of DesR.
Table I.
Kinetic Parameters
| Protein | Km (mM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| Wild-type | 9.1 ± 0.3 | 13.4 ± 0.6 | 1.5 × 103 |
| D273N | 20.4 ± 2.7 | 0.0017 ± 0.00012 | 8.3 × 10−2 |
Comparison of DesR with other GH3 glycoside hydrolases
Until the structure of the β-d-glucan glucohydrolase from Hordeium vulgare (barley) was reported, nothing was known regarding the three-dimensional architecture of a GH3 family member.19 From that elegant investigation, the basic core structure of a GH3 family member was defined and shown to contain two domains, a (β/α)8 TIM barrel and a six-stranded β-sandwich, much like the DesR core. A superposition of the DesR structure onto that of barley glucohydrolase is displayed in Figure 5(a). Excluding Domains A and B of DesR, the two proteins correspond with a root-mean-square deviation of 1.5 Å for 456 equivalent α-carbons.
Figure 5.
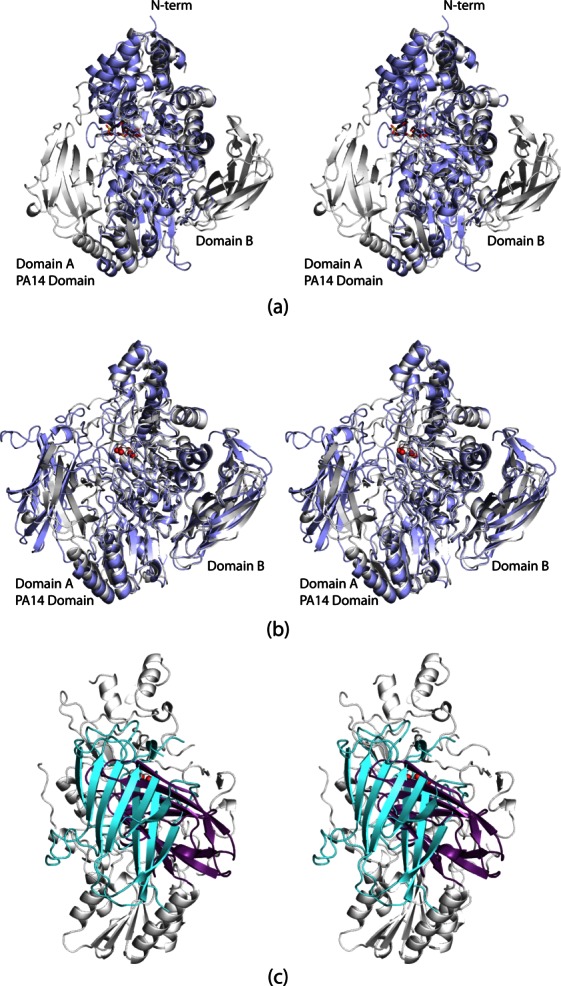
Comparison of DesR to other GH3 family members. The barley β-d-glucan glucohydrolase structure was the first to be reported for a GH3 family member. A superposition of it (light blue) onto DesR (white) is shown in (a). In panel (b), the K. marxianus β-glucosidase (light blue) is shown superimposed upon DesR (white). Whereas their core and immunoglobulin domains match well, the PA14 domains adopt completely different orientations. This is highlighted in (c) where the PA14 domains for K. marxianus β-glucosidase and DesR are depicted in light cyan and violet, respectively. The view shown is rotated by 90° from that displayed in (b).
The recent report of the K. marxianus β-glucosidase structure in 2010 highlighted the fact that the “basic” GH3 family member core can be decorated by two additional motifs.20 This structural analysis was the first to demonstrate the insertion of a PA14 domain into the core unit of a GH3 glycoside hydrolase. Unlike DesR, the K. marxianus β-glucosidase functions on disaccharides and, to a limited extent, on trisaccharides.20 A superposition of the DesR and the K. marxianus β-glucosidase structures is shown in Figure 5(b). Both structures were solved in the presence of d-glucose, and the carbohydrate moieties are situated in nearly identical positions in the active sites of the two enzymes. The overall cores and immunoglobulin domains of the two enzymes superimpose well. Specifically, the root-mean-square deviation between the two enzymes, excluding the PA14 domains, is 1.3 Å for 604 structurally equivalent α-carbons.
