

Abstract
Two variations of the Overman rearrangement have been developed for the highly selective synthesis of anti-vicinal amino alcohol natural products. A MOM ether-directed palladium(II)-catalyzed rearrangement of an allylic trichloroacetimidate was used as the key step for the preparation of the protein kinase C inhibitor d-erythro-sphinganine and the antitumor agent (+)-spisulosine, whereas the Overman rearrangement of chiral allylic trichloroacetimidates generated by the asymmetric reduction of an α,β-unsaturated methyl ketone allowed rapid access both to d-ribo-phytosphingosine and l-arabino-phytosphingosine.
Introduction
Sphingoid-type bases that contain vicinal amino alcohols are key structural and functional components of the plasma membranes of nearly all eukaryotic cells. The sphingolipids that they form play a crucial role in many physiological processes1 and are implicated in many common human diseases, including diabetes,2 cancer,3 and various neurological syndromes.4 The parent sphingoid-type bases also display potent biological activity. For example, d-erythro-sphingosine (1) and d-erythro-sphinganine (2) both strongly inhibit protein kinase C,5 whereas d-ribo-phytosphingosine (3) is a potential heat-stress signal in yeast cells.6 More simple structures from this family of natural products also display potent biological properties. Spisulosine (4), originally isolated from the clam Spisula polynyma, has been shown to have significant cytotoxic activity by the disassembly of actin stress fibers.7
Because of their biological significance and the complicated isolation of sphingolipids from natural sources, methods for the asymmetric synthesis of these compounds and their derivatives have received considerable attention.8−11 A common approach involves the preparation of the anti-vicinal amino-alcohol motif using starting materials from the chiral pool, such as carbohydrates and amino acids. In particular, several members of the sphingoid family of natural products have been synthesized by the stereoselective addition of nucleophiles to aldehydes derived from serine.9b,9d,9g,10d,11d Recently, more specific methods, including the addition of allenylzinc complexes to chiral sulfinyl imines,9k an asymmetric Henry reaction involving a chiral copper complex,11h and the nucleophilic ring opening of chiral aziridine-derived sulfamates,11g have all been used as the key step for the preparation of the anti-vicinal amino-alcohol moiety in these natural products.
Figure 1.

d-erythro-Sphingosine (1), d-erythro-sphinganine (2), d-ribo-phytosphingosine (3), and (+)-spisulosine (4).
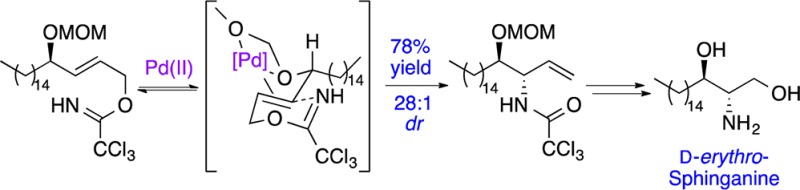
In 2005, we reported a new approach for the preparation of anti-vicinal amino alcohols using a palladium(II)-catalyzed ether-directed Overman rearrangement.12 Mechanistic studies with a range of substrates showed that an ether oxygen atom adjacent to an allylic trichloroacetimidate can direct the facial complexation of the Pd(II)-catalyst and the subsequent rearrangement, resulting in the preparation of allylic anti-vicinal amino alcohols in high diastereoselectivity (Scheme 1).13 Since then, we have shown that the products of this process can be transformed into various natural products, including β-hydroxy-α-amino acids,14 the alkaloids α-conhydrine15 and (+)-monanchorin16 as well as several members of the clavaminol family.17 In our continuing studies of how the stereogenic centers within a molecule can influence the outcome of rearrangement reactions, we were interested in investigating the use of directed Overman rearrangements for the general preparation of sphingoid-type bases. We now report the synthesis of d-erythro-sphinganine (2) and the antitumor agent (+)-spisulosine (4)7,11 using a MOM ether-directed Pd(II)-catalyzed Overman rearrangement to create the key anti-vicinal amino-alcohol moiety. We also describe a novel strategy for the preparation of d-ribo-phytosphingosine (3) and l-arabino-phytosphingosine (35) using an Overman rearrangement of a chiral allylic trichloroacetimidate.
Scheme 1. Ether-Directed Overman Rearrangement for the Preparation of anti-Vicinal Amino Alcohols.

Results and Discussion
Our investigation began with the preparation of a suitable allylic-alcohol substrate for the ether-directed Overman rearrangement that would ultimately yield both d-erythro-sphinganine (2) and (+)-spisulosine (4). Initially, a chiral diol was prepared by a Sharpless asymmetric dihydroxylation of 1-heptadecene.18 As with other long-chain terminal alkenes, dihydroxylation of 5 gave diol 6 in only modest enantiomeric excess (86%).19 However, recrystallization from ethyl acetate allowed isolation of 6 in >99% enantiomeric excess and in 87% yield. The primary and secondary hydroxyl groups of 6 were then selectively protected using TBDMS and MOM ethers, respectively, under standard conditions, giving 7 in quantitative yield. Removal of the TBDMS protecting group was followed by a one-pot Swern oxidation/Horner–Wadsworth–Emmons reaction of 8 with triethyl phosphonoacetate under Masamune–Roush conditions.20 This gave exclusively E-α,β-unsaturated ester 9. DIBAL-H reduction of 9 completed the seven-step synthesis of allylic alcohol 10 in 68% overall yield (Scheme 2).
Scheme 2. Synthesis of Allylic Alcohol 10.

Allylic alcohol 10 was transformed into corresponding allylic trichloroacetimidate 11 using trichloroacetonitrile and DBU, and this was treated with bis(acetonitrile)palladium(II) chloride (10 mol %) to affect the key Overman rearrangement (Scheme 3).21p-Benzoquinone was also added to the directed rearrangement reaction because previous studies have shown that this prevents the side reaction that forms the [1,3]-product via a Pd(0)-catalyzed process.13−17 Analysis of the 1H NMR spectrum of the crude material from this reaction showed the presence of the erythro- and threo-allylic trichloroacetamides in a 28:1 diastereomeric ratio, respectively. Purification by column chromatography allowed the isolation of major erythro-diastereomer 12 in 78% yield from allylic alcohol 10. To complete the synthesis of d-erythro-sphinganine (2), alkene 12 was subjected to ozonolysis followed by a reductive workup, which gave alcohol 13 in 78% yield.22 Removal of both protecting groups under acid-mediated conditions completed the 11-step synthesis of d-erythro-sphinganine (2) in 41% overall yield.
Scheme 3. Synthesis of d-erythro-Sphinganine (2).

Alcohol 13 was also used as an intermediate for the synthesis of the antitumor agent (+)-spisulosine (4) (Scheme 4). Activation of the primary alcohol as the mesylate followed by displacement with sodium bromide gave bromide 14 in good yield. Cleavage of the C–Br bond by hydrogenation also resulted in the reduction of the trichloromethyl group to give N-acetyl derivative 15 in 68% yield. Hydrolysis of the N-acetyl group and removal of the MOM protecting group by treatment with 6 M hydrochloric acid gave (+)-spisulosine (4) in quantitative yield.
Scheme 4. Synthesis of (+)-Spisulosine (4).

Having successfully completed the synthesis of d-erythro-sphinganine (2) and (+)-spisulosine (4) using a highly diastereoselective MOM ether-directed Overman rearrangement as the key step, a similar strategy was proposed for the synthesis of d-ribo-phytosphingosine (3). A nine-step synthesis was developed from d-ribose (16) to allylic alcohol 24 (a precursor deemed suitable for a directed Overman rearrangement; Scheme 5). d-Ribose (16) was converted under standard conditions to 2,3-isopropylidine-1-methoxy-d-ribofuranoside (17).23 Iodination of 17 then allowed Vasella fragmentation in the presence of zinc and acetic acid to give aldehyde 19.24 Attempts at the isolation of 19 led to the formation of a mixture of diastereomers as well as substantial decomposition. Therefore, before isolation the aldehyde was reduced at 0 °C with sodium borohydride to give alcohol 20 in 89% yield over the two steps. Installation of the lipid side chain was then achieved by cross metathesis of 20 with 1-tetradecene using Grubbs second-generation catalyst (5 mol %). Hydrogenation of the resulting alkene and purification gave 22 in 82% yield over the two steps. The synthesis of allylic alcohol 24 was then completed using a one-pot Swern oxidation/Horner–Wadsworth–Emmons reaction of alcohol 22, followed by DIBAL-H reduction of the ester. Optimization of this nine-step route allowed the scalable synthesis of allylic alcohol 24 in 40% overall yield.
Scheme 5. Synthesis of Allylic Alcohol 24.

With allylic alcohol 24 in hand, a directed Overman rearrangement was attempted. Initially, allylic alcohol 24 was converted to allylic trichloroacetimidate 25 using trichloroacetonitrile and DBU (Scheme 6). The 1H NMR spectrum of the crude material from this reaction confirmed a complete conversion. However, the attempted rearrangement using various Pd(II) catalysts, including bis(acetonitrile)palladium(II) chloride, gave none of the desired allylic trichloroacetamide products. Instead, substantial decomposition of the starting material was observed resulting from the loss of the acetonide protecting group. The steric bulk surrounding the alkene moiety of 25 likely results in a slow rearrangement and allows other competing pathways to come to prominence, such as the Pd(II)-mediated hydrolysis of the acetonide group.25,26 Overman rearrangement of 25 was achieved under thermal conditions. Although this process did give allylic trichloroacetamides 26 and 27 in 70% yield over the two steps, the reaction took 60 h to complete and gave undesired diastereomer 26 as the major product (26/27, 1.7:1). Scheme 6 shows the proposed chairlike intermediates in which the side chains adopt a conformation that minimizes allylic 1,3-strain.27 Inspection of the intermediate leading to allylic trichloroacetamide 26 shows that a hydrogen bond between the imidate hydrogen atom and the adjacent acetonide oxygen may account for its preferred formation.
Scheme 6. Overman Rearrangement of Allylic Trichloroacetimidate 25.

Because of the problems associated with the rearrangement of allylic trichloroacetimidate 25, an alternative approach for the synthesis of d-ribo-phytosphingosine (3) was devised. It was proposed that the chiral amino group could be incorporated via an Overman rearrangement of a chiral allylic secondary alcohol. This was prepared in a two-stage approach from previously synthesized alcohol 22. Swern oxidation followed by a Horner–Wadsworth–Emmons reaction with dimethyl 2-oxopropylphosphonate gave E-α,β-unsaturated ketone 28 in 79% yield (Scheme 7). Various methods were then investigated for the stereoselective reduction of 28. The most effective transformation was found using the CBS oxazaborolidines.28 The reduction of 28 using 1 equiv of (R)-CBS-Me in the presence of borane–THF gave (2S)-allylic alcohol 29 as a single diastereomer in 99% yield. Alternatively, the reduction of 28 under the same conditions using (S)-CBS-Me gave (2R)-allylic alcohol 30 in 64% yield.
