

Abstract
The carboxy terminal tail of epidermal growth factor receptor (EGFR) plays a critical role in the regulation of the enzyme activity of the kinase. There is a good structural model for the mechanism by which the C-terminal tail proximal to the kinase domain contributes to the negative regulation of the activity. Its conformation in the active state, conversely, has remained elusive due to its dynamic nature. A recently published structure of EGFR kinase domain shows the conformation of the proximal C-terminal tail in the active kinase. Analysis of this conformational state of the C-terminal tail is presented, and some of the mutagenesis data is revisited. © 2013 The Protein Society
Keywords: EGFR, kinase, C-terminal tail, regulation, conformation
Epidermal Growth Factor Receptor (EGFR) belongs to the ErbB family of transmembrane receptor tyrosine kinases.1 It has a ligand binding extracellular region (residues 25–641), a transmembrane helix (642–668), and an intracellular chain that consists of membrane proximal juxtamembrane segment (669–705) followed by the kinase domain (706–979) and over 200 residue long C-terminal tail (980–1210). There are models from structural data for most of the protein,2–6 except a large part of the C-terminal tail. The C-terminal tail is rich in proline residue, its secondary structure content is predicted to be low and it has five phosphorylation sites. The sixth phosphorylation site downstream of the juxtamembrane region is in the activation loop of the kinase domain.7 However, phosphorylation is not essential for the enzyme activity.8 Instead, EGFR is activated allosterically by asymmetric dimerization of its kinase domain.9 The juxtamembrane segment plays a critical role in stabilizing asymmetric dimers and thus in its activation.4,6,10–13
In this model of dimerization driven activation of the kinase domain, the C-terminal lobe of one molecule (activator) packs against the N-terminal lobe of the other (receiver) thereby stabilizing the active conformation of the kinase domain. The regions flanking the kinase domain play important role in the enzyme activity (Fig. 1). The juxtamembrane segment of the receiver kinase wraps around the C-terminal lobe of the activator, and is proposed to interact with the juxtamembrane segment from the activator, to enhance the interaction between the partners.4,10 Specific mutations at the asymmetric dimer interface compromise catalytic activity; the crystal structure of one such mutant, V948R, showed Src/CDK-like inactive state of the enzyme (Fig. 1).10
Figure 1.
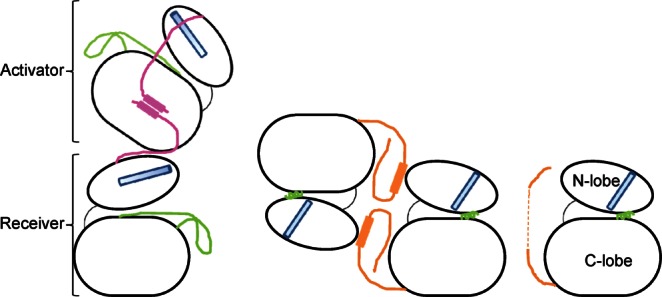
Cartoon representation of the major conformational states of the EGFR kinase domain seen in the crystal structures. Catalytically important regions are highlighted in color: αC helix in cyan, activation loop in green, juxtamembrane region in pink and the C-terminal tail in orange. On the left is the asymmetric dimer of the EGFR kinase domain, partially mediated by the juxtamembrane region. Juxtamembrane region of the activator molecule is modeled based on the NMR data.4,10 In the middle is the inactive symmetric dimer seen with V948R mutant EGFR kinase domain.10 On the right is the monomeric inactive state seen with V948R mutant structure in a different crystal form,9 and with the lapatinib bound conformation of the wild-type enzyme.14 [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The Src/CDK-like inactive state of V948R mutant protein showed the beginning of the C-terminal tail in a unique conformation, at the interface of a symmetric dimer that would preclude juxtamembrane segment mediated activation (see below). This suggested a negative regulatory role for the C-erminal region. The inhibitory role of the C-terminal tail has also been shown by studies on EGFR-ErbB3 chimera.15 This is consistent with the observation that EGFRvIV mutants that lack part of the C-terminal tail, immediately adjacent to the kinase domain, induce transforming potential by virtue of ligand-independent constitutive activation.16 Besides, the C-terminal tail has phosphorylation sites which specifically recruit signaling molecules involved in cellular events.17 Molecular dynamics simulation has suggested correlation between the conformational state of the C-terminal tail and that of the kinase domain as the enzyme cycles through its active and inactive states,18 which is also clearly mirrored in the available crystal structures of major conformational states. Collectively, these data underscore the importance of the C-terminal tail in normal functioning of EGFR as well as in certain disease states. The conformational state of this region of the C-terminal tail has remained somewhat uncertain, however, in the catalytically competent state of the enzyme despite extensive structural characterization of the active kinase domain.
