

Abstract
Polyunsaturated fatty acids (PUFAs) are made in some strains of deep-sea bacteria by multidomain proteins that catalyze condensation, ketoreduction, dehydration, and enoyl-reduction. In this work, we have used the Udwary-Merski Algorithm sequence analysis tool to define the boundaries that enclose the dehydratase (DH) domains in a PUFA multienzyme. Sequence analysis revealed the presence of four areas of high structure in a region that was previously thought to contain only two DH domains as defined by FabA-homology. The expression of the protein fragment containing all four protein domains resulted in an active enzyme, while shorter protein fragments were not soluble. The tetradomain fragment was capable of catalyzing the conversion of crotonyl-CoA to β-hydroxybutyryl-CoA efficiently, as shown by UV absorbance change as well as by chromatographic retention of reaction products. Sequence alignments showed that the two novel domains contain as much sequence conservation as the FabA-homology domains, suggesting that they too may play a functional role in the overall reaction. Structure predictions revealed that all domains belong to the hotdog protein family: two of them contain the active site His70 residue present in FabA-like DHs, while the remaining two do not. Replacing the active site His residues in both FabA domains for Ala abolished the activity of the tetradomain fragment, indicating that the DH activity is contained within the FabA-homology regions. Taken together, these results provide a first glimpse into a rare arrangement of DH domains which constitute a defining feature of the PUFA synthases.
Keywords: polyunsaturated fatty acids, dehydratase, polyketide synthase, Udwary-Merski Algorithm
Introduction
Long-chain polyunsaturated fatty acids (PUFAs) are made in some species of deep-sea bacteria by a multidomain enzyme complex that resembles the well-studied polyketide synthases (PKSs).1,2 This widely conserved PUFA synthase complex consists of five genes which have been found to be sufficient for the production of PUFAs in otherwise non-producing Escherichia coli.3,4 The conserved genes are pfaA, pfaB, pfaC, pfaD, which encode PUFA synthases containing enzyme domains for acyl tranferases, keto-acyl synthase (KS), acyl carrier protein (ACP), keto-acyl reductase (KR), enoyl reductase (ER), and dehydratase (DH) activities (Fig. 1). The full reconstitution of PUFA biosynthesis in E. coli also requires pfaE, which encodes a required phosphopantetheine transferase essential for the activation of ACP domains through chemical modification (Fig. 1). Although some of the partial functions of enzyme components from this pathway have been described in the literature, the overall activity and programming of reactions remains unexplored.5–8
Figure 1.
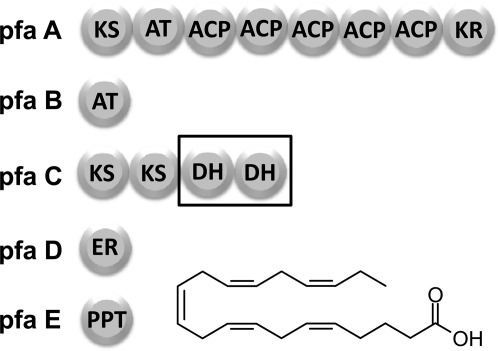
Multienzyme complex for the anaerobic production of PUFAs in deep-sea bacteria. The PKS multienzyme for the anaerobic production of eicosapentaenoic acid (EPA) in Photobacterium profundum contains five required genes (Pfa A,B,C,D, and E). The DH domains identified by sequence homology to FabA/Z are arranged in tandem and housed within the PfaC multienzyme. This arrangement of domains is well conserved among deep-sea bacteria that produce PUFAs.
One of the defining features of this class of PUFA synthases is the presence of a conserved pair of DH domains which are thought to introduce cis double bonds into the final structure of the PUFA product via the dehydration of the β-hydroxyacyl-CoA intermediate, with a subsequent isomerization step.1 It is currently not known how these two DH domains act in concert to generate the pattern of double bonds seen in PUFAs.
DH domains can be easily identified by their sequence similarity to FabA and FabZ, the two DH enzymes involved in fatty acid biosynthesis in E. coli.9,10 The structures for FabA and FabZ reveal homodimeric DH enzymes with active sites containing amino acids contributed by both subunits.11 However, when the DH domains are located within a PKS or fatty acid synthase (FAS) multienzyme, instead of forming homodimers, they exist as a pair of contiguous hotdog domains in which the FabA-homology region is followed by an additional C-terminal hotdog “pseudo-domain” that stabilizes the overall double-hotdog structure and contributes to its activity.12,13
The identification of domain boundaries remains an area of great interest in the study of multidomain proteins precisely because boundary regions may serve as appropriate sites for the insertion, excision, or swapping of enzyme domains.14–16 Also, the identification of boundary sites is important in the expression of embedded domains as stand-alone proteins to be used in mechanistic or structural studies. Toward this identification of domain boundaries, computational strategies have been implemented for the analysis of sequence regions that corresponds to structured domains and differentiate them from unstructured linkers.16
In this work, we have used the Udwary-Merski Algorithm (UMA) to define the domain boundaries for the DH domains from the PUFA synthase from Photobacterium profundum in an effort to generate independent DH domains that could be further interrogated both functionally and structurally. The UMA sequence analysis revealed the presence of two additional domains that have the same degree of sequence conservation as the FabA-homology regions. These two new domains were included in a recombinant protein fragment that was competent to catalyze the hydration of a surrogate substrate. Taken together, the results show that reconstitution of DH activity requires the presence of two additional domains of unknown function located immediately N-terminal from each FabA domain in what appears to be an alternative and unprecedented arrangement of domains and pseudo-domains in a conserved multienzyme.