Scheme 7. Synthesis and Stereoselective Reduction of α,β-Unsaturated Ketone 28.

The stereogenic center formed by reduction of ketone 28 was then used to install the amino group required for d-ribo-phytosphingosine (3) (Scheme 8). (2S)-Allylic alcohol 29 was converted to the corresponding allylic trichloroacetimidate, and various conditions were explored for the Overman rearrangement.29 It was found that standard thermal conditions (K2CO3, p-xylene, and 140 °C) gave after 12 h allylic trichloroacetamide 31 as a single diastereomer in 71% yield over the two steps. However, the use of microwave heating at the same temperature allowed the preparation of 31 in similar yield after only 15 min. The ozonolysis of 31 followed by a reductive workup with sodium borohydride gave alcohol 32 in 73% yield. The synthesis of d-ribo-phytosphingosine (3) was then completed using a two-step approach involving the basic hydrolysis of the trichloroacetamide group and acid-mediated removal of the acetonide protecting group.30 This gave d-ribo-phytosphingosine (3) after 13-steps in 19% overall yield. Using (2R)-allylic alcohol 30, the formation of the allylic trichloroacetimidate and Overman rearrangement using microwave heating gave (4R)-diastereomer 33 in a similar manner (Scheme 8). Conversion to alcohol 34 by ozonolysis was then followed by the two-step deprotection approach, which completed the synthesis of l-arabino-phytosphingosine (35) after 13-steps in 12% overall yield.
Scheme 8. Synthesis of d-ribo-Phytosphingosine (3) and l-arabino-Phytosphingosine (35).

Conclusions
New synthetic routes have been developed for the preparation of allylic alcohols bearing chiral hydroxyl side chains, and these have been used to study the diastereoselective outcome of the Overman rearrangement for the selective preparation of anti-vicinal amino alcohols. In particular, the use of a MOM ether-directed palladium(II)-catalyzed Overman rearrangement gave the corresponding (3S,4R)-allylic trichloroacetamide in a diastereomeric ratio of 28:1, and this synthetic intermediate was converted to the natural products d-erythro-sphinganine (2) and (+)-spisulosine (4). Alternatively, the asymmetric reduction of an α,β-unsaturated methyl ketone using the CBS oxazaborolidines followed by Overman rearrangement of the resulting allylic trichloroacetimidate under microwave heating allowed rapid access to both d-ribo-phytosphingosine (3) and l-arabino-phytosphingosine (35). This work has generated further insight into the scope and limitations of the Overman rearrangement of allylic trichloroacetimidates bearing stereogenic centers using either metal-catalyzed or thermal conditions. Current studies are underway to investigate additional applications of these highly selective rearrangements for use in natural product synthesis.
Experimental Section
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a solvent purification system. All reactions were performed under an atmosphere of argon unless otherwise mentioned. Brine refers to a saturated solution of sodium chloride. Flash column chromatography was performed using silica gel 60 (35–70 μm). Aluminum-backed plates precoated with silica gel 60F254 were used for thin layer chromatography and were visualized with a UV lamp or by staining with potassium permanganate. 1H NMR spectra were recorded on a NMR spectrometer at either 400 or 500 MHz, and data are reported as follows: chemical shift in ppm relative to tetramethylsilane as the internal standard, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of nonequivalent resonances, integration). 13C NMR spectra were recorded on a NMR spectrometer at either 101 or 126 MHz, and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as the internal standard (CDCl3, δ 77.0 ppm or CD3OD, δ 44.0 ppm), multiplicity with respect to proton (deduced from DEPT experiments, C, CH, CH2 or CH3). The infrared spectra were recorded on a FTIR spectrometer; wavenumbers are indicated in cm–1. Mass spectra were recorded using electron-impact, chemical-ionization, electrospray, or fast-atom-bombardment techniques. HRMS spectra were recorded using a dual-focusing magnetic analyzer mass spectrometer. Melting points are uncorrected. The optical rotations were determined as solutions irradiating with the sodium D line (λ = 589 nm) using a polarimeter. The [α]D values are given in units of 10–1 deg cm2 g–1. The chiral HPLC method was calibrated with the corresponding racemic mixture. The microwave reactions were conducted using a CEM Discover Explorer Synthesis unit and performed in glass tubes (capacity 10 mL) sealed with a septum. Temperatures of the reaction mixtures were monitored by an internal infrared temperature-control probe.
(2R)-Heptadecane-1,2-diol (6)19a
A solution of AD-mix-β (6.00 g) in tert-butanol/water (1:1) (150 mL) was stirred vigorously at room temperature for 0.5 h. The reaction mixture was cooled to 0 °C, and 1-heptadecene (5) (1.16 g, 4.86 mmol) was added. The reaction mixture was then stirred vigorously for 40 h. The reaction was quenched by the addition of sodium sulfite (6.00 g), stirred for 1 h, and extracted with ethyl acetate (3 × 100 mL). The combined extracts were dried (Na2SO4) and concentrated to give a solid residue. This was purified by dry flash column chromatography using hexane/ethyl acetate (2:1) (300 mL). Concentration gave (2R)-heptadecane-1,2-diol (6) as a white solid. Recrystallization from ethyl acetate gave (2R)-heptadecane-1,2-diol (6) as a crystalline white solid (1.15 g, 87%). Chiral HPLC (Chiralcel IB column) analysis using 10% isopropanol in hexane as the elution solvent indicated >99% ee: mp 78–80 °C (lit.19a mp 77.8–78.2 °C); [α]D23 −1.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.25–1.44 (m, 28H), 1.81 (dd, 1H, J 6.4, 5.2 Hz), 1.95 (d, 1H, J 4.4 Hz), 3.44 (ddd, 1H, J 10.8, 7.6, 5.2 Hz), 3.66 (ddd, 1H, J 10.8, 6.4, 3.2 Hz), 3.68–3.74 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.5 (CH2), 29.4 (2 × CH2), 29.5 (2 × CH2), 29.6 (3 × CH2), 29.7 (3 × CH2), 31.9 (CH2), 33.3 (CH2), 66.9 (CH2), 72.3 (CH); MS (CI) m/z 273 (MH+, 3), 255 (100), 237 (5), 123 (5), 69 (6).
(2R)-1-(tert-Butyldimethylsilyloxy)heptadecan-2-ol
A mixture of (2R)-heptadecane-1,2-diol (6) (5.40 g, 19.85 mmol), tert-butyldimethylsilyl chloride (4.49 g, 29.78 mmol), and imidazole (2.02 g, 29.78 mmol) was dissolved in tetrahydrofuran (300 mL), and the solution was stirred overnight at room temperature. A white precipitate was removed by filtration and washed with diethyl ether (200 mL). The combined filtrate was concentrated and purified by flash column chromatography using (diethyl ether/petroleum ether 1:15), which gave (2R)-1-(tert-butyldimethylsilyloxy)heptadecan-2-ol (7.80 g, 100%) as a colorless oil: IR (NaCl) 3449, 2925, 1464, 1254, 1097, 836, 778 cm–1; [α]D23 −5.9 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.07 (s, 6H), 0.88 (t, 3H, J 7.2 Hz), 0.91 (s, 9H), 1.24–1.47 (m, 28H), 2.40 (d, 1H, J 3.2 Hz), 3.39 (dd, 1H, J 10.4, 8.4 Hz), 3.59–3.65 (m, 2H); 13C NMR (101 MHz, CDCl3) δ −5.4 (CH3), −5.3 (CH3), 14.1 (CH3), 18.3 (C), 22.7 (CH2), 25.6 (CH2), 25.9 (3 × CH3), 29.4 (CH2), 29.6 (3 × CH2), 29.6 (CH2), 29.7 (3 × CH2), 29.7 (CH2), 31.9 (CH2), 32.8 (CH2), 67.3 (CH2), 71.9 (CH); MS (CI) m/z 387 (MH+, 100), 369 (49), 329 (19), 295 (3), 111 (2). Anal. Calcd for C23H50O2Si: C, 71.50; H, 12.95. Found: C, 71.51; H, 13.10.
(2R)-1-(tert-Butyldimethylsilyloxy)-2-(methoxymethoxy)heptadecane (7)
A solution of (2R)-1-(tert-butyldimethylsilyloxy)heptadecan-2-ol (8.59 g, 22.2 mmol) was dissolved in dichloromethane (400 mL), and the solution was cooled to 0 °C. Diisopropylethylamine (11.6 mL, 66.7 mmol) was added followed by bromomethyl methyl ether (3.72 mL, 45.6 mmol). The solution was stirred for 0.5 h at 0 °C and then heated under reflux overnight. The reaction mixture was cooled to room temperature, acidified with 1.0 M hydrochloric acid (15 mL), and extracted with dichloromethane (3 × 200 mL). After removal of the solvent under reduced pressure, the resulting material was purified by flash column chromatography (petroleum ether/diethyl ether 25:1) to give (2R)-1-(tert-butyldimethylsilyloxy)-2-(methoxymethoxy)heptadecane (7) as a colorless oil (9.48 g, 100%): IR (NaCl) 2923, 1463, 1250, 1105, 1036, 835, 774 cm–1; [α]D23 +17.1 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.85–0.91 (m, 12H), 1.24–1.52 (m, 28H), 3.38 (s, 3H), 3.54–3.64 (m, 3H), 4.65 (d, 1H, J 6.8 Hz), 4.78 (d, 1H, J 6.8 Hz); 13C NMR (101 MHz, CDCl3) δ −5.4 (CH3), −5.3 (CH3), 14.1 (CH3), 18.3 (C), 22.7 (CH2), 25.4 (CH2), 25.9 (3 × CH3), 29.4 (CH2), 29.6 (3 × CH2), 29.6 (CH2), 29.7 (3 × CH2), 29.7 (CH2), 29.8 (CH2), 31.8 (CH2), 31.9 (CH2), 55.4 (CH3), 65.9 (CH2), 78.3 (CH), 96.3 (CH2); MS m/z 431 (MH+, 41), 399 (100), 369 (72), 343 (12), 285 (5), 161 (8), 107 (10), 73 (21); HRMS (CI) calcd for C25H55O3Si (MH+), 431.3920; found, 431.3922.