Most protein constructs used for the structural characterization of the EGFR kinase domain include over 40 residues (980–1022) of the C-terminal tail immediately downstream of the kinase domain. Almost all active kinase domain structures show discontinuous electron density for this region. The stretch of the polypeptide chain approximately from residue 990–1005, which show the largest thermal fluctuations in the MD simulation,18 are disordered in the crystal structures. This poses an interesting challenge of modeling residues 1006–1018 that are ordered with clear electron density. Crystallographically, there are at least three different locations within reasonable distance of the rest of the protein where this ordered region can be modeled, but only one would be physically relevant; others are likely to belong to neighboring symmetry related molecules. The majority of structures show the ordered region of residues 1006–1018 modeled in contact with the β3 strand of the N-lobe of the same kinase domain, near the back of the hinge region [Fig. 2(A)]. But a recently published structure of the EGFR kinase domain in complex with afatinib suggests that this may not be the case. It shows uninterrupted trace of the C-terminal tail packing against the N-lobe of the neighboring molecule in the lattice [Fig. 2(A,B)].19
Figure 2.
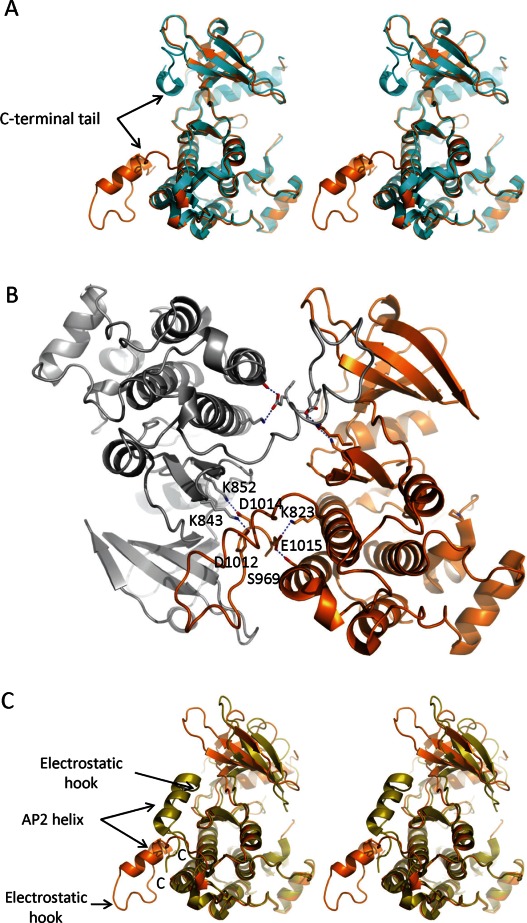
Different models of the EGFR C-terminal tail. (A) Comparison of the different models for the C-terminal tail in stereo view. The revised model for the C-terminal tail (PDBID: 4G5J) is shown in orange; the model in cyan is representative of the most structures of the active EGFR kinase domain in the public domain. (B) Revised model of the C-terminal tail shows extensive intra- and intermolecular interactions. The C-terminal tail of one molecule extensively interacts with the N-lobe of the symmetry-mate. The symmetrymates in the crystal lattice are shown in orange and gray. Some of the specific reciprocal interactions are highlighted. (C) Differences between the C-terminal tails of active and inactive states of the EGFR kinase domain shown in the stereo view. Src/CDK-like inactive state is highlighted in olive (PDBID: 3GT8) and the active state in orange (4G5J). The AP2 helix is maintained in both states, but in opposite orientations. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Afatinib (BIBW 2992) is an ATP-competitive covalent inhibitor of EGFR and HER2 kinases.20 The structure reported by Solca et al.19 was obtained by soaking wild-type EGFR kinase domain crystals in the afatinib solution. This resulted in fortuitous binding of two molecules of the ligand near the C-terminal tail, in addition to the expected covalent binding of the ligand in the ATP binding site. Apparent recognition of the additional two molecules of afatinib is unlikely to be of physiological significance because the protein–ligand contacts seem to be limited, and dominated by nonspecific van der Waals interactions. Instead, the two ligand molecules along with their two symmetrymates make a stack of tetramers to which the crystal lattice seems to play the host. These additional molecules of the ligand intercalate in the lattice, thereby contributing to the well-ordered conformation of the C-terminal tail which is unprecedented for the EGFR kinase domain structures in the active state. The new model shows the contiguous C-terminal tail extending away from the kinase domain and interacting with the neighboring molecule in the lattice [Fig. 2(B)].
The orientation of the ordered C-terminal tail in the active state of the kinase domain is quite different compared with that in the inactive state. It is flipped in the active enzyme relative to the inactive symmetric dimer of V948R enzyme [Fig. 2(C)]. The AP-2 helix, encompassing the FYRAL motif (residues 997–1001) for the recognition of clathrin-associated protein complex AP-2,21 is largely maintained in both structures, albeit in nearly opposite orientations. In the inactive symmetric dimer of V948R mutant structure, each AP-2 helix makes intimate contact with the back of the N-lobe of the dimeric partner (Fig. 1). In the process, it precludes the reorientation of the N-lobe required for activation as well as interaction of the AP-2 recognition motif with the clathrin-associated protein complex AP-2.10 Similarly, residues 1003–1005 from the region referred to as the “electrostatic hook” [Fig. 2(C)] are implicated in maintaining the enzyme in its inactive state by virtue of their specific interactions with the basic side chains on the αC-β4 loop in V948R mutant. However the remodeled C-terminal tail of the active kinase domain suggests that the AP-2 helix is mostly solvent exposed, and the residues 997–1001 are available for interaction with AP-2. Likewise, the residues of the electrostatic hook are removed from previously observed intramolecular contacts, and are solvent exposed. This disposition of the C-terminal tail is maintained by some specific intramolecular contacts with the C-lobe of the kinase domain. These include Asn996 in hydrogen bond interaction with the backbone oxygen of Ala972, and Glu1015 in similar interactions with Ser969 and Lys823. Additionally, guanido groups of Arg999 and Arg973 seem to stack on each other. At the same time, the specific interaction between Arg999 Ne and Val1010 backbone oxygen within the C-terminal tail seems to contribute to the specific conformation.
