

Abstract
Recent advances in cancer research highlighted the importance of target-specific drug discovery. In view of these advances, the most important mechanism in tumour growth is its ability to stimulate the formation of blood capillaries around itself called tumour-driven angiogenesis. Hence targeting the angiogenesis, inhibits the growth of blood vessels around it and responsible for death of the tumour due to starvation and accumulation of toxic waste. The therapy, thus, indirectly cytotoxic to the tumour cells by targeting newly developing blood vessels. In this review, we summarised the various antiangiogenic agents with their clinical uses and current status.
Keywords: Angiogenesis, cancer, inhibitors of angiogenesis, small molecules
Angiogenesis is the phenomenon of formation of new blood capillaries from existing small blood vessels. It usually accounts for normal physiological and pathological changes involved in normal functioning of body, like development of endometrium during menstrual cycle or during wound healing. A tumour consists of a group of cancerous cell, which are bodies own cells but have lost their ability to divide in controlled fashion. For their growth they require oxygen and nutrients, this demand can be fulfilled by the angiogenesis. Hence, it was hypothesised that angiogenesis is a factor enabling malignant tumour growth in cancer.[1]
ANGIOGENESIS AND CANCER
In order to get oxygen and nutrients, tumour cells secrete growth factors. These growth factors penetrate into surrounding tissue and stimulate the endothelial cells of the parent blood vessels to release proteases. This enzyme responsible for digestion of basement membrane present on the same cells and allows movement of these endothelial cells from their original place and allows the cells to migrate in the direction of secretion of growth factor to form a sprout. This sprout then forms a loop followed by formation of full-fledged vessel after maturation.[2] The migrations of endothelial cells are stimulated by tumour, hence the phenomenon is also known as tumour driven angiogenesis. In this way a nonvascular tumour gets converted in to highly vascularised tumour. Some important growth factors responsible for tumour-driven angiogenesis include acidic fibroblast growth factor (aFGF), basic FGF (bFGF), angiogenin, placental growth factor, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), transforming growth factor-α, tissue necrosis factor-α and vascular endothelial growth factor (VEGF).
Effects of growth factors:
As shown in fig. 1, growth factors like VEGF or bFGF are first trascripted inside the tumour cell by the hypoxia inducible factor-α, followed by their translation and secretion. When they encounter endothelial cells, they bind with specific receptors know as receptor tyrosine kinase (RTK) present on the endothelial cells. This binding causes the receptor to undergo autophosphorylation, which stimulates Mitogen-activated protein kinases (MAPK) pathway. This pathway causes the secretion of proteases, like matrix metalloproteinases (MMPs). This group of enzymes is responsible for the degradation of the basement membrane present on the endothelial cells of blood vessel and making them to migrate towards the stimulus.[3]
Fig. 1.
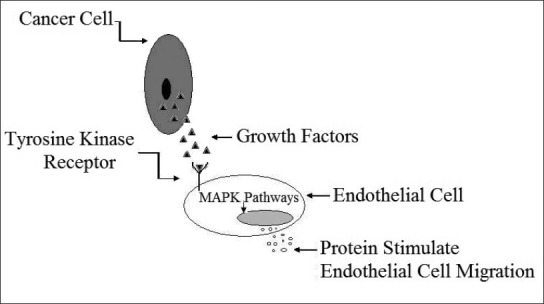
Effects of growth factors secreted by tumour cells on endothelial cell.
ANTIANGIOGENIC AGENTS
By considering the fact that tumour stimulates the formation of blood vessels, development of antiangiogenic agents will have potential anticancer activity. Antiangiogenic agents induce their effect by inhibiting the enzymes responsible for proliferation, or sequestering the growth factor or inhibiting the receptor responsible for binding with growth factors. Most of the available literature points out that newly formed blood vessels are highly selective towards low dose of antiangiogenic agents.[4,5] These agents include humanised monoclonal antibodies (HMABs) against growth factors, matrix metalloproteinases inhibitors (MMPIs), and small molecule inhibitor of tyrosine kinase.