(2R)-2-(Methoxymethoxy)heptadecane-1-ol (8)
A solution of tetrabutylammonium fluoride (1.0 M in tetrahydrofuran, 22.00 mL, 22.00 mmol) was added to a solution of (2R)-1-(tert-butyldimethylsilyloxy)-2-(methoxymethoxy)heptadecane (7) (7.88 g, 18.32 mmol) in tetrahydrofuran (400 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was concentrated, and the resulting residue was resuspended in diethyl ether (200 mL). The solution was washed with water (100 mL), and the aqueous layer was extracted with diethyl ether (3 × 150 mL). The combined organic extracts were dried (MgSO4), concentrated, and purified by flash column chromatography (petroleum ether/diethyl ether 7:2) to give (2R)-2-(methoxymethoxy)heptadecane-1-ol (8) as a white solid (5.91 g, 91%): mp 32–34 °C; IR (NaCl) 2929, 1216, 1032, 753, 667 cm–1; [α]D23 −27.6 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.25–1.53 (m, 28H), 3.04 (dd, 1H, J 8.8, 3.6 Hz), 3.43 (s, 3H), 3.46–3.62 (m, 3H), 4.69 (d, 1H, J 6.8 Hz), 4.74 (d, 1H, J 6.8 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.6 (CH2), 29.4 (2 × CH2), 29.6 (2 × CH2), 29.6 (4 × CH2), 29.7 (2 × CH2), 31.7 (CH2), 31.9 (CH2), 55.6 (CH3), 65.7 (CH2), 82.4 (CH), 97.0 (CH2); MS (CI) m/z 317 (MH+, 9), 285 (93), 255 (80), 181 (2), 139 (3), 97 (4). Anal. Calcd for C19H40O3: C, 72.15; H, 12.65. Found: C, 72.34; H, 12.80.
Ethyl (2E,4R)-4-(Methoxymethoxy)nonadecan-2-enoate (9)
Dimethyl sulfoxide (2.52 mL, 35.6 mmol) was added to a stirred solution of oxalyl chloride (1.74 mL, 19.93 mmol) in dichloromethane (350 mL) at −78 °C. The reaction mixture was stirred for 0.3 h before (2R)-2-(methoxymethoxy)heptadecane-1-ol (8) (4.50 g, 14.24 mmol) in dichloromethane (50 mL) was slowly added. The reaction mixture was stirred for an additional 0.3 h before triethylamine (9.90 mL, 71.20 mmol) was added. This reaction mixture was stirred for 0.5 h at −78 °C, allowed to warm to room temperature, and stirred for another 2 h. Meanwhile, a solution of lithium chloride (1.10 g, 25.63 mmol), triethyl phosphonoacetate (4.23 mL, 21.36 mmol), and 1,8-diazabicyclo[5,4,0]undec-7-ene (3.22 mL, 21.36 mmol) in acetonitrile (350 mL) was prepared and stirred for 1.0 h. The Swern solution was concentrated in vacuo, the Horner–Wadsworth–Emmons solution was added, and the reaction mixture was stirred at room temperature overnight. The reaction was quenched by the addition of a saturated solution of ammonium chloride (50 mL) and concentrated to give an orange residue, which was extracted with diethyl ether (4 × 100 mL). The organic layers were combined, dried (MgSO4), and concentrated to give an orange oil. Purification by flash column chromatography (diethyl ether/petroleum ether 1:20) gave ethyl (2E,4R)-4-(methoxymethoxy)nonadecan-2-enoate (9) (5.48 g, 100%) as a yellow oil: IR (NaCl) 2922, 1723, 1265, 1152, 1030, 921 cm–1; [α]D23 +40.6 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.25–1.68 (m, 31H), 3.38 (s, 3H), 4.16–4.23 (m, 3H), 4.58 (d, 1H, J 6.8 Hz), 4.63 (d, 1H, J 6.8 Hz), 5.97 (dd, 1H, J 15.6, 1.2 Hz), 6.81 (dd, 1H, J 15.6, 6.4 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 14.2 (CH3), 22.7 (CH2), 25.2 (CH2), 29.4 (2 × CH2), 29.5 (3 × CH2), 29.6 (3 × CH2), 29.7 (2 × CH2), 31.9 (CH2), 34.9 (CH2), 55.6 (CH3), 60.5 (CH2), 75.3 (CH), 94.6 (CH2), 121.8 (CH), 148.0 (CH), 166.3 (C); MS (CI) m/z 385 (MH+, 11), 355 (33), 323 (100), 283 (47), 173 (12), 109 (4). Anal. Calcd for C23H44O4: C, 71.87; H, 11.45. Found: C, 71.90; H, 11.68.
(2E,4R)-4-(Methoxymethoxy)nonadecan-2-ene-1-ol (10)
Ethyl (2E,4R)-4-(methoxymethoxy)nonadecan-2-enoate (9) (4.7 g, 12.24 mmol) was dissolved in diethyl ether (350 mL), and the solution was cooled to −78 °C. DIBAL-H (1 M in hexane, 27 mL, 27.00 mmol) was added dropwise, and the reaction mixture was stirred at −78 °C for 3 h before warming to room temperature. The solution was cooled to 0 °C, quenched by the addition of a saturated solution of ammonium chloride (50 mL), and warmed to room temperature with vigorous stirring over 1 h, producing a white precipitate. The precipitate was filtered through a pad of Celite and washed with diethyl ether (400 mL). The filtrate was dried (MgSO4) and concentrated in vacuo. Flash chromatography (diethyl ether/petroleum ether 2:5) gave (2E,4R)-4-(methoxymethoxy)nonadecan-2-ene-1-ol (10) (3.60 g, 86%) as a white solid: mp 38–40 °C; IR (NaCl) 3390, 2913, 2849, 1471, 1093, 1002, 914, 716 cm–1; [α]D23 +61.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.24–1.62 (m, 29H), 3.37 (s, 3H), 4.02 (q, 1H, J 7.2 Hz), 4.16 (td, 2H, J 5.6, 1.6 Hz), 4.53 (d, 1H, J 6.8 Hz), 4.70 (d, 1H, J 6.8 Hz), 5.57 (ddt, 1H, J 15.6, 7.2, 1.6 Hz), 5.81 (dt, 1H, J 15.6, 5.6 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.4 (CH2), 29.4 (2 × CH2), 29.6 (3 × CH2), 29.6 (2 × CH2), 29.7 (3 × CH2), 31.9 (CH2), 35.6 (CH2), 55.4 (CH3), 63.0 (CH2), 76.2 (CH), 93.7 (CH2), 131.7 (CH), 131.9 (CH); MS (CI) m/z 343 (MH+, 3), 319 (11), 281 (81), 263 (14), 157 (8), 113 (7), 85 (100). Anal. Calcd for C21H42O3: C, 73.68; H, 12.28. Found: C, 73.81; H, 12.47.
(3S,4R)-3-(2′,2′,2′-Trichloromethylcarbonylamino)-4-(methoxymethoxy)nonadecan-1-ene (12)
(2E,4R)-4-(Methoxymethoxy)nonadecan-2-ene-1-ol (10) (3.01 g, 8.80 mmol) was dissolved in dichloromethane (300 mL), and the solution was cooled to 0 °C. 1,8-Diazabicyclo[5.4.0]undec-7-ene (0.33 mL, 2.20 mmol) was added to the solution followed by trichloroacetonitrile (1.32 mL, 13.20 mmol). The reaction mixture was warmed to room temperature, stirred for 2 h, filtered through a short pad of silica gel, and washed with diethyl ether (200 mL). The resulting filtrate was concentrated to give allylic trichloroacetimidate 11, which was used without further purification. Resulting allylic trichloroacetimidate 11 was dissolved in toluene (300 mL), and bis(acetonitrile)palladium(II) chloride (0.22 g, 0.88 mmol) and p-benzoquinone (1.90 g, 17.60 mmol) were added. The reaction mixture was stirred at 45 °C for 24 h, concentrated, and purified by flash column chromatography (diethyl ether/petroleum ether 1:30) to give (3S,4R)-3-(2′,2′,2′-trichloromethylcarbonylamino)-4-(methoxymethoxy)nonadecan-1-ene (12) as a yellow oil (3.22 g, 78% over 2 steps): IR (NaCl) 3292, 2922, 1717, 1516, 1465, 1236, 1033, 819, 721 cm–1; [α]D23 −5.1 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.23–1.65 (m, 28H), 3.43 (s, 3H), 3.56 (ddd, 1H, J 8.4, 4.8, 2.0 Hz), 4.35–4.42 (m, 1H), 4.65 (d, 1H, J 6.8 Hz), 4.74 (d, 1H, J 6.8 Hz), 5.30–5.36 (m, 2H), 5.85 (ddd, 1H, J 17.2, 10.4, 6.8 Hz), 8.28 (br d, 1H, J 7.6 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.7 (CH2), 29.4 (2 × CH2), 29.5 (3 × CH2), 29.5 (CH2), 29.6 (2 × CH2), 29.7 (2 × CH2), 31.9 (CH2), 33.0 (CH2), 55.9 (CH), 56.7 (CH3), 83.8 (CH), 93.0 (C), 98.1 (CH2), 118.9 (CH2), 131.6 (CH), 161.4 (C); MS (CI) m/z 486 (MH+, 42), 454 (100), 416 (31), 392 (23), 356 (18), 263 (38), 214 (28), 113 (17), 85 (71). Anal. Calcd for C23H42Cl3NO3: C, 56.79; H, 8.64; N, 2.88. Found: C, 56.87; H, 8.75; N, 2.79.
(2S,3R)-2-(2′,2′,2′-Trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecan-1-ol (13)
(3S,4R)-3-(2′,2′,2′-Trichloromethylcarbonylamino)-4-(methoxymethoxy)nonadecan-1-ene (12) (1.46 g, 3.00 mmol) was dissolved in a mixture of dichloromethane (60 mL) and methanol (20 mL), and the solution was cooled to −78 °C. Ozone was bubbled through the reaction mixture until the solution turned slightly blue. After excess ozone was purged with argon, sodium borohydride (0.11 g, 3.00 mmol) was added in portions. The solution was stirred for 1.5 h at 0 °C and acidified with 1 M hydrochloric acid (10 mL). The reaction was quenched by the addition of water (20 mL) and extracted with diethyl ether (4 × 75 mL). The organic layers were combined, dried (MgSO4), and concentrated to give an orange oil. Purification by flash column chromatography using (diethyl ether/petroleum ether 1:1) yielded (2S,3R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecan-1-ol (13) (1.15 g, 78% over 2 steps) as a colorless oil: IR (NaCl) 2922, 1705, 1521, 1467, 1030, 819, 822 cm–1; [α]D23 −24.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.24–1.70 (m, 28H), 2.69 (dd, 1H, J 9.2, 3.4 Hz), 3.43 (s, 3H), 3.70–3.81 (m, 2H), 3.92 (dq, 1H, J 8.4, 3.4 Hz), 4.03 (dt, 1H, J 12.0, 3.4 Hz), 4.63 (d, 1H, J 6.8 Hz), 4.71 (d, 1H, J 6.8 Hz), 7.96 (br d, 1H, J 8.4 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.7 (CH2), 29.4 (2 × CH2), 29.5 (2 × CH2), 29.6 (4 × CH2), 29.7 (CH2), 30.3 (CH2), 31.9 (CH2), 32.9 (CH2), 54.8 (CH), 56.1 (CH3), 61.2 (CH2), 82.5 (CH), 92.7 (C), 97.9 (CH2), 162.2 (C); MS (CI) m/z 490 (MH+, 21), 458 (18), 428 (12), 372 (100), 340 (22), 255 (5), 137 (6), 73 (16). Anal. Calcd for C22H42Cl3NO4: C, 53.87; H, 8.57; N, 2.85. Found: C, 53.93; H, 8.71; N, 2.95.