The new model of the the C-terminal tail relative to the kinase domain shows reciprocal intermolecular interactions between the neighboring molecules in the crystal lattice via swapping of their C-terminal tails [Fig. 2(B)]. As a result, whereas the previous structures of EGFR kinase domain have residues 1006–1018 modeled such that they make intramolecular interactions with the N-terminal lobe of the same molecule, the revised model suggests that it is actually involved in intermolecular contacts with the N-lobe of a neighboring symmetry-related molecule. The two molecules with reciprocal interactions form the symmetric dimer, burying in excess of 3000 Å2 of surface area, almost all of which is contributed by C-terminal tail interactions [Fig. 2(B)]. Some of these reciprocal interactions are specific such as between Asp1012–Lys846, Asp1014–Lys852, and Tyr1016–Glu736. Asp1008 and Asp1009 are within hydrogen bonding distance of Lys739. Tyr998 is making a polar contact with the backbone oxygen from Leu792. Interestingly, these intermolecular contacts are all contributed by the part of the C-terminal tail that has mostly been ordered in the previously reported structures. Presumably these are important contacts for crystal lattice formation since all EGFR kinase domain constructs that lack the C-terminal tail have failed to crystallize, at least in our hands. The reciprocal interactions involving the C-terminal tail may provide critical impetus for crystal lattice building but are not likely to be of physiological importance. Mutagenesis data suggest that the symmetric dimer of the EGFR kinase domain may not play a significant role in its activation.9
Most mutations at the dimer interface had little to no impact on the phosphorylation pattern of the full-length receptor in the cell. However, K823E was an exception; it greatly reduced the EGFR phosphorylation. The new model for the C-terminal tail addressed here suggests that Lys823 is involved in an intramolecular ion-pair interaction with Glu1015 [Fig. 2(B)], and thereby stabilizes the particular conformation. Glu1015 is located in an acidic stretch of the C-terminal tail where three of the four sidechains are acidic. Hence, it is conceivable that Lys823 provides a strong and essential counter-charge to stabilize the region. Its mutation may perturb the conformation of this segment of the C-terminus, making it more dynamic, which could impact enzyme activity by a mechanism that is yet to be determined. A charge reversal double mutation E1015K/K823E was expected to restore the ion-pair, and hence the autophosphorylation, but it did not. This may be due to the location of Glu1015 in the EGFR sequence. Because of the acidic region where Glu1015 is located, mutation of Glu1015 to Lys could result in an alternate ion-pair, with Asp1012 for example [Fig. 2(B)], making it unavailable to interact with Glu823. This would explain why charge reversal double mutant failed to show the autophosphorylation activity of the wild-type enzyme.
As mentioned above, the currently accepted model of EGFR activation suggests that it proceeds through asymmetric dimerization aided by the intermolecular interaction of the juxtamembrane segment of the receiver molecule with the C-lobe of the activator kinase domain (Fig. 1).6,9 The ordered C-terminal tail of the inactive V948R EGFR kinase domain, on the other hand, showed its intramolecular interactions with the C-lobe. Comparison of these interactions in the active and inactive states of the kinase provided the rationale for the role of the ordered C-terminal tail in ablating these interactions and precluding its activation.
Ironically, the newly modeled conformation of the C-terminal tail of the active kinase would also preclude it from serving the role of an activator in its asymmetric dimer state. This is because, in the context of the activator molecule, the observed conformation of the C-terminal tail seems to compete for the same region of the C-lobe that is occupied by the juxtamembrane region of the receiver molecule in the catalytically competent dimer (Fig. 3). This implies that if the observed conformation of the C-terminal tail is of physiological significance, it may be representative of only the receiver molecule of the asymmetric dimer (This is also true for the the known conformation of the juxtamembrane region: the crystal structure of the EGFR kinase domain showing the conformation of the juxtamembrane region is suggested to be representative only of the receiver molecule). The C-terminal tail (like the juxtamembrane region) of the activator molecule must take on a yet unknown conformation in its catalytically competent state, and the MD simulations have suggested that it may have lower thermal disorder than that for the activator molecule.18 It then follows that if the remodeled conformation of the C-terminal tail is of physiological significance, it may only be representative of the receiver half of the active dimer.
Figure 3.