Humanised monoclonal antibodies:
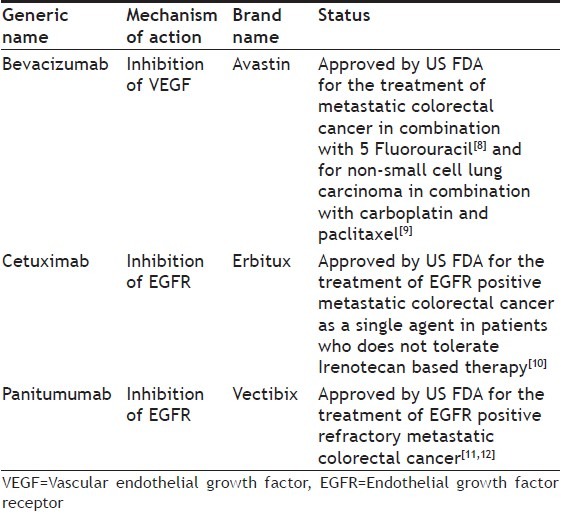
HMABs act by either directly blocking the receptor or exfoliating growth factors.[6] Both the mechanisms prevent the activation of the receptor, thereby blocking the signalling cascade responsible for secretion of MMPs.[7] These agents are specific for particular protein, e.g. bevacizumab is humanised monoclonal antibody against VEGF, cetuximab and panitumumab against endothelial growth factor receptor (EGFR) (Table 1).
Table 1.
DIFFERENT HUMANISED MONOCLONAL ANTIBODIES WITH THEIR CURRENT STATUS
Matrix metalloproteinases inhibitors:
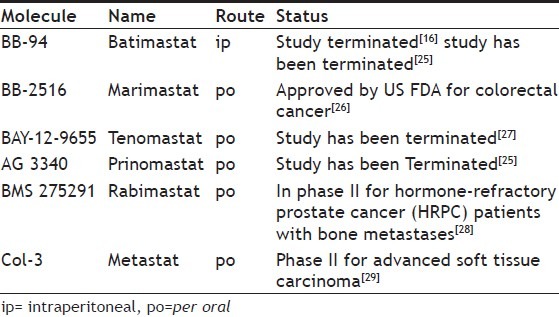
MMPs are a group of enzymes, which are destructive in nature and require zinc ion (Zn+2) for their activity. They play an important role in normal turnover and remodelling of extracellular matrix. It has been reported that, MMPs are over expressed in a variety of malignant tumour types and this is associated with tumour growth and metastasis potential. MMPs degrade the basement membrane and extracellular matrix present on parent blood vessel, making endothelial cells to migrate from their original place and play a pivotal role in angiogenesis. Inhibition of MMPs prevents metastasis and progression of angiogenesis, therefore, can actively peruse as an antiangiogenic agent. Table 2 gives the current status of the MMPIs as antiangiogenic agents. Inhibitors of MMPIs are categorised as peptidomimetic inhibitors, nonpeptidomimetic inhibitors, modified tetracycline derivatives.[13]
Table 2.
TYPES OF MATRIX METALLOPROTEINASES INHIBITORS WITH THEIR GENERIC NAME AND CURRENT STATUS
Compounds of peptidomimetic inhibitors (first generation MMPIs) class are hydroxamic acid derivatives designed to mimic the structure of collagen at the site where the MMPs are thought to bind for the cleavage. Compounds like marimastat and batimastat (fig. 2)bind reversibly at the active site of the enzyme in stereospecific manner and chelate the zinc ion present on the enzyme active site.[14]
Fig. 2.
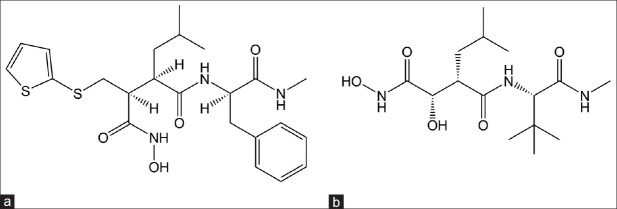
Peptidomimetic matrix metalloproteinases inhibitors.
(a) Batimastat, (b) marimastat.