(2S,3R)-2-Aminooctadecane-1,3-diol Hydrochloride (2)10c
(2S,3R)-2-(2′,2′,2′-Trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecan-1-ol (13) (0.06 g, 0.12 mmol) was dissolved in 6 M hydrochloric acid (10 mL), and the solution was heated at 60 °C for 24 h. The reaction mixture was cooled to room temperature and washed with diethyl ether (3 × 10 mL). The aqueous layer was concentrated to give (2R,3S)-2-aminooctadecane-1,3-diol hydrochloride (2) (0.038 g, 100%) as a colorless oil: IR (NaCl) 3348, 2924, 1600, 1467, 1051 cm–1; [α]D23 +8.4 (c 3.0, MeOH), lit.10c [α]D +8.1 (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ 0.80 (t, 3H, J 6.8 Hz), 1.17–1.46 (m, 28H), 3.10 (dt, 1H, J 8.6, 4.4 Hz), 3.60 (dd, 1H, J 11.6, 8.6 Hz), 3.65–3.72 (m, 1H), 3.74 (dd, 1H, J 11.6, 4.4 Hz); 13C NMR (101 MHz, CD3OD) δ 14.5 (CH3), 23.8 (CH2), 27.0 (CH2), 30.5 (2 × CH2), 30.6 (3 × CH2), 30.7 (3 × CH2), 30.7 (CH2), 30.8 (CH2), 33.1 (CH2), 34.2 (CH2), 58.5 (CH), 58.9 (CH2), 70.3 (CH); MS (CI) m/z 302 (MH+, 90), 282 (17), 267 (23), 241 (36), 137 (11), 107 (22), 73 (100).
(2S,3R)-1-Methanesulfonate-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane
A solution of (2S,3R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecan-1-ol (13) (1.08 g, 2.20 mmol) was dissolved in dichloromethane (50 mL). Methanesulfonyl chloride (0.37 mL, 3.30 mmol), triethylamine (0.67 mL, 4.84 mmol), and 4-dimethylaminopyridine (0.03 g, 0.22 mmol) were added at 0 °C, and the solution was stirred at room temperature for 24 h. The reaction mixture was washed with water (20 mL) and extracted with dichloromethane (3 × 40 mL). The organic layer was dried (MgSO4) and concentrated to give the crude product. Purification by flash column chromatography (diethyl ether/petroleum ether 2:3) gave (2S,3R)-1-methanesulfonate-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane (1.17 g, 93%) as a colorless oil: IR (NaCl) 3343, 2916, 1686, 1521, 1349, 1171, 1034, 895 cm–1; [α]D23 −22.0 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 7.2 Hz), 1.24–1.68 (m, 28H), 3.06 (s, 3H), 3.45 (s, 3H), 3.62 (ddd, 1H, J 8.0, 4.6, 2.8 Hz), 4.23–4.29 (m, 1H), 4.34 (dd, 1H, J 10.8, 7.6 Hz), 4.50 (dd, 1H, J 10.8, 4.6 Hz), 4.63 (dd, 1H, J 6.8 Hz), 4.73 (d, 1H, J 6.8 Hz), 8.13 (br d, 1H, J 8.4 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.9 (CH2), 29.4 (CH2), 29.4 (2 × CH2), 29.5 (2 × CH2), 29.6 (2 × CH2), 29.7 (CH2), 30.0 (2 × CH2), 31.9 (CH2), 32.9 (CH2), 37.8 (CH3), 53.2 (CH), 56.0 (CH3), 66.4 (CH2), 82.3 (CH), 92.6 (C), 98.2 (CH2), 162.2 (C); MS (CI) m/z 568 (MH+, 4), 536 (10), 502 (8), 474 (20), 438 (100), 404 (89), 370 (31), 356 (21), 241 (9), 137 (15), 97 (19). Anal. Calcd for C23H44Cl3NO6S: C, 48.59; H, 7.74; N, 2.46. Found: C, 48.64; H, 7.85; N, 2.58.
(2R,3R)-1-Bromo-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane (14)
(2S,3R)-1-Methanesulfonate-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane (0.20 g, 0.35 mmol) was dissolved in dimethyl sulfoxide (10 mL), and sodium bromide (0.18 g, 1.76 mmol) was added. The reaction mixture was heated under reflux overnight. The reaction mixture was cooled and concentrated in vacuo. The resulting residue was dissolved in diethyl ether (20 mL) and washed with water (2 × 30 mL). The organic layer was dried and concentrated in vacuo. Purification by column chromatography (petroleum ether/diethyl ether 14:1) gave (2R,3R)-1-bromo-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane (14) (0.14 g, 73%) as a colorless oil: IR (NaCl) 3330, 2921, 1718, 1517, 1030, 819 cm–1; [α]D23 +30.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.8 Hz), 1.25–1.62 (m, 28H), 3.44 (s, 3H), 3.57 (dd, 1H, J 11.2, 7.6 Hz), 3.63–3.68 (m, 2H), 4.26 (ddt, 1H, J 9.2, 7.6, 4.0 Hz), 4.65 (d, 1H, J 6.8 Hz), 4.73 (d, 1H, J 6.8 Hz), 7.69 (br d, 1H, J 9.2 Hz); 13C NMR (101 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2), 25.4 (CH2), 29.4 (2 × CH2), 29.5 (2 × CH2), 29.5 (CH2), 29.6 (2 × CH2), 29.7 (2 × CH2), 29.7 (CH2), 31.4 (CH2), 31.9 (CH2), 32.4 (CH2), 54.6 (CH), 56.0 (CH3), 81.6 (CH), 92.7 (C), 97.8 (CH2), 161.8 (C); MS m/z 552 (MH+, 10), 522 (71), 492 (38), 472 (16), 438 (24), 285 (100), 239 (33), 189 (12), 151 (12), 117 (11); HRMS (CI) calcd for C22H4279Br35Cl3NO3 (MH+), 552.1414; found, 552.1416.
(2S,3R)-2-Acetylamino-3-(methoxymethoxy)octadecane (15)
To a solution of (2R,3R)-1-bromo-2-(2′,2′,2′-trichloromethylcarbonylamino)-3-(methoxymethoxy)octadecane (14) (0.02g, 0.04 mmol) in methanol (5 mL) was added 10% palladium on carbon (0.03 g) and triethylamine (0.03 mL, 0.21 mmol). The reaction mixture was allowed to stir under an atmosphere of hydrogen at room temperature for 18 h. The reaction mixture was filtered through a short pad of Celite, which was washed with methanol (50 mL) and concentrated in vacuo. Purification by flash column chromatography (petroleum ether/diethyl ether 1:6) gave (2S,3R)-2-acetylamino-3-(methoxymethoxy)octadecane (15) (0.01 g, 68%) as a white solid: mp 35–37 °C; IR (NaCl) 3370, 2925, 1647, 1540, 1372, 1034, 920 cm–1; [α]D23 −45.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.81 (t, 3H, J 6.4 Hz), 1.02 (d, 3H, J 6.8 Hz), 1.18–1.51 (m, 28H), 1.93 (s, 3H), 3.35–3.38 (m, 4H), 3.90–3.97 (m, 1H), 4.55 (d, 1H, J 6.8 Hz), 4.65 (d, 1H, J 6.8 Hz), 6.69 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 13.9 (CH3), 14.1 (CH3), 22.7 (CH2), 23.5 (CH3), 25.9 (CH2), 29.3 (CH2), 29.5 (2 × CH2), 29.5 (4 × CH2), 29.6 (2 × CH2), 29.7 (CH2), 31.9 (CH2), 32.7 (CH2), 47.2 (CH), 55.7 (CH3), 84.4 (CH), 98.1 (CH2), 169.1 (C); MS m/z 372 (MH+, 100), 340 (31), 310 (4), 174 (3), 113 (4), 71 (10); HRMS (CI) calcd for C22H46NO3 (MH+), 372.3478; found, 372.3475.
(2S,3R)-2-Aminooctadecan-3-ol Hydrochloride (4)11g
(2S,3R)-2-Acetylamino-3-(methoxymethoxy)octadecane (15) (0.01 g, 0.03 mmol) was dissolved in 6 M hydrochloric acid (1.5 mL), and the solution was heated at 60 °C for 48 h. The reaction mixture was cooled and washed with diethyl ether (3 × 10 mL). The aqueous layer was concentrated to give (2S,3R)-2-aminooctadecan-3-ol hydrochloride (4) (0.008 g, 100%) as a white solid: mp 64–66 °C; [α]D23 +7.5 (c 0.3, MeOH), lit.11g [α]D +7.15 (c 0.42, MeOH); 1H NMR (400 MHz, CD3OD) δ 0.80 (t, 3H, J 5.6 Hz), 1.12 (d, 3H, J 5.2 Hz), 1.19–1.35 (m, 28H), 3.14–3.20 (m, 1H), 3.57–3.62 (m, 1H); 13C NMR (101 MHz, CD3OD) δ 12.1 (CH3), 14.5 (CH3), 23.8 (CH2), 27.0 (CH2), 30.5 (CH2), 30.7 (CH2), 30.7 (2 × CH2), 30.8 (4 × CH2), 30.8 (2 × CH2), 33.1 (CH2), 34.0 (CH2), 52.6 (CH), 71.7 (CH); MS (CI) m/z 286 (MH+, 100), 268 (16), 241 (8), 197 (12), 113 (100), 85 (41).