As can be seen in Figure 5(c), however, the PA14 domains adopt strikingly different orientations with respect to the main bodies of the enzymes. They are related to one another by a rotation of ∼116°. The PA14 domain in K. marxianus β-glucosidase is larger by ∼60 amino acid residues. It contains an additional β-hairpin motif at the N-terminus [Fig. 2(b)], and the surface loops are considerably longer than those found in DesR. The observed orientations of the PA14 domains may be a function of the differences in substrate specificities between the two enzymes. It is also possible that the PA14 domain of DesR moves upon binding the macrolide antibiotic. Likewise, the structure of the K. marxianus β-glucosidase was solved only in the presence of a monosaccharide, and the PA14 domain, which was implicated in sugar binding via site-directed mutagenesis, may adopt a different orientation when a disaccharide enters the active site cleft.20
The DesR structure described here represents the first model of a GH3 family member that functions on macrolide antibiotics. Given that, we aligned the DesR amino acid sequence against those for the homologs from the narbomycin, chalcomycin, oleandomycin, lankamycin, and erythromycin biosynthetic clusters using ClustalW221 and the available GenBank sequences (Supporting Information Fig. 1).22 All of the cis-peptides are conserved as well as the disulfide bridge. In addition, the amino acid residues involved in d-glucose binding, as highlighted in Figure 3(b), are strictly conserved.
A recent report has shown that DesR is inhibited by both erythromycin and clarithromycin.18 As such, these types of natural products may provide a new avenue for the development of glycosidase inhibitors. Improperly functioning glycoside hydrolases have been implicated in a variety of diseases including cancer, diabetes, and viral infections, among others.23 The structure of DesR described here provides a molecular scaffold for understanding the binding modes of both its substrates and competitive inhibitors, and thus, it will aid in the future development of novel glycoside hydrolase inhibitors.
Materials and Methods
Cloning, expression, and purification
The desR gene was PCR-amplified from genomic DNA isolated from S. venezuelae (ATCC 15439) such that the forward primer 5′-AAAACATATGGCTCCCGGCGCCGCCGACACGGCC and the reverse primer 5′-AAAACTCGAGTCACCAGACGTTGACCGTGGCGCTGCCCCG added NdeI and XhoI cloning sites, respectively. The forward primer started at amino acid residue 38 in the protein so that the final PCR product did not include the leader sequence. The purified PCR product was A-tailed and ligated into a pGEM-T (Promega) vector for screening and sequencing. A DesR-pGEM-T vector construct of the correct sequence was then appropriately digested and ligated into a pET28(b+) (Novagen) plasmid that had been previously modified to include a TEV cleavage site for the production of protein with an N-terminal His6-tag.24
The pET28JT-DesR plasmid was used to transform Rosetta 2 (DE3) Escherichia coli cells (Novagen). Cultures were grown with shaking at 37°C in lysogeny broth supplemented with kanamycin and chloramphenicol until optical densities of 0.8 at 600 nm were reached. The flasks were then cooled to 16°C, and the cells were subsequently induced with the addition of 1 mM isopropyl-β-d-thiogalactopyranoside. Protein expression was allowed to occur at 16°C for 18 h following induction. DesR was purified by standard procedures using Ni-nitrilotriacetic acid resin (Qiagen). The hexahistidine tag was cleaved using TEV protease at room temperature for two days, and cleaved DesR was purified using the same Ni-nitrilotriacetic acid resin column. The purified protein was dialyzed against 10 mM Tris (pH 8.0) and 200 mM NaCl, concentrated to ∼20 mg mL−1, and flash-frozen in liquid nitrogen. Analysis by analytical ultracentrifugation showed the oligomerization state of the protein to be monomeric (University of Wisconsin–Madison Biophysics Instrumentation Facility, Supporting Information Fig. 2).
For kinetic studies, DesR was expressed and purified as described above but dialyzed against 50 mM sodium dihydrogen phosphate (pH 8.0) and 300 mM NaCl to avoid a possible glucosidase inhibition effect caused by the Tris buffer.25
Crystallization of DesR
Crystallization conditions for DesR were initially surveyed by the hanging drop method of vapor diffusion using a sparse matrix screen developed in the laboratory. Small crystals were observed growing against a well-solution containing 1.5 M ammonium sulfate, 100 mM MOPS, and 2% 2-methyl-2,4-pentanediol at pH 7.0. Larger X-ray diffraction quality crystals were subsequently grown by streak seeding into hanging drops. These drops were prepared by mixing in a 1:1 ratio the enzyme solution and the precipitant, which included 25 mMd-glucose, 1.3 M ammonium sulfate, 100 mM MOPS, and 300 mM KCl. The crystals reached maximum dimensions of 0.3 × 0.3 × 0.1 mm3 in about 1 month. They belonged to the space group P21212 with unit cell dimensions of a = 131.3 Å, b = 198.3 Å, and c = 67.0 Å. The asymmetric unit contained two monomers.
Structural analysis of DesR
Before flash cooling, the DesR crystals were transferred to a cryoprotectant solution containing 2.0 M ammonium sulfate, 400 mM KCl, 200 mM NaCl, 900 mM glucose, and 5% ethylene glycol for 15–20 s. An X-ray data set from a wild-type DesR crystal was collected at the Structural Biology Center Beamline 19-ID at a wavelength of 0.979 Å (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). The dataset was processed and scaled with HKL3000.26 Relevant X-ray data collection statistics are listed in Table II.