First generation MMPIs are nonselective and also exhibits poor oral bioavailability due to peptide nature. The second generation MMPIs (Nonpeptidomimetic inhibitors) lacks the peptide backbone and shows improved selectivity towards MMPs[15] (fig. 3). Examples include BAY-12-9655, AG 3340 and BMS 275291. Phase III studies showed that continuous administration of BAY-12-9655 with placebo in patients with small cell lung carcinoma did not improve the outcome of chemotherapy. It also showed inferior survival of patients treated with BAY-12-9655. Hence, clinical development of BAY-12-9655 has been suspended.[16] AG 3340 is a nonpeptide, collagen mimicking MMPIs. It shows inhibition of tumour growth and angiogenesis in xenograft models of prostate, colon, breast and non-small cell lung carcinoma.[17,18] In vitro, BMS 275291 exhibits antiangiogenic effects in renal assay and in vivo it inhibited the growth of B16F10 murine melanoma and reduced the size and metastases of the rat HOSP-1 mammary carcinoma.[19,20]
Fig. 3.
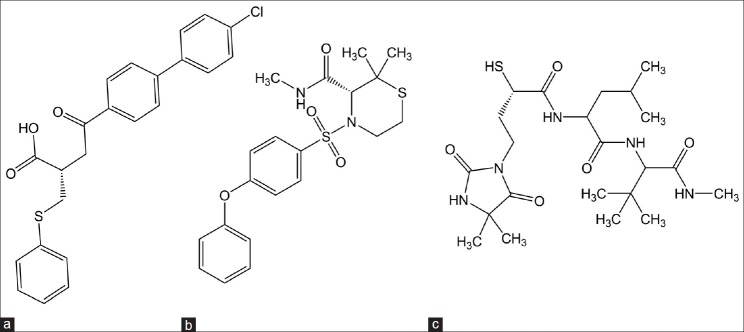
Nonpeptidomimetic matrix metalloproteinases inhibitors.
(a) BAY-12-9655, (b) AG 3340, (c) BMS 275291.
Modified tetracycline derivatives not only inhibit the activity, but also the production of MMPs. Newer tetracycline analogues are chemically modified tetracyclines (CMT), which lack their antibacterial properties. This is achieved by removal of dimethyl amino group from C-4 of ring A.[21] These agents are thought to be act by, blocking the activity of MMPs by chelation of zinc atom at the enzyme’s active site, interfering with the proteolytic activation of pro-MMP into their active form, reducing the expression of MMPs, protecting MMPs from proteolytic and oxidative degradation.[22–24] There are many CMT from CMT-1 to CMT-10, but out of which CMT-3 called as Col-3 is most active in the series. Table 2 describes the details of the MMPIs inhibitors.
Small molecule tyrosine kinase inhibitors:
Tyrosine kinase inhibitors (TKIs) are the synthetic agents that target enzyme tyrosine kinase linked to the receptor of growth factors like VEGF, EGF and PDGF. Depending upon the type of enzyme targeted by the agents they are divided into following categories: endothelial growth factor RTK inhibitors (EGFR TKI), vascular endothelial growth factor receptor (VEGFR) TKIs, multiple TKIs.
Endothelial growth factor receptor tyrosine kinase inhibitors were developed with the lead molecule 4-anilinoquinazoline. Structure-activity relationship (SAR) studies proved that quinazoline moiety is absolutely essential for activity. Sixth and seventh position of the quinazoline moiety must be substituted with electron withdrawing substitutients. Second, seventh and eighth position must remain unsubstituted. The anilinic nitrogen must be secondary for optimal activity. The SAR studies on the lead structure led to compounds viz. gefitinib and erlotinib. Table 3 and fig. 4 give the details of the clinical applications and the current status of EGFR TKIs.[30]
Table 3.
TYPES OF ENDOTHELIAL GROWTH FACTOR RECEPTOR TYROSINE KINASE INHIBITORS WITH THEIR GENERIC NAME AND CURRENT STATUS
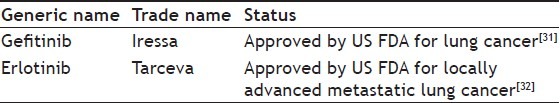
Fig. 4.