2,3-Isopropylidine-1-methoxy-d-ribofuranoside (17)23
Concentrated hydrochloric acid (34–36%, 0.02 mL) was added to a stirred solution of d-ribose (16) (0.20 g, 1.34 mmol) in acetone (3 mL) and methanol (3 mL). The solution was heated under reflux for 2 h and cooled to room temperature. The reaction was neutralized by the addition of a saturated solution of sodium hydrogen carbonate (10 mL) and extracted with ethyl acetate (2 × 50 mL). The organic layers were combined and washed with water (40 mL) and brine (40 mL), dried (MgSO4), and concentrated in vacuo to yield 2,3-isopropylidine-1-methoxy-d-ribofuranoside (17) (0.23 g, 84%) as a colorless oil (the spectroscopic data were as reported in the literature23): [α]D25 −52.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.32 (s, 3H), 1.48 (s, 3H), 3.24 (dd, 1H, J 10.5, 2.7 Hz), 3.43 (s, 3H), 3.61 (ddd, 1H, J 12.8, 10.5, 2.7 Hz), 3.70 (dt, 1H, J 12.8, 2.7 Hz), 4.43 (t, 1H, J 2.7 Hz), 4.59 (d, 1H, J 5.9 Hz), 4.84 (d, 1H, J 5.9 Hz), 4.97 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 24.7 (CH3), 26.3 (CH3), 55.5 (CH3), 64.0 (CH2), 81.5 (CH), 85.8 (CH), 88.4 (CH), 110.0 (CH), 112.1 (C); MS (CI) m/z 205 (MH+, 14), 173 (100), 113 (5), 85 (10), 69 (14).
5-Deoxy-5-iodo-2,3-isopropylidine-1-methoxy-d-ribofuranoside (18)31
Iodine (0.34 g, 1.34 mmol) was added to a stirred solution of 2,3-isopropylidine-1-methoxy-d-ribofuranoside (17) (0.23 g, 1.12 mmol), imidazole (0.11 g, 1.68 mmol), and triphenylphosphine (0.35 g, 1.34 mmol) in toluene (15 mL) and acetonitrile (5 mL). The solution was heated under reflux for 0.1 h and cooled to room temperature. Iodine was added in 10 mg portions until the solution remained dark-brown. The solution was diluted with diethyl ether (40 mL) and washed with a saturated solution of sodium thiosulfate (80 mL), water (100 mL), and brine (70 mL). The organic layer was dried (MgSO4), concentrated in vacuo, and filtered through a short plug of silica (ethyl acetate/petroleum ether 1:20) to yield 5-deoxy-5-iodo-2,3-isopropylidine-1-methoxy-d-ribofuranoside (18) (0.33 g, 94%) as a colorless oil: [α]D25 −65.7 (c 1.0, CHCl3), lit.31 [α]D −68.3 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.33 (d, 3H, J 0.4 Hz), 1.48 (d, 3H, J 0.4 Hz), 3.16 (t, 1H, J 10.0 Hz), 3.29 (dd, 1H, J 10.0, 6.0 Hz), 3.37 (s, 3H), 4.44 (ddd, 1H, J 10.0, 6.0, 0.7 Hz), 4.63 (d, 1H, J 5.9 Hz), 4.76 (dd, 1H, J 5.9, 0.7 Hz), 5.05 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 6.8 (CH2), 25.2 (CH3), 26.6 (CH3), 55.4 (CH3), 83.2 (CH), 85.5 (CH), 87.6 (CH), 109.9 (CH), 112.8 (C); MS (CI) m/z 315 (MH+, 8), 279 (7), 127 (8), 113 (38), 85 (52), 73 (100).
(2S,3R)-2,3-(O-Isopropylidene)pent-4-ene-1,2,3-triol (20)32
Zinc (2.44 g, 35.66 mmol) was added to a solution of 5-deoxy-5-iodo-2,3-isopropylidine-1-methoxy-d-ribofuranoside (18) (2.24 g, 7.14 mmol) and a catalytic amount of glacial acetic acid (12 drops) in methanol (40 mL). The suspension was heated under reflux for 1 h and cooled to 0 °C. A saturated solution of sodium borohydride in ethanol (140 mL) was added dropwise with stirring to the reaction mixture, stirred at 0 °C for 0.5 h, and warmed to room temperature over 3 h. The mixture was concentrated in vacuo. The resulting residue was dissolved in diethyl ether (50 mL) and filtered through Celite. The organic layer was washed with water (2 × 50 mL) and brine (2 × 50 mL), dried (MgSO4), and concentrated in vacuo to yield (2S,3R)-2,3-(O-isopropylidene)pent-4-ene-1,2,3-triol (20) (1.01 g, 89%) as a colorless oil: [α]D25 −40.0 (c 1.5, CHCl3), lit.32 [α]D −45.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.40 (d, 3H, J 0.4 Hz), 1.51 (d, 3H, J 0.4 Hz), 1.81 (t, 1H, J 6.2 Hz), 3.59 (dd, 2H, J 6.2, 5.9 Hz), 4.26 (dt, 1H, J 6.6, 5.9 Hz), 4.65 (tt, 1H, J 6.6, 1.1 Hz), 5.28 (ddd, 1H, J 10.3, 1.5, 1.1 Hz), 5.40 (ddd, 1H, J 17.5, 1.5, 1.1 Hz), 5.87 (ddd, 1H, J 17.5, 10.3, 6.6 Hz); 13C NMR (101 MHz, CDCl3) δ 25.3 (CH3), 27.8 (CH3), 62.1 (CH2), 78.3 (CH), 78.4 (CH), 108.9 (C), 119.0 (CH2), 133.1 (CH); MS (CI) m/z 159 (MH+, 71), 141 (11), 101 (100), 83 (12).
(2S,3R)-2,3-(O-Isopropylidene)heptadecane-1,2,3-triol (22)
A solution of Grubbs second-generation catalyst (0.14 g, 0.17 mmol) in dichloromethane (20 mL) was added to a solution of (2S,3R)-2,3-(O-isopropylidene)pent-4-ene-1,2,3-triol (20) (0.54 g, 3.41 mmol) and 1-tetradecene (1.01 g, 5.12 mmol) in dichloromethane (20 mL) and heated under reflux for 16 h. The reaction mixture was then concentrated in vacuo and filtered through a plug of silica first with petroleum ether (500 mL) followed by 25% ethyl acetate in petroleum ether (1000 mL). The second wash was concentrated in vacuo to yield cross metathesis product 21 as a clear oil, which was used without further purification. Alkene 21 was dissolved in ethyl acetate (40 mL), and a suspension of 10% palladium on carbon (0.16 g, 30% w/w) in ethyl acetate (10 mL) was added. The mixture was degassed, purged with hydrogen gas, and left to stir at room temperature under a hydrogen atmosphere for 2 h. The suspension was filtered through Celite with ethyl acetate (100 mL) and purified by column chromatography (ethyl acetate/petroleum ether 1:5) to yield (2S,3R)-2,3-(O-isopropylidene)heptadecane-1,2,3-triol (22) (0.92 g, 82% over two steps) as a colorless oil: IR (neat) 3437, 2923, 2854, 1466, 1379, 1217, 1042 cm–1; [α]D24 −10.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.87 (t, 3H, J 6.8 Hz), 1.21–1.53 (m, 32H), 1.95 (br s, 1H), 3.55–3.63 (m, 2H), 4.10–4.19 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 14.3 (CH3), 22.8 (CH2), 25.7 (CH3), 26.8 (CH3), 28.4 (CH2), 29.0 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (3 × CH2), 29.8 (2 × CH2), 29.8 (CH2), 32.1 (CH2), 62.0 (CH2), 77.2 (CH), 78.1 (CH), 108.2 (C); MS m/z 329 (MH+, 100), 313 (13), 271 (60), 253 (10), 235 (5), 159 (3), 69 (7); HRMS (CI) calcd for C20H41O3 (MH+), 329.3056; found, 329.3051.
Ethyl (2E,4S,5R)-4,5-(O-Isopropylidene)-4,5-dihydroxynonadec-2-enoate (23)
Dimethyl sulfoxide (0.68 g, 8.66 mmol) was added to a stirred solution of oxalyl chloride (0.56 g, 4.34 mmol) in dichloromethane (15 mL) at −78 °C. The reaction mixture was stirred for 0.3 h, and (2S,3R)-2,3-(O-isopropylidene)heptadecane-1,2,3-triol (22) (0.95 g, 2.89 mmol) in dichloromethane (15 mL) was slowly added. The mixture was stirred for an additional 0.3 h, and triethylamine (1.46 g, 14.44 mmol) was added. This reaction mixture was stirred for 0.5 h at −78 °C and warmed to room temperature for a further 2 h. Meanwhile, a solution of lithium chloride (0.24 g, 5.78 mmol), triethyl phosphonoacetate (0.75 g, 4.91 mmol), and 1,8-diazabicyclo[5.4.0]undec-7-ene (1.10 g, 4.91 mmol) in acetonitrile (30 mL) was prepared and stirred for 1.0 h. The Swern solution was concentrated in vacuo and the Horner–Wadsworth–Emmons solution was added. The reaction mixture was stirred at room temperature overnight, quenched by the addition of a saturated solution of ammonium chloride (20 mL), and concentrated to give an orange residue that was extracted with diethyl ether (2 × 30 mL). The organic layers were combined, washed with water (50 mL) and brine (50 mL), dried (MgSO4), and concentrated to give a yellow oil. Purification by filtration through a pad of silica (ethyl acetate/petroleum ether 1:5) gave ethyl (2E,4S,5R)-4,5-(O-isopropylidene)-4,5-dihydroxynonadec-2-enoate (23) (0.95 g, 83%) as a clear yellow oil: IR (neat) 2924, 2853, 1722, 1466, 1370, 1252, 1161, 1038 cm–1; [α]D26 −5.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J 6.9 Hz), 1.23–1.33 (m, 27H), 1.35–1.49 (m, 8H), 4.16–4.26 (m, 3H), 4.63 (td, 1H, J 6.3, 1.4 Hz), 6.06 (dd, 1H, J 15.6, 1.4 Hz), 6.84 (dd, 1H, J 15.6, 6.3 Hz); 13C NMR (101 MHz, CDCl3) δ 14.3 (CH3), 14.4 (CH3), 22.8 (CH2), 25.7 (CH3), 26.5 (CH2), 28.2 (CH3), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.8 (2 × CH2), 30.6 (CH2), 32.1 (CH2), 60.6 (CH2), 77.6 (CH), 78.7 (CH), 108.9 (C), 123.2 (CH), 144.0 (CH), 166.2 (C); MS (CI) m/z 397 (MH+, 88), 379 (12), 339 (100), 293 (8), 143 (13), 81 (12). Anal. Calcd for C24H44O4: C, 72.68; H, 11.18. Found: C, 72.48; H, 11.29.