Results
Protein sequence analysis
Preliminary analysis of the amino acid sequence for PfaC (GenBank Accession No. AF409100.1) using the Conserved Domain Search application from NCBI resulted in the identification of the four expected functional domains: two ketosynthase (KS) domains and two DH domains.17,18 From this analysis, it was clear that, in addition to the four recognizable enzyme domains, the sequence contained two additional long stretches of amino acids located directly N-terminal to the FabA-homology domains. These long stretches were not recognized by the NCBI algorithm as conserved or as previously described domains.
To gain some understanding about the nature of these two “linker” or unstructured sequences, we implemented the UMA software that was developed precisely for highlighting unknown domains within multienzymes.16 Analysis of the PfaC protein sequence by the UMA software revealed a total of six regions of high probability of forming a structured domain as suggested by their high UMA score (Fig. 2). These six regions included the two KS domains and the two FabA-like DH domains that had been recognized in the initial analysis. In addition to these four expected domains, there were two more regions with a high UMA score corresponding to amino acids 1096–1305 and 1498–1755, which we have termed pseudo-domains DH1′ and DH2′.
Figure 2.
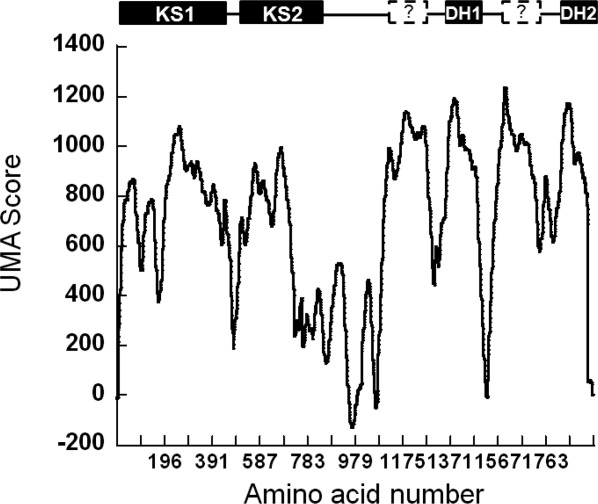
The PfaC sequence was analyzed using the UMA algorithm to detect the presence of additional structured domains within the DH region. The areas of high UMA score are typically considered to coincide with structured domains. The data shows that in addition to the four known domains detected by sequence alone (shown on bar above the graph), there are two previously uncharacterized regions of high UMA score directly N-terminal to the DH domains. These two new regions of predicted structure were termed DH′ pseudo-domains. This exercise in primary sequence analysis resulted in the design of DH1-DH2-UMA, a tretradomain fragment with activity.
A possible functional role for the DH1′ and DH2′ pseudo-domains may be reflected in their degree of sequence conservation. The multiple sequence alignment for the DH′ pseudo-domains revealed a total of 24 amino acids that were conserved among the 32 species included in the alignment [Fig. 3(B)]. This number is comparable to the 28 conserved amino acids in the sequence alignment of the known FabA/Z-homology domains among the same 32 species [Fig. 3(A); a complete list of the sequences analyzed can be found in the supplementary material]. Although the number of identical amino acids in the alignments is similar for both FabA domains and the DH′ pseudo-domains, it is clear that the distribution of conserved amino acids is different in each. While conserved amino acids in the FabA-homology domains cluster at the core and include the putative active site histidine [Fig. 3(A)], the conserved amino acids in the DH′ pseudo-domains are more evenly distributed throughout the sequence [Fig. 3(B)]. The absence of a conserved His indicates that the pseudo-domains do not have DH activity of their own.
Figure 3.

Multiple sequence alignment for DH domains and pseudo-domains. The sequences corresponding to the (A) FabA-homology regions and (B) the DH′ pseudo-domains from 32/34 different organisms were aligned using ClustalW. Even though the number of identical residues in the FabA-homology regions was similar to the number of identical residues in the pseudo-domains, their distribution is different. While in the FabA-homology region the conserved amino acids cluster inside a core, in the DH′ pseudo-domains the conserved residues are more evenly distributed.
Secondary structure prediction and 3D model of domains and pseudo-domains
To better predict a possible functional role for the DH′ pseudo-domains, we built three-dimensional (3D) models for domains and pseudo-domains using the Phyre Server19 (http://www.sbg.bio.ic.ac.uk/phyre2). As expected, the FabA-homology DH domains were recognized by the server as hotdog fold DHs, with the His and Glu residues typical of a DH active site (Fig. 4). More striking was the fact that the DH′ pseudo-domains, which do not have the conserved His residue, were also recognized by the server as single hotdog folds. Thus, even though the sequence of the DH′ pseudo-domains does not resemble any known structures, their secondary structure predictions strongly suggest that they belong to the hotdog protein family.