Endothelial growth factor receptor tyrosine kinase inhibitors.
(a) 4-anilinoquinazoline, (b) gefitinib, (c) erlotinib.
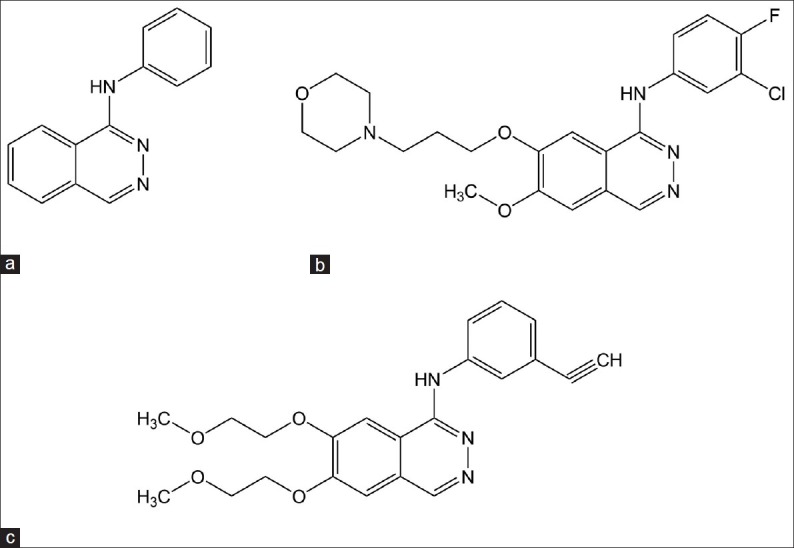
According to SAR studies of VEGFR TKIs (fig. 5) 4-anilinoquinazoline moiety is of extreme importance for activity. A small lipophilic, electron deficient substitution at C-2 position and a large lipophilic electron withdrawing substitution at C-4 position on anilinic phenyl ring are better for activity. Furthermore, the introduction of basic, nitrogen containing side chain at C-7 position improves activity. It may include morpholine, piperidine, or pyrolidine however, most active compounds were observed when C-7 substitution was N-methylpiperidine. When attached directly, the N-methylpiperidinoxy to C-7 quinazoline ring showed weaker activity, however, insertion of a methylene group in between with C-6 position substituted with methoxy group gave most active compound ZD 6474.[33]
Fig. 5.
Vascular endothelial growth factor receptor tyrosine kinase inhibitors.
(a) 4-anilinoquinazoline, (b) 4-anilinopthalazine, (c) PTK-787, (d) ZD 6474.
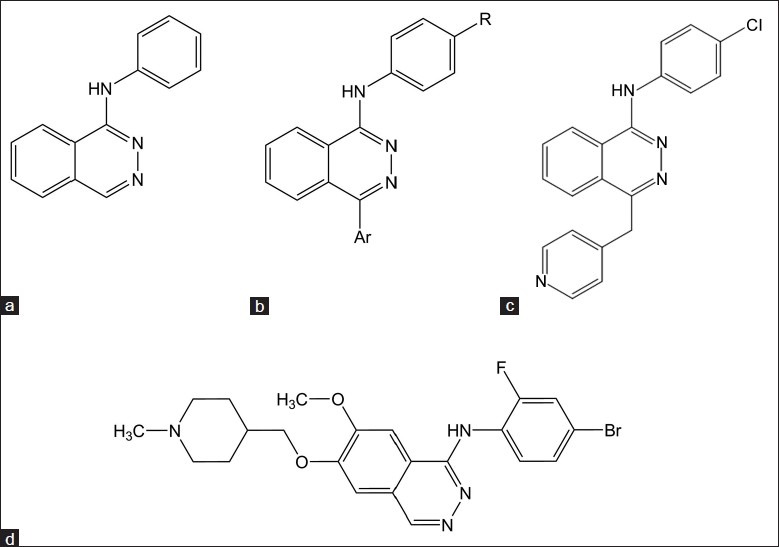
Molecules developed from 4-anilinopthalazine by varying substitution on first and fourth positions on pthalazine template has lead to the establisment of the SAR for this group of compounds. Anilinic NH is required for good inhibitory activity. Also ethoxy and thio ether derivatives shows activity but secondary nitrogen exhibited maximum activity. Lipophilic substitution like chloro or bromo on fourth position of anilinic phenyl ring attributed to good inhibitory activity. When chloro was replaced by tert-butyl group, it retained the activity. Three substituted or 3,4-disubstituted anilinic phenyl ring also shows good inhibitory activity. Position four of pthalazine template is crucial for VEGFR inhibitory activity. When substituted with 4-pyridyl methyl group, the compound was found to be most active as nitrogen of 4-pyridine is involved in hydrogen bonding with Lys 1060 amino acid residue of the kinase pocket. Hence, 2-pyridyl and 3-pyridyl containing substitution was found to be inactive. Thus, by varying the substitution on suitable position, most active molecule was reported as 1-(4-chlorophenyl)-4-(4-pyridylmethyl)phthalazine and known as PTK 787.[34]