Table II.
X-ray Data Collection Statistics
| Enzyme in complex with d-glucose | |
|---|---|
| Resolution limits | 30.0–1.4 (1.45–1.4)b |
| Number of independent reflections | 324,839 (32,626) |
| Completeness (%) | 94.1 (96.7) |
| Redundancy | 6.4 (4.2) |
| Avg I/avg σ(I) | 36.9 (2.8) |
| Rsyma | 0.081 (0.38) |
Rsym = (∑|∑I − Ī|/∑I).
Statistics for the highest resolution bin.
The initial structure of DesR was determined via molecular replacement with PHASER.27 The search probe used was the monomer of the β-glucosidase from K. marxianus with all ligands and waters removed.20 The quality of the electron density map was further improved by molecular averaging and solvent flattening with the software DM.28 Alternate cycles of manual model building with Coot29 and refinement with Refmac30 reduced the Rworking and Rfree values to 17.3 and 19.9%, respectively. Model refinement statistics are listed in Table III.
Table III.
Refinement Statistics
| Enzyme in complex with d-glucose | |
|---|---|
| Resolution limits (Å) | 30.0–1.4 |
| R-factor (overall)/number of reflectionsa | 0.173/324,820 |
| R-factor (working)/number of reflections | 0.172/308,445 |
| R-factor (free)/number of reflections | 0.199/16,375 |
| Number of protein atoms | 11,566 |
| Number of heteroatoms | 1667 |
| Average B values | |
| Protein atoms (Å2) | 20.0 |
| Ligand (Å2) | 15.1 |
| Solvent (Å2) | 29.9 |
| Weighted RMS deviations from ideality | |
| Bond lengths (Å) | 0.012 |
| Bond angles (º) | 1.95 |
| Planar groups (Å) | 0.010 |
| Ramachandran regions (%)b | |
| Most favored | 91.7 |
| Additionally allowed | 8.3 |
| Generously allowed | 0.1 |
| Disallowed | 0.0 |
R-factor = (∑|Fo − Fc|/∑|Fo|) where Fo is the observed structure factor amplitude, and Fc is the calculated structure factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.31
Production of a site-directed mutant protein
The D273N mutation was introduced using methods described within the QuikChange site-directed mutagenesis kit (Stratagene). The mutant protein was expressed, purified, and dialyzed in a manner identical to that for the wild-type enzyme.
Measurement of enzymatic activity
To assay the glucosidase activity of wild-type DesR, reactions were set up using 4-nitrophenyl β-d-glucopyranoside (pNP-Glc) as the substrate in concentrations ranging from 1.5 to 25 mM. Reactions were preincubated at 25°C and initiated upon the addition of 100 μL DesR (final concentration: 0.5 μM). The glucosidase activity of DesR was monitored spectrophotometrically by following the increase in absorbance at 400 nm due to pNP-Glc hydrolysis, which leads to free p-nitrophenol. Kinetic parameters were determined similarly for the D273N mutant enzyme, except with a final protein concentration of 10 μM. For both the wild-type enzyme and the D273N mutant protein, the reactions were performed in triplicate, and the kinetic parameters presented in Table I are the averages. The data were fitted to the equation v0 = (Vmax[S])/(KM + [S]). The kcat values were calculated according to the equation kcat = Vmax/[ET].
Acknowledgments
The authors thank Grover L. Waldrop for helpful comments. A portion of the research described in this manuscript was performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. The authors gratefully acknowledge Norma E. C. Duke for assistance during the X-ray data collection at Argonne.