Figure 4.
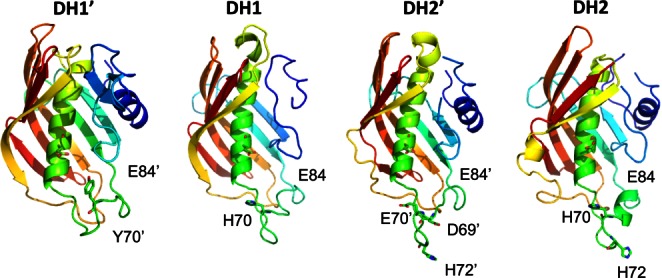
3D models for DH domains and DH′ pseudo-domains. A comparison of the 3D models obtained for the FabA-homology regions (DH1 and DH2) and for the uncharacterized pseudo-domains (DH1′ and DH2′) using the Phyre Server anticipate that the pseudo-domains will likely consist of a hotdog fold with differences in conserved amino acids in key positions. While the DH models contain a conserved His in the active site (number 70 in FabA) which has been implicated in DH activity in other DH domains, the DH′ pseudo-domains do not. Figures were made using PyMol (Delano Scientific). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Enzyme expression
Initially, a number of DNA sequences corresponding to DH domains were amplified by PCR and cloned into a vector for recombinant expression in E. coli. The list of attempted fragments is summarized in Figure 5(A). Two “short” fragments, DH1S (H1318-S1491) and DH2S (I1787-C-term) were designed to include only the conserved FabA-homology sequence. Two additional “longer” fragments, DH1L (F1249-S1491) and DH2L (S1733-C-term), were designed to include the conserved sequence N-terminal to the FabA-homology region. Finally, to explore the possibility that the two FabA-homology regions stabilized one another, we also generated protein fragments which contained both FabA-homology regions, DH1-DH2S (H1318-C-Term) and DH1-DH2L (F1249-C-Term). All of these protein fragments were expressed as GST fusion proteins or as His-tagged proteins in E. coli and all were found to be insoluble as evidenced by their presence in the lysis pellet (data not shown).
Figure 5.
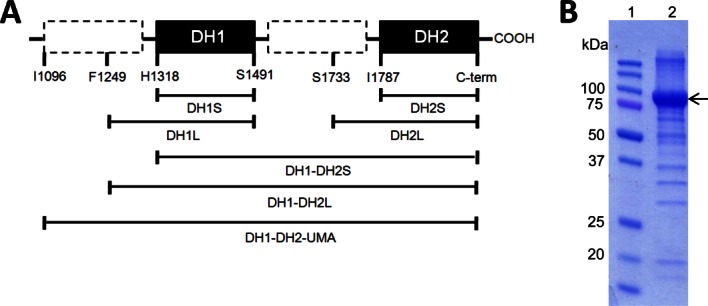
Design of soluble and active DH constructs. (A) A number of protein constructs were designed to contain single and multiple DH domains. Of the fragments that were cloned, only the DH1-DH2-UMA, which was made according to the UMA results, was found to be soluble and stable. (B) SDS-PAGE gel shows that DH1-DH2-UMA protein can be made with a high yield (1 mg pure protein per L of culture) and purity. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Based on the UMA analysis and on the Phyre prediction, fragment DH1-DH2-UMA (I1096-C-Term) was designed and expressed as a His-tagged protein in soluble form. After nickel resin purification and size exclusion chromatography, a total yield of 1.0 mg of pure protein was obtained per liter of culture [Fig. 5(B)].
Enzyme activity
The purified DH1-DH2-UMA enzyme was incubated with either β-hydroxybutyryl-CoA, crotonyl-CoA, or crotonyl-N-acetylcysteamine (NAC thioester) to detect enzyme activity in either the forward or the reverse direction. Only the conversion of crotonyl-CoA to β-hydroxybutyryl-CoA (hydration reaction) was observed as evidenced by the decrease in substrate absorbance at 260 nm [Fig. 6(A)]. The activity of DH1-DH2-UMA against crotonyl-NAC was substantially decreased, as was the activity in the direction of the dehydration of β-hydroxybutyryl-CoA [Fig. 6(B)]. The lower activity against the NAC crotonyl thioester suggests a substantial degree of molecular recognition of the pantetheine arm by the enzyme.
Figure 6.