(2E,4S,5R)-4,5-(O-Isopropylidene)nonadec-2-ene-1,4,5-triol (24)
Diisobutylaluminium hydride (0.55 mL, 5.50 mmol, 1 M in hexanes) was added dropwise to a stirred solution of ethyl (2E,4S,5R)-4,5-(O-isopropylidene)-4,5-dihydroxynonadec-2-enoate (23) (0.95 g, 2.39 mmol) in diethyl ether (50 mL) at −78 °C. The solution was stirred at −78 °C for 3 h and warmed to room temperature over 18 h. The reaction mixture was quenched with a saturated solution of ammonium chloride (30 mL), filtered through a pad of Celite, and extracted with diethyl ether (30 mL). The organic layer was washed with water (50 mL) and brine (50 mL). The organic layer was dried (MgSO4) and concentrated in vacuo to give a colorless oil. Purification by column chromatography (ethyl acetate/petroleum ether 1:5) gave (2E,4S,5R)-4,5-(O-isopropylidene)nonadec-2-ene-1,4,5-triol (24) (0.71 g, 83%) as a colorless oil: IR (neat) 3417, 2922, 2853, 1456, 1369, 1216, 1095, 1027, 974 cm–1; [α]D24 –42.0 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.87 (t, 3H, J 7.0 Hz), 1.22–1.52 (m, 32H), 1.62 (br s, 1H), 4.12 (ddd, 1H, J 8.7, 6.3, 4.7 Hz), 4.16 (dd, 2H, J 5.2, 1.5 Hz), 4.50 (dd, 1H, J 7.9, 6.3 Hz), 5.68 (ddt, 1H, J 15.5, 7.9, 1.5 Hz), 5.88 (dtd, 1H, J 15.5, 5.2, 0.6 Hz); 13C NMR (126 MHz, CDCl3) δ 14.2 (CH3), 22.8 (CH2), 25.8 (CH3), 26.4 (CH2), 28.4 (CH3), 29.5 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.8 (3 × CH2), 30.5 (CH2), 32.1 (CH2), 63.0 (CH2), 78.5 (CH), 79.1 (CH), 108.2 (C), 127.5 (CH), 133.5 (CH); MS m/z 355 (MH+, 26), 337 (20), 297 (41), 279 (100), 251 (23), 225 (90), 111 (69), 73 (71); HMRS (CI) calcd for C22H43O3 (MH+), 355.3212; found, 355.3211.
(3R,4S,5R)-3-(2′,2′,2′-Trichloromethylcarbonylamino)-4,5-(O-isopropylidene)nonadec-1-ene-4,5-diol (26) and (3S,4S,5R)-3-(2′,2′,2′-Trichloromethylcarbonylamino)-4,5-(O-isopropylidene)nonadec-1-ene-4,5-diol (27)
1,8-Diazabicycloundec-7-ene (0.07 g, 0.49 mmol) was added to a stirred solution of (2E,4S,5R)-4,5-(O-isopropylidene)nonadec-2-en-1,4,5-triol (24) (0.19 g, 0.54 mmol) and trichloroacetonitrile (0.12 g, 0.82 mmol) at 0 °C in dichloromethane (30 mL). The solution was stirred at 0 °C for 0.5 h and warmed to room temperature over 3 h. The reaction mixture was filtered through a pad of silica with dichloromethane (200 mL) and concentrated in vacuo to yield crude allylic trichloroacetimidate 25, which was used without further purification. Allylic trichloroacetimidate 25 was dissolved in p-xylene (4 mL), and the solution was added to a pressure tube loaded with a stirrer bar and potassium carbonate (0.01 g, 0.72 mmol). The tube was sealed under argon and heated with stirring at 160 °C for 60 h. The reaction mixture was cooled and filtered through Celite to yield a yellow viscous oil. Purification by column chromatography (ethyl acetate/petroleum ether 1:20) gave (3R,4S,5R)-3-(2′,2′,2′-trichloromethylcarbonylamino)-4,5-(O-isopropylidene)nonadec-1-ene-4,5-diol (26) (0.12 g, 44%) as a colorless oil followed by (3S,4S,5R)-3-(2′,2′,2′-trichloromethylcarbonylamino)-4,5-(O-isopropylidene)nonadec-1-ene-4,5-diol (27) (0.07 g, 26%) as a colorless oil. For 26: IR (neat) 3427, 2922, 2853, 1721, 1496, 1211, 819 cm–1; [α]D27 −10.5 (c 1.4, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 7.0 Hz), 1.22–1.33 (m, 24H), 1.38 (s, 3H), 1.46–1.58 (m, 5H), 4.21–4.27 (m, 2H), 4.37–4.42 (m, 1H), 5.27 (d, 1H, J 10.4 Hz), 5.30 (d, 1H, J 17.1 Hz), 5.83 (ddd, 1H, J 17.1, 10.4, 5.8 Hz), 7.19 (d, 1H, J 7.7 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 22.9 (CH2), 24.3 (CH3), 27.2 (CH3), 27.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.6 (CH2), 29.7 (2 × CH2), 29.8 (2 × CH2), 29.8 (CH2), 29.8 (CH2), 32.1 (CH2), 53.2 (CH), 77.6 (CH), 78.4 (CH), 92.9 (C), 108.1 (C), 117.2 (CH2), 134.8 (CH), 160.8 (C); MS m/z 498 (MH+, 14), 440 (31), 406 (28), 297 (18), 161 (35), 113 (52), 73 (100); HRMS (CI) calcd for C24H4335Cl3NO3 (MH+), 498.2309; found, 498.2307. For 27: IR (neat) 3324, 2922, 2853, 1694, 1515, 1370, 1217, 1056, 822 cm–1; [α]D −48.0 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 7.0 Hz), 1.24–1.33 (m, 24H), 1.36 (s, 3H), 1.48 (s, 3H), 1.50–1.64 (m, 2H), 4.13 (dd, 1H, J 6.3, 3.8 Hz), 4.23 (ddd, 1H, J 9.7, 6.3, 3.8 Hz), 4.55 (dddt, 1H, J 8.7, 5.2, 3.8, 1.7 Hz), 5.26–5.33 (m, 2H), 6.00 (ddd, 1H, J 17.4, 10.5, 5.2 Hz), 6.92 (d, 1H, J 8.7 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 22.8 (CH2), 25.3 (CH3), 27.0 (CH3), 27.1 (CH2), 29.0 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.8 (CH2), 29.9 (CH2), 32.1 (CH2), 54.2 (CH), 77.6 (CH), 79.0 (CH), 92.8 (C), 108.7 (C), 118.0 (CH2), 134.0 (CH), 161.2 (C); MS m/z 498 (MH+, 5), 464 (13), 440 (100), 406 (81), 394 (22), 329 (21), 297 (20), 271 (9), 113 (7), 73 (12); HRMS (CI) calcd for C24H4335Cl3NO3 (MH+), 498.2309; found, 498.2304.
(3E,5S,6R)-5,6-(O-Isopropylidene)-5,6-dihydroxyicos-3-en-2-one (28)
Dimethyl sulfoxide (0.68 g, 8.66 mmol) was added to a stirred solution of oxalyl chloride (0.56 g, 4.34 mmol) in dichloromethane (15 mL) at −78 °C. The reaction mixture was stirred for 0.3 h, and (2S,3R)-2,3-(O-isopropylidene)heptadecane-1,2,3-triol (22) (0.95 g, 2.89 mmol) in dichloromethane (15 mL) was slowly added. The mixture was stirred for an additional 0.3 h, and triethylamine (1.46 g, 14.44 mmol) was added. This reaction mixture was stirred for 0.5 h at −78 °C, allowed to warm to room temperature, and stirred for a further 2 h. Meanwhile, a solution of lithium chloride (0.24 g, 5.78 mmol), dimethyl 2-oxopropylphosphonate (0.82 g, 4.91 mmol), and 1,8-diazabicyclo[5.4.0]undec-7-ene (1.10 g, 4.91 mmol) in acetonitrile (30 mL) was prepared and stirred for 1.0 h. The Swern solution was concentrated in vacuo, and the Horner–Wadsworth–Emmons solution was added. The reaction mixture was stirred at room temperature overnight, quenched by the addition of a saturated solution of ammonium chloride (20 mL), and concentrated to give an orange residue that was extracted with diethyl ether (2 × 30 mL). The organic layers were combined, washed with water (50 mL) and brine (50 mL), dried (MgSO4), and concentrated to give a yellow oil. Purification by filtration through a pad of silica (ethyl acetate/petroleum ether 1:5) gave (3E,5S,6R)-5,6-(O-isopropylidene)-5,6-dihydroxyicos-3-en-2-one (28) (0.84 g, 79%) as a colorless oil: IR (neat) 2916, 2848, 1697, 1676, 1632, 1373, 1246, 1217, 1102, 1036 cm–1; [α]D21 −2.3 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.87 (t, 3H, J 6.9 Hz), 1.21–1.34 (m, 24H), 1.38 (s, 3H), 1.41–1.49 (m, 2H), 1.51 (s, 3H), 2.28 (s, 3H), 4.23 (ddd, 1H, J 8.7, 6.4, 4.6 Hz), 4.64 (td, 1H, J 6.4, 1.3 Hz), 6.28 (dd, 1H, J 15.9, 1.3 Hz), 6.65 (dd, 1H, J 15.9, 6.4 Hz); 13C NMR (101 MHz, CDCl3) δ 14.3 (CH3), 22.8 (CH2), 25.6 (CH3), 26.4 (CH2), 27.7 (CH3), 28.2 (CH3), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.8 (2 × CH2), 30.7 (CH2), 32.1 (CH2), 77.8 (CH), 78.6 (CH), 109.0 (C), 131.9 (CH), 142.6 (CH), 198.0 (C); MS m/z 367 (MH+, 71), 349 (17), 309 (45), 239 (4), 141 (100), 113 (16), 81 (16); HRMS (CI) calcd for C23H43O3 (MH+), 367.3212; found, 367.3216.
(2S,3E,5S,6R)-5,6-(O-Isopropylidene)icos-3-ene-2,5,6-triol (29)
(R)-(+)-2-Methyl-CBS-oxazaborolidine (0.60 mL, 0.60 mmol, 1 M solution in toluene) was added dropwise with stirring to a solution of (3E,5S,6R)-5,6-(O-isopropylidene)-5,6-dihydroxyicos-3-en-2-one (28) (0.20 g, 0.55 mmol) in dry tetrahydrofuran (20 mL) at 0 °C. The solution was stirred for 0.5 h at 0 °C, borane (1.64 mL, 1.64 mmol, 1 M in tetrahydrofuran) was added dropwise, and stirring was continued at 0 °C for 7.5 h. The reaction was quenched with methanol (10 mL), warmed to room temperature, and concentrated in vacuo. The resulting residue was dissolved in diethyl ether (30 mL) and washed with 1 M citric acid (3 × 50 mL), water (50 mL), and brine (50 mL). The organic layer was dried (MgSO4) and purified by column chromatography (ethyl acetate/petroleum ether 1:6, 1% triethylamine) to yield (2S,3E,5S,6R)-5,6-(O-isopropylidene)icos-3-ene-2,5,6-triol (29) (0.20 g, 99%) as a colorless oil: IR (neat) 3397, 2982, 2922, 1456, 1368, 1215, 1032 cm–1; [α]D22 +4.5 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 7.0 Hz), 1.23–1.32 (m, 27H), 1.36 (s, 3H), 1.38–1.53 (m, 5H), 4.12 (ddd, 1H, J 8.6, 6.3, 4.7 Hz), 4.35 (quin, 1H, J 5.9 Hz), 4.49 (dd, 1H, J 7.9, 6.3 Hz), 5.65 (ddd, 1H, J 15.5, 7.9, 1.2 Hz), 5.81 (ddd, 1H, J 15.5, 5.9, 0.7 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 22.8 (CH2), 23.4 (CH3), 25.8 (CH3), 26.3 (CH2), 28.4 (CH3), 29.5 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.8 (CH2), 29.8 (2 × CH2), 30.6 (CH2), 32.1 (CH2), 68.2 (CH), 78.5 (CH), 79.0 (CH), 108.2 (C), 125.9 (CH), 138.4 (CH); HRMS (ESI) calcd for C23H44NaO3 (MNa+), 391.3183; found, 391.3169.