Multiple tyrosine kinase inhibitors (Table 4 and fig. 6) like SU 5416 was one of the initial, and probably the most extensively studied kinase insert domain receptor (KDR, VEGFR-2) inhibitor, and the first specific synthesized inhibitor of VEGF-RTK activity. It showed growth inhibition in mouse xenotransplants of human tumours; also demonstrated promising antiangiogenic and antitumour effects in animal models.[35] SU 11248 is a small molecule inhibitor of multiple tyrosine kinases, including VEGFRs 1, 2, and 3; stem cell factor receptor (c-KIT); platelet-derived growth factor receptors (PDGFRs) a and h; Fms-like tyrosine kinase 3 (FLT3); colony stimulating factor receptor type 1 receptor (CSF-1R); and the glial cell line-derived neurotrophic factor receptor (RET).[36] BAY-43-9006: It’s a multikinase inhibitor with activity against Raf kinase and several RTKs, including VEGFR2, PDGFR, FLT3, RET, and c-Kit.[37] ABT-86 is a structurally novel, RTK inhibitor that is a potent inhibitor of members of the VEGF and PDGFR families but has much less activity against unrelated RTKs, soluble tyrosine kinases, or serine/threonine kinases. The inhibition profile of ABT-869 is evident in cellular assays of RTK phosphorylation (IC50 = 2, 4, and 7 nmol/l for PDGFR-β, KDR, and CSF-1R, respectively) and VEGF-stimulated proliferation (IC50 = 0.2 nmol/l for human endothelial cells).[38] AMG 706 a novel nicotinamide derivative was identified as a potent, orally bioavailable inhibitor of the VEGFR1/Flt1, VEGFR2/kinase domain receptor/Flk-1, VEGFR3/Flt4, PDGFR, and Kit receptors in preclinical models. AMG 706 inhibited human endothelial cell proliferation induced by VEGF, but not by basic FGF in vitro, as well as vascular permeability induced by VEGF in mice.[39]
Table 4.
SMALL MOLECULE INHIBITORS OF TYROSINE KINASE WITH THEIR GENERIC NAME AND CURRENT STATUS

Fig. 6.

Multiple tyrosine kinase inhibitors.
(a) SU 5416, (b) SU11248, (c) BAY-43-9006, (d) ABT-869, (e) AMG 706.