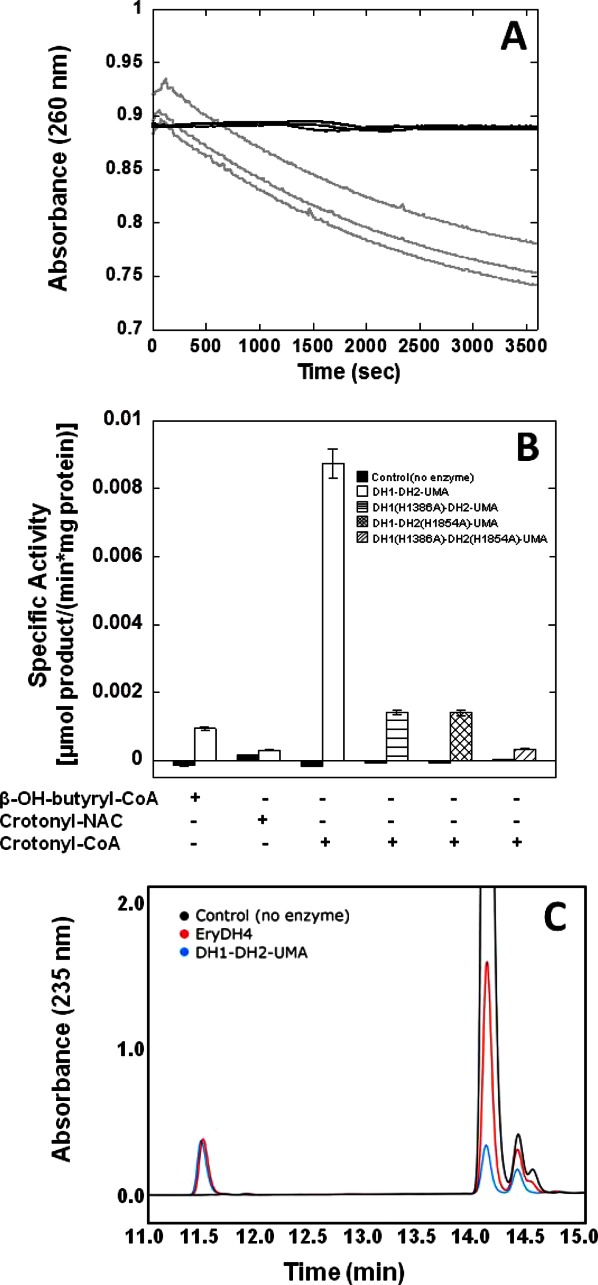
Enzymatic activity of DH1-DH2-UMA. (A) The decrease in absorbance at 260 nm was monitored as the substrate, crotonyl-CoA, was hydrated to β-hydroxybutyryl-CoA. The light gray line denotes the disappearance of the substrate in the presence of DH1-DH2-UMA and the dark gray lines denote the control with no enzyme. (B) The rates of the enzyme reaction were measured toward the substrates β-hydroxybutyryl-CoA, crotonyl-NAC, and crotonyl-CoA and converted to units of specific activity. Additionally, the activity of active site mutants DH1(H1386A)-DH2-UMA and DH1-DH2(H1854A)-UMA, as well as the activity of the double mutant was measured toward crotonyl-CoA. (C) The formation of hydrated product of crotonyl-pantetheine was confirmed by reverse phase HPLC. The black trace corresponds to the substrate alone with a major peak eluting at 14 min. In the presence of DH1-DH2-UMA (blue trace), the formation of a peak which elutes at 11.5 min can be appreciated. This peak is also observed in the presence of DH4 from the erythromycin gene cluster whose activity is well established (red trace).20
The chemical nature of the hydration product was ascertained by reverse phase HPLC [Fig. 6(C)]. In these experiments, crotonyl-S-pantetheine was used as a substrate. In the chromatogram, the unreacted substrate eluted with a retention time of 14 min. After a 2-h incubation with DH1-DH2-UMA, a peak with a retention time of 11.5 min appears. This is the same peak that is observed after incubation with the EryDH4, a bona fide DH whose activity is well established.20 The simultaneous replacement of both active site His residues in the FabA-homology regions for Ala, nearly abolished the activity of DH1-DH2-UMA (3% residual activity), whereas the single mutants have partial activity of 16% that of the wild type [Fig. 6(B)].
Discussion
The biosynthesis of PUFAs in many deep-sea bacteria is carried out by a family of enzymes that contain a unique and conserved arrangement of enzyme domains.1,2 PUFA synthases have been found in metagenomic DNA from marine samples collected throughout the world,21 indicating that anaerobic PUFA biosynthesis is a widely selected mechanism for microbial adaptation to high-pressure and low temperature environments. The widespread interest in elucidating how the PUFA synthase carries out its function is revealed in the amount of published work on the individual enzymatic activities of PUFA synthases. Bumpus et al.6 showed for the first time the in vitro activity of the enoyl reductase (PfaD) enzyme from Shewanella oneidensis PUFA synthase and Jiang et al.5 interrogated the role of the tandem ACP arrangement, which is a hallmark of PUFA synthases. Additionally, Trujillo et al.7 recently characterized the solution structure of the tandem ACP fragment and Rodríguez-Guilbe et al.8 uncovered a thioesterase associated with fatty acid production in this family of marine organisms. In this work, we have addressed another conserved feature of PUFA synthases, a pair of conserved DH domains arranged in tandem near the C-terminus of the multidomain protein, PfaC.