Glossary
- MOPS
3-(N-morpholino)propanesulfonic acid
- PCR
polymerase chain reaction
- pNP-Glc
4-nitrophenyl β-d-glucopyranoside
- TEV
tobacco etch virus
- Tris
tris-(hydroxymethyl)aminomethane
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Madigan MT, Martinko JM. Biology of Microorganisms, 11 ed. Upper Saddle River, NJ: Pearson, Prentice Hall; 2006. [Google Scholar]
- 2.Baltz RH. Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Ehrlich J, Gottlieb D, Burkholder PR, Anderson LE, Pridham TG. Streptomyces venezuelae, N. Sp., the source of chloromycetin. J Bacteriol. 1948;56:467–477. doi: 10.1128/jb.56.4.467-477.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambalot RH, Cane DE. Isolation and characterization of 10-deoxymethynolide produced by Streptomyces venezuelae. J Antibiot. 1992;45:1981–1982. doi: 10.7164/antibiotics.45.1981. [DOI] [PubMed] [Google Scholar]
- 5.Xue Y, Zhao L, Liu HW, Sherman DH. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: architecture of metabolic diversity. Proc Natl Acad Sci USA. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasaki J, Mizoue K, Morimoto S, Omura S. Microbial glycosylation of macrolide antibiotics by Streptomyces hygroscopicus ATCC 31080 and distribution of a macrolide glycosyl transferase in several Streptomyces strains. J Antibiot. 1996;49:1110–1118. doi: 10.7164/antibiotics.49.1110. [DOI] [PubMed] [Google Scholar]
- 7.Vilches C, Hernandez C, Mendez C, Salas JA. Role of glycosylation and deglycosylation in biosynthesis of and resistance to oleandomycin in the producer organism, Streptomyces antibioticus. J Bacteriol. 1992;174:161–165. doi: 10.1128/jb.174.1.161-165.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez C, Olano C, Mendez C, Salas JA. Characterization of a Streptomyces antibioticus gene cluster encoding a glycosyltransferase involved in oleandomycin inactivation. Gene. 1993;134:139–140. doi: 10.1016/0378-1119(93)90189-a. [DOI] [PubMed] [Google Scholar]
- 9.Quiros LM, Hernandez C, Salas JA. Purification and characterization of an extracellular enzyme from Streptomyces antibioticus that converts inactive glycosylated oleandomycin into the active antibiotic. Eur J Biochem. 1994;222:129–135. doi: 10.1111/j.1432-1033.1994.tb18850.x. [DOI] [PubMed] [Google Scholar]
- 10.Quiros LM, Aguirrezabalaga I, Olano C, Mendez C, Salas JA. Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol Microbiol. 1998;28:1177–1185. doi: 10.1046/j.1365-2958.1998.00880.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhao L, Beyer NJ, Borisova SA, Liu HW. Beta-glucosylation as a part of self-resistance mechanism in methymycin/pikromycin producing strain Streptomyces venezuelae. Biochemistry. 2003;42:14794–14804. doi: 10.1021/bi035501m. [DOI] [PubMed] [Google Scholar]
- 12.Faure D. The family-3 glycoside hydrolases: from housekeeping functions to host-microbe interactions. Appl Environ Microbiol. 2002;68:1485–1490. doi: 10.1128/AEM.68.4.1485-1490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ketudat-Cairns JR, Esen A. beta-Glucosidases. Cell Mol Life Sci. 2010;67:3389–3405. doi: 10.1007/s00018-010-0399-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 15.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 16.Rigden DJ, Mello LV, Galperin MY. The PA14 domain, a conserved all-beta domain in bacterial toxins, enzymes, adhesins and signaling molecules. Trends Biochem Sci. 2004;29:335–339. doi: 10.1016/j.tibs.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Holden HM, Ito M, Hartshorne DJ, Rayment I. X-ray structure determination of telokin, the C-terminal domain of myosin light chain kinase, at 2.8 A resolution. J Mol Biol. 1992;227:840–851. doi: 10.1016/0022-2836(92)90226-a. [DOI] [PubMed] [Google Scholar]
- 18.Sadeghi-Khomami A, Lumsden MD, Jakeman DL. Glycosidase inhibition by macrolide antibiotics elucidated by STD-NMR spectroscopy. Chem Biol. 2008;15:739–749. doi: 10.1016/j.chembiol.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Varghese JN, Hrmova M, Fincher GB. Three-dimensional structure of a barley beta-D-glucan exohydrolase, a family 3 glycosyl hydrolase. Structure. 1999;7:179–190. doi: 10.1016/s0969-2126(99)80024-0. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida E, Hidaka M, Fushinobu S, Koyanagi T, Minami H, Tamaki H, Kitaoka M, Katayama T, Kumagai H. Role of a PA14 domain in determining substrate specificity of a glycoside hydrolase family 3 beta-glucosidase from Kluyveromyces marxianus. Biochem J. 2010;431:39–49. doi: 10.1042/BJ20100351. [DOI] [PubMed] [Google Scholar]
- 21.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 22.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gloster TM. Development of inhibitors as research tools for carbohydrate-processing enzymes. Biochem Soc Trans. 2012;40:913–928. doi: 10.1042/BST20120201. [DOI] [PubMed] [Google Scholar]
- 24.Thoden JB, Timson DJ, Reece RJ, Holden HM. Molecular structure of human galactokinase: implications for Type II galactosemia. J Biol Chem. 2005;280:9662–9670. doi: 10.1074/jbc.M412916200. [DOI] [PubMed] [Google Scholar]
- 25.Larner J, Schliselfeld LH. Studies on amylo-1, 6-glucosidase. Biochim Biophys Acta. 1956;20:53–61. doi: 10.1016/0006-3002(56)90262-1. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowtan K. 'DM:' an automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newslett Prot Cryst. 1994;31:34–38. [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.