CONCLUSION
Angiogenesis is an important and necessary phenomenon in tissue growth or in wound healing. However, cancerous cells stimulate the formation of vasculature around itself called as tumour-driven angiogenesis for the purpose of its survival and growth. This tumour-driven angiogenesis is stimulated by the abnormal activation of RTK by interaction of growth factors with the tyrosine kinase receptor. Inhibitors of angiogenesis thus, help in blocking progression, growth and development of the tumour and can be a class of anticancer agents. The antiangiogenic agents have advantages over conventional anticancer therapy like, the therapy does not directly targeting the tumour but inhibits the development of blood vessels and thus, indirectly cytotoxic, development of multikinase inhibitors can result in better antiangiogenic agents. Better results have been observed when used in combination with other anticancer agents. E.g., Use of ZD-6474 (Zactima) with docetaxel for the patients suffering from nonsmall cell lung cancer. Furthermore, these agents are not cytotoxic to normal somatic cells. However, they have also suffered from few disadvantages which includes the inhibition of the initial phase of tumour growth, interference with the wound healing and menstrual cycle, involves huge research cost and the therapy cost is high. Rather than designing the selective inhibitors of RTK, multiple TKIs can be better candidate for antiangiogenic drug discovery. As the possibility of developing resistance due to selective inhibitors will be less and if developed antiangiogenic effect will be due to the inhibition of other RTKs as well.[46]
Footnotes
Somani and Bhanushali: Antiangiogenic Agents for Cancer Treatment
REFERENCES
- 1.Folkman J. Tumour angiogenesis: Therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. What is the evidence that tumours are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 3.Liekens S, De Clercq E, Neyts J. Angiogenesis: Regulators and clinical applications. Biochem Pharmacol. 2001;61:253–70. doi: 10.1016/s0006-2952(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 4.Wu HC, Huang CT, Chang D. Antiangiogenic therapeutic drugs for treatment of human cancer. J Cancer Mol. 2008;4:37–45. [Google Scholar]
- 5.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 6.Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat Rev Drug Discov. 2007;6:349–56. doi: 10.1038/nrd2241. [DOI] [PubMed] [Google Scholar]
- 7.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 8.Krämer I, Lipp HP. Bevacizumab, a humanized antiangiogenic monoclonal antibody for the treatment of colorectal cancer. J Clin Pharm Ther. 2007;32:1–14. doi: 10.1111/j.1365-2710.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- 9.Sandler A, Herbst R. Combining targeted agents: Blocking the epidermal growth factor and vascular endothelial growth factor pathways. Clin Cancer Res. 2006;12:4421s–5. doi: 10.1158/1078-0432.CCR-06-0796. [DOI] [PubMed] [Google Scholar]
- 10.Derk J, Chris J, Christos SK, Zalecberg JR. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 11.Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: Panitumumab (Vectibix) Oncologist. 2007;12:577–83. doi: 10.1634/theoncologist.12-5-577. [DOI] [PubMed] [Google Scholar]
- 12.Messersmith WA, Hidalgo M. Panitumumab, a monoclonal anti epidermal growth factor receptor antibody in colorectal cancer: Another one or the one? Clin Cancer Res. 2007;13:4664–6. doi: 10.1158/1078-0432.CCR-07-0065. [DOI] [PubMed] [Google Scholar]
- 13.Hoekstra R, Eskens FA, Verweij J. Matrix metalloproteinase inhibitors: Current developments and future perspectives. Oncologist. 2001;6:415–27. doi: 10.1634/theoncologist.6-5-415. [DOI] [PubMed] [Google Scholar]
- 14.Betz M, Huxley P, Davies SJ, Mushtaq Y, Pieper M, Tschesche H, et al. 1.8-A crystal structure of the catalytic domain of human neutrophil collagenase (matrix metalloproteinase-8) complexed with a peptidomimetic hydroxamate primed-side inhibitor with a distinct selectivity profile. Eur J Biochem. 1997;247:356–63. doi: 10.1111/j.1432-1033.1997.00356.x. [DOI] [PubMed] [Google Scholar]