Analysis of the sequence of PfaC protein using the UMA revealed the presence of two new DH′ pseudo-domains located directly N-terminal to the regions of FabA-homology. These pseudo-domains were found to be essential for the proper expression of protein fragments, since only the protein fragments that included both pseudo-domains were soluble, stable, and active. However, given that the DH′ pseudo-domains lack any similarity to any known domains, they would not have been detected by sequence conservation alone. Interestingly, the UMA assigns a score to the sequence according to a mathematical function that incorporates sequence conservation, hydrophobicity and propensity to form secondary structure, thus providing a more robust way of identifying new domains where no structural or functional information exists. Our identification of two new domains confirms the general applicability of the UMA for the identification of structural units of unknown functions within multidomain proteins.22,23
The predicted secondary structure for both DH′ pseudo-domains was that of a hotdog fold, which is also the expected 3D topology of the FabA-homology DH domains. This predicted arrangement of contiguous hotdog folds points towards an overall double hotdog structure, which has become the widely accepted model for embedded DHs based on structural and biochemical evidence.12,13,24,25 However, several differences exist between the PUFA DH arrangement and its FAS and PKS evolutionary cousins. While in FAS/PKS DH the pseudo-domains are located C-terminal to the FabA-homology domain, in the PUFA DH, the pseudo-domains are located N-terminal to the FabA-homology domain. This alternative gene structure of the PUFA DH suggests a tandem gene duplication event that took place independently in terrestrial FAS/PKS and marine PUFA synthase for the generation of functional DH dimers, resulting in two alternative convergent topological solutions. Another difference between FAS/PKS DH and PUFA DH is that, while FAS/PKS DH domains consist of didomains (one FabA-homology domain plus one pseudo-domain), the PUFA DH complex invariably consists of a tetradomain (two FabA-homology plus two pseudo-domains).
The simultaneous replacement of both active site His residues located in the FabA-homology regions abolished hydratase activity, indicating that these domains carry out the dehydration reaction [Fig. 6(B)]. It is not clear from these results whether the pseudo-domains play an active role in the reaction. This report does not address the important question of how the four protein domains are paired in the functional assembly, although that is an area of current and future development. Additional structural characterization of DH1-DH2-UMA will have to be carried out in order to elucidate how the four domains are arranged three-dimensionally to form a functional complex.
Substantial work has been aimed at determining the specific role of pseudo-domains in the activity of the FAS multienzyme beyond that of stabilizing the intermolecular dimeric structure.12,26 DH pseudo-domains in the animal FAS have been shown to contribute catalytic residues that are essential for DH activity.12 Furthermore, animal FAS has been shown to also contain ketoreductase (KR′) pseudo-domains which are not near their cognate KR domain in the linear sequence, but still contribute to KR function.26 This observation, which was originally derived from careful experiments, was later confirmed by the crystal structure of the full-length animal FAS.25 In this report, the multiple sequence alignment generated for the pseudo-domains of the PUFA synthase reveal levels of sequence conservation (26% and 15% identities for DH1′ and DH2′, respectively) that were comparable to the sequence conservation of the FabA-homology domains (22% and 16% identities for DH1 and DH2, respectively). This level of sequence similarity among the pseudo-domains is suggestive of a role in DH function beyond that of a structural scaffold for dimerization.
Methods
Sequence analysis
The sequence for PfaC (GenBank Accession No. AF409100.1) was used as input for the BLAST (NCBI) to identify conserved domains.17,18 The same sequence was analyzed by the UMA program that was kindly provided by Craig Townsend (Johns Hopkins University) and Daniel Udwary (University of Rhode Island).16 The UMA algorithm is a scoring function of three weighted terms that depend on the (i) degree of sequence conservation as evidenced by a multiple sequence alignment, (ii) secondary structure propensity, and (iii) hydrophobicity. A multiple alignment of homologues of PfaC from related organisms was performed in CLUSTALW with an output in “.pir” format and a secondary structure prediction for PfaC was performed using the PSIPRED Server (University College London). The output from the secondary structure prediction was used to generate an “.ss” file. Finally, both the “.pir” alignment and the “.ss” secondary structure prediction were used as inputs for the “uma19.pl” application with the input parameters in Table I. The results of the calculation were visualized using Keleidagraph for Windows.
Table I.
Input Parameters for the UMA Calculations
| Parameter | Value |
|---|---|
| Homology matrix | blosum 30 |
| Gap to gap penalty | 0 |
| Gap to aa penalty | −4 |
| Component averaging (k) | 5 |
| Final averaging (gamma) | 20 |
| Sim score weight | 10 |
| Struc score weight | 1 |
| Hydro score weight | 5 |
The regions of high UMA score used to generate sequence fragments whose 3D model was calculated on the Phyre Server (Imperial College, UK)19 based on sequence similarity and a secondary structure prediction.