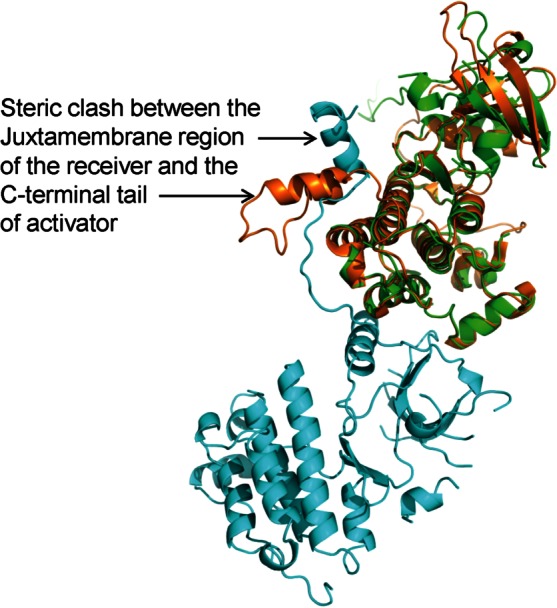
Observed conformation of the C-terminal tail would preclude the kinase domain from serving as the activator. The kinase domain with the revised model for the C-terminal tail (PDBID: 4G5J, in orange) is superposed on the activator (shown in green) of the proposed physiologically relevant EGFR dimer (PDBID: 3GOP) to illustrate the steric clash with the juxtamembrane region of the receiver molecule (shown in cyan). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
In conclusion, ordering of the flexible region (spanning residues 995–1005) due to the presence of two additional molecules of afatinib in the soaked crystal structure suggests that there is a reciprocal exchange of the C-terminal tail between the molecules that form the symmetric crystallographic dimer. Accepted model for the allosteric activation of the enzyme requires that only the receiver half of the asymmetric dimer could adopt such a conformation of the C-terminal tail. Combining this analysis with the known structural information provides complete description for a portion of the juxtamembrane region, kinase domain and part of the C-terminal tail of the receiver molecule of the activated EGFR. It implies then that the structure of the activator half of the asymmetric dimer remains to be determined.
Acknowledgments
Ping Chen & Al Stewart are gratefully acknowledged for their support.
References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Cleveland TEt, Bouyain S, Byrne PO, Longo PA, Leahy DJ. A single ligand is sufficient to activate EGFR dimers. Proc Natl Acad Sci USA. 2012;109:10861–10866. doi: 10.1073/pnas.1201114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, Groves JT, Kuriyan J. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell. 2013;152:543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 6.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y, Li Z, Apetri M, Luo B, Settleman JE, Anderson KS. Temporal resolution of autophosphorylation for normal and oncogenic forms of EGFR and differential effects of gefitinib. Biochemistry. 2012;51:5212–5222. doi: 10.1021/bi300476v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem Biophys Res Commun. 1992;186:768–774. doi: 10.1016/0006-291x(92)90812-y. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, Kuriyan J, Shaw DE. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubbard SR. The juxtamembrane region of EGFR takes center stage. Cell. 2009;137:1181–1183. doi: 10.1016/j.cell.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc Natl Acad Sci USA. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Gilmer TM, Shewchuk L. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 15.Bublil EM, Pines G, Patel G, Fruhwirth G, Ng T, Yarden Y. Kinase-mediated quasi-dimers of EGFR. FASEB J. 2010;24:4744–4755. doi: 10.1096/fj.10-166199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pines G, Huang PH, Zwang Y, White FM, Yarden Y. EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene. 2010;29:5850–5860. doi: 10.1038/onc.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 18.Mustafa M, Mirza A, Kannan N. Conformational regulation of the EGFR kinase core by the juxtamembrane and C-terminal tail: a molecular dynamics study. Proteins. 2011;79:99–114. doi: 10.1002/prot.22862. [DOI] [PubMed] [Google Scholar]
- 19.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 20.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorkin A, Mazzotti M, Sorkina T, Scotto L, Beguinot L. Epidermal growth factor receptor interaction with clathrin adaptors is mediated by the Tyr974-containing internalization motif. J Biol Chem. 1996;271:13377–13384. doi: 10.1074/jbc.271.23.13377. [DOI] [PubMed] [Google Scholar]