- 15.Millar AW, Brown PD, Moore J, Galloway WA, Cornish AG, Lenehan TJ, et al. Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br J Clin Pharmacol. 1998;45:21–6. doi: 10.1046/j.1365-2125.1998.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;93:178–93. doi: 10.1093/jnci/93.3.178. [DOI] [PubMed] [Google Scholar]
- 17.Shalinsky DR, Brekken J, Zou H, McDermott CD, Forsyth P, Edwards D, et al. Broad antitumour and antiangiogenic activities of AG3340, a potent and selective MMP inhibitor undergoing advanced oncology clinical trials. Ann N Y Acad Sci. 1999;878:236–70. doi: 10.1111/j.1749-6632.1999.tb07689.x. [DOI] [PubMed] [Google Scholar]
- 18.Shalinsky DR, Brekken J, Zou H, Bloom LA, McDermott CD, Zook S, et al. Marked antiangiogenic and antitumour efficacy of AG3340 in chemoresistant human non-small cell lung cancer tumours: Single agent and combination chemotherapy studies. Clin Cancer Res. 1999;5:1905–17. [PubMed] [Google Scholar]
- 19.Shoji A, Sakamoto Y, Tsuchiya T, Moriyama K, Kaneko T, Okubo T, et al. Inhibition of tumour promoter activity toward mouse fibroblasts and their in vitro transformation by tissue inhibitor of metalloproteinases-1 (TIMP-1) Carcinogenesis. 1997;18:2093–100. doi: 10.1093/carcin/18.11.2093. [DOI] [PubMed] [Google Scholar]
- 20.Krüger A, Sanchez-Sweatman OH, Martin DC, Fata JE, Ho AT, Orr FW, et al. Host TIMP-1 overexpression confers resistance to experimental brain metastasis of a fibrosarcoma cell line. Oncogene. 1998;16:2419–23. doi: 10.1038/sj.onc.1201774. [DOI] [PubMed] [Google Scholar]
- 21.Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne J, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv Dent Res. 1998;12:12–26. doi: 10.1177/08959374980120010501. [DOI] [PubMed] [Google Scholar]
- 22.Ryan ME, Ramamurthy S, Golub LM. Matrix metalloproteinases and their inhibition in periodontal treatment. Curr Opin Periodontol. 1996;3:85–96. [PubMed] [Google Scholar]
- 23.Golub LM, Ramamurthy N, McNamara TF, Gomes B, Wolff M, Casino A, et al. Tetracyclines inhibit tissue collagenase activity. A new mechanism in the treatment of periodontal disease. J Periodontal Res. 1984;19:651–5. doi: 10.1111/j.1600-0765.1984.tb01334.x. [DOI] [PubMed] [Google Scholar]
- 24.Moore MJ, Hamm P, Eisenberg P, Dagenais M, Hagan K, Fields A. A comparison between gemcitabine (GEM) and the matrix metalloproteinase (MMP) inhibitor BAY12-9566 (9566) in patients (pts) with advanced pancreatic cancer [abstract] Proc Am Soc Clin Oncol. 2000;19:240a. [Google Scholar]
- 25.Gilbertson-Beadling S, Powers EA, Stamp-Cole M, Scott PS, Wallace TL, Copeland J, et al. The tetracycline analogs minocycline and doxycycline inhibit angiogenesis in vitro by a non-metalloproteinase-dependent mechanism. Cancer Chemother Pharmacol. 1995;36:418–24. doi: 10.1007/BF00686191. [DOI] [PubMed] [Google Scholar]
- 26.Primrose JN, Bleiberg H, Daniel F, Van Belle S, Mansi JL, Seymour M, et al. Marimastat in recurrent colorectal cancer: Exploratory evaluation of biological activity by measurement of carcinoembryonic antigen. Br J Cancer. 1999;79:509–14. doi: 10.1038/sj.bjc.6690079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bissett D, O’Byrne KJ, von Pawel J, Gatzemeier U, Price A, Nicolson M, et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005;23:842–9. doi: 10.1200/JCO.2005.03.170. [DOI] [PubMed] [Google Scholar]
- 28.Lara PN, Jr, Stadler WM, Longmate J, Quinn DI, Wexler J, Van Loan M, et al. A randomized phase II trial of the matrix metalloproteinase inhibitor BMS-275291 in hormone-refractory prostate cancer patients with bone metastases. Clin Cancer Res. 2006;12:1556–63. doi: 10.1158/1078-0432.CCR-05-2074. [DOI] [PubMed] [Google Scholar]
- 29.Chu QS, Forouzesh B, Syed S, Mita M, Schwartz G, Cooper J, et al. A phase II and pharmacological study of the matrix metalloproteinase inhibitor (MMPI) COL-3 in patients with advanced soft tissue sarcomas. Invest New Drugs. 2007;25:359–67. doi: 10.1007/s10637-006-9031-6. [DOI] [PubMed] [Google Scholar]