Cloning, expression, and purification
Different DH fragments were PCR-amplified from fosmid 8E1, which was originally isolated and sequenced in Allen and Bartlett,2 and kindly provided by Dr. Eric Allen from the Scripps Institution of Oceanography (GenBank Accession AF409100.1). All restriction endonucleases, polynucleotide kinase, T4 DNA ligase, and alkaline phosphatase were purchased from New England Biolabs. The oligonucleotides used in the PCR amplification of the fragments are summarized in Table II. For cloning into pGEX4T-3 vector (GE Healthcare), the amplified DNA was phosphorylated using polynucleotide kinase and cloned into pUC19 which was previously digested with SmaI and treated with alkaline phosphatase. The ligation mixture was used to transform DH10B cells and clones were selected in LB-agar containing ampicillin (100 μg/mL). Insertion of the DH fragment into pUC19 was confirmed by agarose gel electrophoresis. The resulting plasmid pUC19:DH was digested with BamHI and SmaI and the resulting excised DNA fragment was cloned into the corresponding sites of pGEX4T-3.
Table II.
Oligonucleotides Used to Amplify the Different DH Fragments in This Study
| DH construct | Oligonucleotide sequence |
|---|---|
| DH1S | Fwd: 5′- CATGCATGGGATCCAACTTGCTAGACGCAAATATCGCA -3′ |
| Rv: 5′- CATGCATGCCCGGGTCATGATTCTTCTTTGATCATCACG -3′ | |
| DH1L | Fwd: 5′- CACCTTCTCTTACGAATGTTTCGTTGGC -3′ |
| Rv: 5′- CATGCATGCCCGGGTCATGATTCTTCTTTGATCATCACG -3′ | |
| DH2S | Fwd: 5′- CATGCATGGGATCCAACTTACTGGATAAGAAAGCCGTT -3′ |
| Rv: 5′- TCAGGCTTCTTCAATACAGATTGC -3′ | |
| DH2L | Fwd: 5′- CATGCATGGGATCCTTCAGCTTCGAACTCAGTACCGA -3′ |
| Rv: 5′- TCAGGCTTCTTCAATACAGATTGC -3′ | |
| DH1-DH2-S | Fwd: 5′-CACCAACTTGCTAGACGCAAATATCGCA-3′ |
| Rv: 5′-TCAGGCTTCTTCAATACAGATTGC-3′ | |
| DH1-DH2-L | Fwd: 5′-CACCTTCTCTTACGAATGTTTCGTTGGC-3′ |
| Rv: 5′-TCAGGCTTCTTCAATACAGATTGC-3′ | |
| DH1-DH2 UMA | Fwd: 5′-CACCCGCAAACCTTGTATCTGGGATTA-3′ |
| Rv: 5′-TCAGGCTTCTTCAATACAGATTGC-3′ | |
| DH1(H70A) | Fwd: 5′-ACTTCCCTTGTGCATTCAAAGATGA -3′ |
| Rv: 5′- TCATCTTTGAATGCACAAGGGAAGT-3′ | |
| DH2(H70A) | Fwd: 5′- CTTCCAATTCGCATTCCACCAAG-3′ |
| Rv: 5′-CTTGGTGGAATGCGAATTGGAAG-3′ |
For the cloning of fragments into pET200TOPO, the amplified DNA was gel purified using the QIAquick Gel Extraction Kit (QIagen) and incubated with pET200TOPO (Invitrogen). The resulting clones were selected in LB-agar containing kanamycin (100 μg/mL). All resistant clones were introduced into E. coli strain BL21-DE3-Codon Plus-RIL (Promega) and grown in liquid LB at 37oC until the OD600 = 0.2 at which time the temperature was decreased to 22°C until the OD600 = 0.6 at which time protein expression was induced to a final concentration 1 mM IPTG. After 16 h, the cells were collected and resuspended in lysis buffer (50 mM Na3HPO4 pH 7.4, 300 mM NaCl, 1 mM DTT, 1 mM MgCl2,10% glycerol, 0.1 mg/mL lysozyme, and DNAse) for 1 h, sonicated and centrifuged at a speed of 14,000 rpm at 4oC for 30 min in a J2-21 Beckman centrifuge, JA17 rotor. Samples were collected for the total, supernatant and pellet to assess solubility of the protein products.
For His-tagged soluble proteins, the lysate was collected and poured through a column filled with Ni-NTA resin (Qiagen) equilibrated in 220.0 mM Na3HPO4 pH 7.4, 500 mM NaCl, 10% glycerol, 1.0 mM DTT. The DH fragment was eluted with the same buffer containing 500 mM imidazole.
Eluted protein was infused into a HiPrep Superdex 200 10/300 GL column (GE Healthcare) operated at room temperature and equilibrated in 25 mM Tris pH 8.0, 150 mM NaCl, and 10% glycerol. The proteins eluted in two peaks at 0.5 mL/min; one at 26 min and the other one at 30 min, consistent with the molecular mass of the dimer and the monomer, respectively. Each run was preceded by a run with a mixture of standard proteins [ferritin (440,000 Da), aldolase (158,000 Da), conalbumin (75,000 Da), carbonic anhydrase, (29,000 Da), ribonuclease A (13,700 Da), and apropotin (6500 Da)]. The fraction containing the protein was concentrated and stored at −20°C. Typical yields for all proteins were 1.0 mg of protein per liter of culture, purity ∼80% by SDS-PAGE using 4–15% Mini-PROTEAN® TGX gels (BioRad®).