(2R,3E,5S,6R)-5,6-(O-Isopropylidene)icos-3-ene-2,5,6-triol (30)
The reaction was carried out according to the procedure described above using (3E,5S,6R)-5,6-(O-isopropylidene)-5,6-dihydroxyicos-3-ene-2-one (28) (0.10 g, 0.28 mmol) and (S)-(−)-2-methyl-CBS-oxazaborolidine (0.30 mL, 0.30 mmol, 1 M solution in tetrahydrofuran), yielding (2R,3E,5S,6R)-5,6-(O-isopropylidene)icos-3-en-2,5,6-triol (30) (0.065 g, 64%) as a colorless oil: IR (neat) 3414, 2922, 2853, 1458, 1370, 1217, 1044, 756 cm–1; [α]D24 −3.7 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 6.9 Hz), 1.20–1.31 (m, 27H), 1.35 (s, 3H), 1.38–1.48 (s, 5H), 4.08–4.15 (m, 1H), 4.33 (quintet of doublets, 1H, J 6.5, 0.9 Hz), 4.47 (dd, 1H, J 8.0, 6.2 Hz), 5.62 (ddd, 1H, J 15.5, 8.0, 0.9 Hz), 5.78 (ddd, 1H, J 15.5, 6.5, 0.6 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 22.8 (CH2), 23.3 (CH3), 25.8 (CH3), 26.2 (CH2), 28.4 (CH3), 29.5 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (3 × CH2), 29.8 (CH2), 29.8 (CH2), 30.4 (CH2), 30.5 (CH2), 32.1 (CH2), 68.3 (CH), 78.5 (CH), 79.0 (CH), 108.2 (C), 126.1 (CH), 138.5 (CH); MS m/z 369 (MH+, 21), 351 (37), 309 (14), 293 (100), 225 (28), 183 (39), 125 (88); HRMS (CI) calcd for C23H45O3 (MH+), 369.3369; found, 369.3366.
(2E,4S,5S,6R)-4-(2′,2′,2′-Trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (31)
1,8-Diazabicycloundec-7-ene (0.011 g, 0.07 mmol) was added to a stirred solution of (2S,3E,5S,6R)-5,6-(O-isopropylidene)icos-3-ene-2,5,6-triol (29) (0.053 g, 0.14 mmol) and trichloroacetonitrile (0.031 g, 0.21 mmol) at 0 °C in dichloromethane (3 mL). The solution was stirred at 0 °C for 0.5 h and warmed to room temperature over 3 h. The solution was filtered through a pad of silica with dichloromethane (200 mL) and concentrated in vacuo to yield the allylic trichloroacetimidate as a yellow oil, which was used without further purification. The allylic trichloroacetimidate was dissolved in p-xylene (2 mL) and added to a microwave vial loaded with a silicon carbide bar and potassium carbonate (0.012 g, 6 mg/mL). The vial was sealed under argon and heated at 140 °C for 0.25 h in a microwave reactor. After the reaction was cooled to room temperature, the reaction mixture was concentrated in vacuo. Purification by column chromatography (diethyl ether/petroleum ether 1:10) gave (2E,4S,5S,6R)-4-(2′,2′,2′-trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (31) (0.053 g, 72%) as a white solid: mp 53–58 °C; IR (neat) 3356, 2918, 2851, 1688, 1516, 1219, 959, 818 cm–1; [α]D22 −28.5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 6.9 Hz), 1.23–1.38 (m, 27H), 1.48 (s, 3H), 1.57–1.69 (m, 2H), 1.72 (dd, 3H, J 6.4, 1.4 Hz), 4.11 (dd, 1H, J 6.3, 3.8 Hz), 4.21 (ddd, 1H, J 9.6, 6.3, 3.7 Hz), 4.47–4.51 (m, 1H), 5.60 (ddd, 1H, J 15.5, 5.7, 1.4 Hz), 5.74 (dqd, 1H, J 15.5, 6.4, 1.1 Hz), 6.90 (d, 1H, J 8.7 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 18.2 (CH3), 22.9 (CH2), 25.3 (CH3), 27.0 (CH3), 27.1 (CH2), 29.0 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 29.9 (2 × CH2), 32.1 (CH2), 53.8 (CH), 77.6 (CH), 79.1 (CH), 92.9 (C), 108.5 (C), 126.7 (CH), 129.4 (CH), 161.0 (C); HRMS (ESI) calcd for C25H4435Cl3NNaO3 (MH+), 534.2279; found, 534.2265.
(2S,3S,4R)-2-(2′,2′,2′-Trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (32)
(2E,4S,5S,6R)-4-(2′,2′,2′-Trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (31) (0.057 g, 0.11 mmol) was dissolved in a mixture of dichloromethane (10 mL) and methanol (10 mL), and the solution was cooled to −78 °C. Ozone was bubbled through the reaction mixture until the solution turned deep blue. After excess ozone was purged with oxygen gas, a solution of sodium borohydride (0.013 g, 0.32 mmol) in ethanol (10 mL) was added dropwise with vigorous stirring. The reaction mixture was allowed to slowly return to room temperature over 5 h. The reaction mixture was quenched with a saturated solution of ammonium chloride (10 mL), concentrated in vacuo to an aqueous solution, and extracted with diethyl ether (3 × 15 mL). The combined organic layers were washed with water (20 mL) and brine (20 mL), dried (MgSO4), and concentrated in vacuo to yield the crude product. Purification by column chromatography (diethyl ether/petroleum ether 1:1) gave (2S,3S,4R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (32) (0.041 g, 73%) as a colorless oil: IR (neat) 3307, 2918, 2849, 1687, 1533, 1221, 1047, 822 cm–1; [α]D20 +4.8 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 7.0 Hz), 1.17–1.33 (m, 24H), 1.36 (s, 3H), 1.49 (s, 3H), 1.52–1.58 (m, 2H), 3.73 (dd, 1H, J 11.4, 3.2 Hz), 4.01 (dd, 1H, J 11.4, 3.2 Hz), 4.05 (ddt, 1H, J 8.4, 5.6, 3.2 Hz), 4.19–4.26 (m, 2H), 7.18 (d, 1H, J 8.4 Hz); 13C NMR (126 MHz, CDCl3) δ 14.2 (CH3), 22.8 (CH2), 25.1 (CH3), 26.9 (CH2), 27.6 (CH3), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (CH2), 29.8 (CH2), 29.8 (2 × CH2), 32.1 (CH2), 52.1 (CH), 62.5 (CH2), 77.8 (CH), 77.8 (CH), 92.7 (C), 108.6 (C), 161.7 (C); MS m/z 502 (MH+, 16), 444 (100), 410 (41), 329 (80), 313 (60), 271 (46), 257 (38), 219 (31), 172 (27), 148 (26), 73 (59); HRMS (CI) calcd for C23H4335Cl3NO4 (MH+), 502.2258; found, 502.2264.
(2S,3S,4R)-2-Aminooctadecane-1,3,4-triol Hydrochloride (3)10j
(2S,3S,4R)-2-(2′,2′,2′-Trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (32) (0.026 g, 0.05 mmol) was dissolved in a mixture of dichloromethane (2 mL) and methanol (2 mL). Sodium hydroxide (4 M, 4 mL) was added, and the reaction mixture was stirred for 60 h. The solution was extracted with dichloromethane (3 × 20 mL), washed with water (3 × 20 mL), and concentrated in vacuo. The crude oil was dissolved in a mixture of dichloromethane (2 mL) and methanol (2 mL). To this solution was added 6 M hydrochloric acid (2 mL), and the resulting solution was stirred for 3 h at room temperature. The reaction was concentrated in vacuo and washed with petroleum ether (3 × 10 mL) to yield (2S,3S,4R)-2-aminooctadecane-1,3,4-triol hydrochloride (3) as a white solid (0.015 g, 82%) (the spectroscopic data were as previously reported10j): IR (neat) 3304 (OH), 2916 (CH), 2849 (CH), 1688, 1470, 1015 cm–1; [α]D21 +4.8 (c 0.8, MeOH); 1H NMR (500 MHz, CD3OD) δ 0.90 (t, 3H, J 7.0 Hz), 1.28–1.42 (m, 24H), 1.50–1.57 (m, 1H), 1.64–1.72 (m, 1H), 3.38–3.45 (m, 2H), 4.11 (ddd, 1H, J 8.8, 5.9, 3.2 Hz), 4.39 (t, 1H, J 8.8 Hz), 4.46 (dd, 1H, J 8.8, 5.9 Hz); 13C NMR (126 MHz, CD3OD) δ 14.3 (CH3), 23.6 (CH2), 26.6 (CH2), 30.4 (CH2), 30.7 (2 × CH2), 30.7 (5 × CH2), 30.8 (CH2), 33.0 (CH2), 34.8 (CH2), 55.6 (CH), 67.4 (CH2), 74.0 (CH), 75.8 (CH); HRMS (ESI) calcd for C18H40NO3 (MH+), 318.3003; found, 318.2990.