- 30.Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, et al. Studies leading to the identification of ZD1839 (IRESSA): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–4. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- 31.Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8:303–6. doi: 10.1634/theoncologist.8-4-303. [DOI] [PubMed] [Google Scholar]
- 32.Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R. FDA drug approval summary: Erlotinib (Tarceva) tablets. Oncologist. 2005;10:461–6. doi: 10.1634/theoncologist.10-7-461. [DOI] [PubMed] [Google Scholar]
- 33.Hennequin LF, Thomas AP, Johnstone C, Stokes ES, Plé PA, Lohmann JJ, et al. Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J Med Chem. 1999;42:5369–89. doi: 10.1021/jm990345w. [DOI] [PubMed] [Google Scholar]
- 34.Bold G, Altmann KH, Frei J, Lang M, Manley PW, Traxler P, et al. New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumour-driven angiogenesis. J Med Chem. 2000;43:2310–23. doi: 10.1021/jm9909443. [DOI] [PubMed] [Google Scholar]
- 35.Kuenen BC, Tabernero J, Baselga J, Cavalli F, Pfanner E, Conte PF, et al. Efficacy and toxicity of the angiogenesis inhibitor SU5416 as a single agent in patients with advanced renal cell carcinoma, melanoma, and soft tissue sarcoma. Clin Cancer Res. 2003;9:1648–55. [PubMed] [Google Scholar]
- 36.Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, et al. Sunitinib in patients with metastatic renal cell carcinoma. JAMA. 2006;295:2516–24. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 37.Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumour cells and tyrosine kinases VEGFR/PDGFR in tumour vasculature. Methods Enzymol. 2006;407:597–612. doi: 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- 38.Albert DH, Tapang P, Magoc TJ, Pease LJ, Reuter DR, Wei RQ, et al. Preclinical activity of ABT-869, a multitargeted receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2006;5:995–1006. doi: 10.1158/1535-7163.MCT-05-0410. [DOI] [PubMed] [Google Scholar]
- 39.Polverino A, Coxon A, Starnes C, Diaz Z, DeMelfi T, Wang L, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumour xenografts. Cancer Res. 2006;66:8715–21. doi: 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- 40.Morabito A, Piccirillo MC, Falasconi F, De Feo G, Del Giudice A, Bryce J, et al. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: Current status and future directions. Oncologist. 2009;14:378–90. doi: 10.1634/theoncologist.2008-0261. [DOI] [PubMed] [Google Scholar]
- 41.Los M, Roodhart JM, Voest EE. Target practice: Lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist. 2007;12:443–50. doi: 10.1634/theoncologist.12-4-443. [DOI] [PubMed] [Google Scholar]
- 42.Fury MG, Zahalsky A, Wong R, Venkatraman E, Lis E, Hann L, et al. A Phase II study of SU5416 in patients with advanced or recurrent head and neck cancers. Invest New Drugs. 2007;25:165–72. doi: 10.1007/s10637-006-9011-x. [DOI] [PubMed] [Google Scholar]
- 43.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, et al. Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumours and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–73. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 44.Kane RC, Farrell AT, Madabushi R, Booth B, Chattopadhyay S, Sridhara R, et al. Sorafenib for the treatment of unresectable hepatocellular carcinoma. Oncologist. 2009;14:95–100. doi: 10.1634/theoncologist.2008-0185. [DOI] [PubMed] [Google Scholar]
- 45.Toh H, Chen P, Carr BI, Knox JJ, Gill S, Steinberg J. A phase II study of ABT869 in hepatocellular carcinoma (HCC): Interim analysis. J Clin Oncol. 2009;27:15s. [Google Scholar]
- 46.Claret L, Lu J, Bruno R, Sikorski RS, Hei Y, Sun Y. Simulation of phase III studies of motesanib 125 mg once daily (QD) plus carboplatin/paclitaxel (CPM) or bevacizumab plus carboplatin/paclitaxel (CPB) versus carboplatin/paclitaxel (CP) in first-line non-small cell lung cancer (NSCLC) using a public domain drug-disease modelling framework and phase II data. J Clin Oncol. 2010;28:e18089. [Google Scholar]