Activity assays
Both hydratase and DH activities were measured for DH1-DH2-UMA by incubation of the enzyme with either crotonyl-CoA (Sigma), β-hydroxybutyryl-CoA (Sigma) or crotonyl-NAC as substrates. Crotonyl-NAC was synthesized from crotonic acid (Sigma) and NAC (Sigma) using a DCC coupling strategy as describes by others27 and purified by flash column chromatography on silica gel using 1:1 ethyl acetate: ethyl ether. Enzymatic reactions were followed spectrophotometrically by monitoring the absorbance at 260 nm in a 96-well plate format on a Spectramax 190 instrument (Molecular Devices). The total volume was 200 μL (25 mM Tris, 150 mM NaCl, 10% glycerol, pH 8.0, 26 μM DH1-DH2-UMA, and 234 μM of substrate). Control reactions with no enzyme, but containing the same substrate and buffer concentration, were performed. The values for the absorbance slope (given in mAU/min) were converted to units of μmole of product per minute by using the following equation:
| (1) |
where the slope is given by the instrument in units of milli-absorbance (mAU) per minute, b is the path length measured to be 0.89 cm for a Voltotal = 200 μL in our 96-well plates. The ε is the molar extinction coefficient resulting from the loss of a double bond as defined by the difference in absorbance between crotonyl-CoA and β-hydroxybutyryl-CoA at a particular wavelength. The extinction coefficient was calculated to be ε = 969.9 M−1 cm−1 for the reaction monitored at 260 nm and ε = 790.7 M−1 cm−1 for the reaction monitored at 235 nm.
HPLC analysis of reaction products
Synthesis and characterization of crotonyl-pantetheine from crotonyl-chloride and (D)-pantetheine was carried out as previously described.28 The following reaction was initiated: 10 μM DH1-DH2-UMA or EryDH4, 2 mM crotonyl-pantetheine, 10% (v/v) glycerol, 50 mM NaCl, 150 mM HEPES, pH 7.5 for 16 h. The reaction was then injected onto C18 reversed-phase HPLC column, and monitored at 235 nm (100% water to 100% MeOH containing 0.1% TFA in 20 min). A mock reaction was also performed with no enzyme as a control.
Site-directed mutagenesis of active site histidines
The presumed active site histidines in positions H1386 and H1854 of the PfaC protein were replaced with alanine residues by site directed mutagenesis. The “mega-primer” method was carried out as described previously29 but using oligonucleotide sequences in Table II. Two overlapping PCR reactions which encompass the mutagenesis site were carried out using a combination of mutagenic and flanking primers. The two resulting PCR products which contain the mutation were subjected to additional cycles of amplification with the flanking or outside primers to generate the single mutants DH1(H1386A)-DH2-UMA and DH1-DH2(H1854A)-UMA, and the double mutant DH1(H1386A)-DH2(H1854A)-UMA which were all cloned into pET200TOPO as described previously. Successful amino acid replacement was ascertained by DNA sequencing of the resulting clones by the Sanger method.
Acknowledgments
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The authors thank Dr. Eric Allen from the Scripps Institution of Oceanography for providing Photobacterium profundum clones, Dr. Daniel Udwary from University of Rhode Island and Dr. Craig Townsend from Johns Hopkins University for providing the code for UMA. The authors also thank Dr. Adrian Keatinge-Clay from UT-Austin and Dr. Pearl Akamine for helpful suggestions and discussions. Dr. Rafael Arce Nazario (UPR-Rio Piedras) helped with the implementation of UMA and Mr. Angel Colón and Mr. Wallace Schroeder helped with running the UMA application.
Glossary
- ACP
acyl carrier protein
- AT
acyl tranferases
- CoA
coenzyme-A
- DH
dehydratase
- ER
enoyl reductase
- FAS
fatty acid synthase
- KR
keto-acyl reductase
- KS
keto-acyl synthase
- NAC
N-acetylcysteamine
- PKS
polyketide synthase
- PUFA
polyunsaturated fatty acids
- rpHPLC
reverse phase high-performance liquid chromatography
- UMA
Udwary-Merski Algorithm
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, Yazawa K, Knauf V, Browse J. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science. 2001;293:290. doi: 10.1126/science.1059593. [DOI] [PubMed] [Google Scholar]
- 2.Allen EE, Bartlett DH. Structure and regulation of the omega-3 polyunsaturated fatty acid synthase genes from the deep-sea bacterium Photobacterium profundum strain SS9. Microbiology. 2002;148:1903–1913. doi: 10.1099/00221287-148-6-1903. [DOI] [PubMed] [Google Scholar]
- 3.Orikasa Y, Nishida T, Hase A, Watanabe K, Morita N, Okuyama H. A phosphopantetheinyl transferase gene essential for biosynthesis of n−3 polyunsaturated fatty acids from Moritella marina strain MP-1. FEBS Lett. 2006;580:4423–4429. doi: 10.1016/j.febslet.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Okuyama H, Orikasa Y, Nishida T, Watanabe K, Morita N. Bacterial genes responsible for the biosynthesis of eicosapentaenoic and docosahexaenoic acids and their heterologous expression. Appl Environ Microbiol. 2007;73:665–670. doi: 10.1128/AEM.02270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang H, Zirkle R, Metz JG, Braun L, Richter L, Van Lanen SG, Shen B. The role of tandem acyl carrier protein domains in polyunsaturated fatty acid biosynthesis. J Am Chem Soc. 2008;130:6336–6337. doi: 10.1021/ja801911t. [DOI] [PubMed] [Google Scholar]
- 6.Bumpus SB, Magarvey NA, Kelleher NL, Walsh CT, Calderone CT. Polyunsaturated fatty-acid-like trans-enoyl reductases utilized in polyketide biosynthesis. J Am Chem Soc. 2008;130:11614–11616. doi: 10.1021/ja8040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trujillo U, Vazquez-Rosa E, Oyola-Robles D, Stagg LJ, Vassallo DA, Vega IE, Arold ST, Baerga-Ortiz A. Solution structure of the tandem acyl carrier protein domains from a polyunsaturated fatty acid synthase reveals beads-on-a-string configuration. PLoS ONE. 2013;8:e57859. doi: 10.1371/journal.pone.0057859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodríguez-Guilbe M, Oyola-Robles D, Schreiter ER, Baerga-Ortiz A. Structure, activity and substrate selectivity of the Orf6 thioesterase from Photobacterium profundum. J Biol Chem. 2013;288:10841–10848. doi: 10.1074/jbc.M112.446765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kass LR, Brock DJH, Bloch K. β-hydroxydecanoyl thioester dehydrase. J Biol Chem. 1967;242:4418–4431. [PubMed] [Google Scholar]
- 10.Brock DJH, Kass LR, Bloch K. β-hydroxydecanoyl thioester dehydrase. J Biol Chem. 1967;242:4432–4440. [PubMed] [Google Scholar]
- 11.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure. 1996;4:253–264. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 12.Pasta S, Witkowski A, Joshi AK, Smith S. Catalytic residues are shared between two pseudosubunits of the dehydratase domain of the animal fatty acid synthase. Chem Biol. 2007;14:1377–1385. doi: 10.1016/j.chembiol.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Keatinge-Clay A. Crystal structure of the erythromycin polyketide synthase dehydratase. J Mol Biol. 2008;384:941–953. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yadav G, Gokhale RS, Mohanty D. SEARCHPKS: a program for detection and analysis of polyketide synthase domains. Nucleic Acids Res. 2003;31:3654–3658. doi: 10.1093/nar/gkg607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yadav G, Gokhale RS, Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J Mol Biol. 2003;328:335–363. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 16.Udwary DW, Merski M, Townsend CA. A method for prediction of the locations of linker regions within large multifunctional proteins, and application to a Type I polyketide synthase. J Mol Biol. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 20.Valenzano CR, You Y, Garg A, Keatinge-Clay A, Khosla C, Cane DE. Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J Am Chem Soc. 2010;132:14697–14699. doi: 10.1021/ja107344h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shulse CN, Allen EE. Diversity and distribution of microbial long-chain fatty acid biosynthetic genes in the marine environment. Environ Microbiol. 2011;13:684–695. doi: 10.1111/j.1462-2920.2010.02373.x. [DOI] [PubMed] [Google Scholar]
- 22.Crawford JM, Vagstad AL, Ehrlich KC, Udwary DW, Townsend CA. Acyl carrier protein-phosphopantetheinyltransferase partnerships in fungal fatty acid synthases. ChemBioChem. 2008;9:1559–1563. doi: 10.1002/cbic.200700659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeckh V, Scherlach K, Nützmann H, Shelest E, Schmidt-Heck W, Schuemann J, Martin K, Hertweck C, Brakhage AA. Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Nat Acad Sci USA. 2009;106:14558–14563. doi: 10.1073/pnas.0901870106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Crystal structures of dehydratase domains from the curacin polyketide biosynthetic pathway. Structure. 2010;18:94–105. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 26.Witkowski A, Joshi AK, Smith S. Characterization of the B-carbon processing reactions of the mammalian cytosolic fatty acid synthase: role of the central core. Biochemistry. 2004;43:10458–10466. doi: 10.1021/bi048988n. [DOI] [PubMed] [Google Scholar]
- 27.Neises B, Steglich W. Simple method for the esterification of carboxylic acids. Angew Chem Intl Ed. 1978;17:522–524. [Google Scholar]
- 28.Zheng J, Gay DC, Demeler B, White MA, Keatinge-Clay AT. Divergence of multimodular polyketide synthases revealed by a didomain structure. Nat Chem Biol. 2012;8:615–621. doi: 10.1038/nchembio.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.