(2E,4R,5S,6R)-4-(2′,2′,2′-Trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (33)
The reactions were carried out according to the procedure described for (2E,4S,5S,6R)-4-(2′,2′,2′-trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (31) using (2R,3E,5S,6R)-5,6-(O-isopropylidene)icos-3-ene-2,5,6-triol (30) (0.029 g, 0.08 mmol). This yielded (2E,4R,5S,6R)-4-(2′,2′,2′-trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (33) (0.029 g, 71%) as a yellow oil: IR (neat) 3426, 2922, 2853, 1719, 1495, 1213, 964, 820 cm–1; [α]D22 −12.6 (c 0.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (t, 3H, J 7.0 Hz), 1.20–1.33 (m, 24H), 1.38 (s, 3H), 1.45–1.57 (m, 5H), 1.72 (ddd, 3H, J 6.5, 1.6, 0.9 Hz), 4.16–4.24 (m, 2H), 4.30–4.37 (m, 1H), 5.45 (ddq, 1H, J 15.3, 7.3, 1.6 Hz), 5.75 (dqd, 1H, J 15.3, 6.5, 0.9 Hz), 7.18 (d, 1H, J 7.0 Hz); 13C NMR (126 MHz, CDCl3) δ 14.3 (CH3), 18.0 (CH3), 22.9 (CH2), 24.4 (CH3), 27.2 (CH3), 27.3 (CH2), 29.3 (CH2), 29.5 (CH2), 29.6 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (2 × CH2), 29.8 (CH2), 29.8 (CH2), 29.8 (CH2), 32.1 (CH2), 52.8 (CH), 77.6 (CH), 78.9 (CH), 93.0 (C), 108.0 (C), 127.8 (CH), 129.0 (CH), 160.6 (C); MS m/z 512 (MH+, 32), 478 (84), 442 (100), 408 (55), 297 (20), 189 (34), 113 (14); HRMS (CI) calcd for C25H4535Cl3NO3 (MH+), 512.2465; found, 512.2458.
(2R,3S,4R)-2-(2′,2′,2′-Trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (34)
The reactions were carried out according to the procedure described for (2S,3S,4R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (32) using (2E,4R,5S,6R)-4-(2′,2′,2′-trichloromethylcarbonylamino)-5,6-(O-isopropylidene)icos-2-ene-5,6-diol (33) (0.09 g, 0.18 mmol). This yielded (2R,3S,4R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (34) (0.058 g, 66%) as a colorless oil: IR (neat) 3419, 2922, 2853, 1717, 1502, 1211, 1050, 820 cm–1; [α]D26 −20.5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.90 (t, 3H, J 6.9 Hz), 1.21–1.33 (m, 24H), 1.40 (s, 3H), 1.41–1.60 (m, 5H), 3.75 (dd, 1H, J 11.1, 6.4 Hz), 3.81 (dd, 1H, J 11.1, 5.1 Hz), 3.91–3.99 (m, 1H), 4.22–4.30 (m, 1H), 4.42 (dd, 1H, J 7.1, 1.4 Hz), 7.27 (d, 1H, J 7.4 Hz); 13C NMR (126 MHz, CDCl3) δ 14.2 (CH3), 22.8 (CH2), 24.5 (CH3), 27.2 (CH2), 27.2 (CH3), 29.4 (CH2), 29.5 (CH2), 29.5 (CH2), 29.6 (CH2), 29.6 (CH2), 29.7 (CH2), 29.7 (CH2), 29.8 (CH2), 29.8 (CH2), 29.8 (CH2), 32.1 (CH2), 52.7 (CH), 63.9 (CH2), 76.0 (CH), 77.7 (CH), 92.9 (C), 108.3 (C), 161.9 (C); MS m/z 502 (MH+, 18), 486 (14), 444 (100), 419 (34), 391 (42), 342 (37), 297 (68), 149 (11); HRMS (CI) calcd for C23H4335Cl3NO4 (MH+), 502.2258; found, 502.2252.
(2R,3S,4R)-2-Aminooctadecane-1,3,4-triol Hydrochloride (35)10j
The reactions were carried out as described for d-ribo-phytosphingosine hydrochloride (3) using (2R,3S,4R)-2-(2′,2′,2′-trichloromethylcarbonylamino)-3,4-(O-isopropylidene)octadecane-1,3,4-triol (34) (0.033 g, 0.07 mmol). This gave (2R,3S,4R)-2-aminooctadecane-1,3,4-triol hydrochloride (35) as a white solid (0.020 g, 87%) (the spectroscopic data were as previously reported10j): IR (neat) 3335 (NH/OH), 2916 (CH), 2849 (CH), 1472, 1418, 1034, 718 cm–1; [α]D20 −12.2 (c 0.9, MeOH); 1H NMR (500 MHz, CD3OD) δ 0.90 (t, 3H, J 7.0 Hz), 1.27–1.40 (m, 24H), 1.49–1.58 (m, 1H), 1.75 (td, 1H, J 9.4, 3.4 Hz), 3.19 (dd, 1H, J 8.1, 4.2 Hz), 3.42 (td, 1H, J 8.1, 3.4 Hz), 4.13 (ddd, 1H, J 8.9, 6.3, 4.2 Hz), 4.29 (dd, 1H, J 8.9, 6.3 Hz), 4.48 (t, 1H, J 8.9 Hz); 13C NMR (126 MHz, CD3OD) δ 14.3 (CH3), 23.6 (CH2), 26.5 (CH2), 30.4 (CH2), 30.7 (CH2), 30.7 (CH2), 30.7 (5 × CH2), 30.8 (CH2), 33.0 (CH2), 35.1 (CH2), 55.9 (CH), 69.2 (CH2), 73.9 (CH), 76.3 (CH); HRMS (ESI) calcd for C18H40NO3 (MH+), 318.3003; found, 318.2990.
Acknowledgments
The authors are grateful to the EPSRC (studentship to E.D.D.C), Libyan People’s Bureau, London (studentship to A.M.Z.), and the University of Glasgow for funding.
Supporting Information Available
1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Snook C. F.; Jones J. A.; Hannun Y. A. Biochim. Biophys. Acta 2006, 1761, 927. [DOI] [PubMed] [Google Scholar]
- Summers S. A.; Nelson D. H. Diabetes 2005, 54, 591. [DOI] [PubMed] [Google Scholar]
- Modrak D. E.; Gold D. V.; Goldenberg D. M. Mol. Cancer Ther. 2006, 5, 200. [DOI] [PubMed] [Google Scholar]
- Kolter T.; Sandhoff K. Biochim. Biophys. Acta 2006, 1758, 2057. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lafrasse C.; Rousson R.; Valla S.; Antignac P.; Louisot P.; Vanier M. T. Biochem. J. 1997, 325, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dickson R. C.; Nagiec E. E.; Skrzypek M.; Tillman P.; Wells G. B.; Lester R. L. J. Biol. Chem. 1997, 272, 30196. [DOI] [PubMed] [Google Scholar]; b Schneiter R. BioEssays 1999, 21, 1004. [DOI] [PubMed] [Google Scholar]
- a Rinehart K. L.; Fregeau N. L.; Warwick R. A.; Garcia Gravalos D.; Avila J.; Faircloth G. T.. WO 9952521A, 1999;; Chem. Abstr. 1999, 131, 295576 [Google Scholar]; b Cuadros R.; Montejo de Garcini E.; Wandosell F.; Faircloth G.; Fernández-Sousa J. M.; Avila J. Cancer Lett. 2000, 152, 23. [DOI] [PubMed] [Google Scholar]
- For reviews on the syntheses of sphingoid-type bases, see:; a Koskinen P. M.; Koskinen A. M. P. Synthesis 1998, 1075. [Google Scholar]; b Howell A. R.; So R. C.; Richardson S. K. Tetrahedron 2004, 60, 11327. [Google Scholar]; c Morales-Serna J. A.; Llaveria J.; Díaz Y.; Matheu M. I.; Castillón S. Curr. Org. Chem. 2010, 14, 2483. [Google Scholar]
- For recent syntheses of d-erythro-sphinganine, see:; a Fernandes R. A.; Kumar P. Eur. J. Org. Chem. 2000, 3447. [Google Scholar]; b Sengupta S.; Das D.; Mondal S. Synlett 2001, 1464. [Google Scholar]; c Hertweck C.; Sebek P.; Svatos A. Synlett 2001, 1965. [Google Scholar]; d Ndakala A. J.; Hashemzadeh M.; So R. C.; Howell A. R. Org. Lett. 2002, 4, 1719. [DOI] [PubMed] [Google Scholar]; e Cook G. R.; Pararajasingham K. Tetrahedron Lett. 2002, 43, 9027. [Google Scholar]; f Enders D.; Müller-Hüwen A. Eur. J. Org. Chem. 2004, 1732. [Google Scholar]; g So R. C.; Ndonye R.; Izmirian D. P.; Richardson S. K.; Guerrera R. L.; Howell A. R. J. Org. Chem. 2004, 69, 3233. [DOI] [PubMed] [Google Scholar]; h Ndonye R. M.; Izmirian D. P.; Dunn M. F.; Yu K. O. A.; Porcelli S. A.; Khurana A.; Kronenberg M.; Richardson S. K.; Howell A. R. J. Org. Chem. 2005, 70, 10260. [DOI] [PubMed] [Google Scholar]; i Abraham E.; Davies S. G.; Millican N. L.; Nicholson R. L.; Roberts P. M.; Smith A. D. Org. Biomol. Chem. 2008, 6, 1655. [DOI] [PubMed] [Google Scholar]; j Kokatla H. P.; Sagar R.; Vankar Y. D. Tetrahedron Lett. 2008, 49, 4728. [Google Scholar]; k Séguin C.; Ferreira F.; Botuha C.; Chemla F.; Pérez-Luna A. J. Org. Chem. 2009, 74, 6986. [DOI] [PubMed] [Google Scholar]; l Ait-Youcef R.; Moreau X.; Greck C. J. Org. Chem. 2010, 75, 5312. [DOI] [PubMed] [Google Scholar]
- For recent syntheses of d-ribo-phytosphingosine analogs, see:; a Luo S.-Y.; Thopate S. R.; Hsu C.-Y.; Hung S.-C. Tetrahedron Lett. 2002, 43, 4889. [Google Scholar]; b Naidu S. V.; Kumar P. Tetrahedron Lett. 2003, 44, 1035. [Google Scholar]; c Cai Y.; Ling C.-C.; Bundle D. R. Org. Biomol. Chem. 2006, 4, 1140. [DOI] [PubMed] [Google Scholar]; d Lombardo M.; Capdevila M. G.; Pasi F.; Trombini C. Org. Lett. 2006, 8, 3303. [DOI] [PubMed] [Google Scholar]; e Yoon H. J.; Kim Y.-W.; Lee B. K.; Lee W. K.; Kim Y.; Ha H.-J. Chem. Commun. 2007, 79. [DOI] [PubMed] [Google Scholar]; f Llaveria J.; Díaz Y.; Matheu M. I.; Castillón S. Org. Lett. 2009, 11, 205. [DOI] [PubMed] [Google Scholar]; g Cai Y.; Ling C.-C.; Bundle D. R. Carbohydr. Res. 2009, 344, 2120. [DOI] [PubMed] [Google Scholar]; h Liu Z.; Byun H.-S.; Bittman R. J. Org. Chem. 2010, 75, 4356. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Martinková M.; Gonda J.; Pomikalová K.; Kožíšek J.; Kuchár J. Carbohydr. Res. 2011, 346, 1728. [DOI] [PubMed] [Google Scholar]; j Perali R. S.; Mandava S.; Chalapala S. Tetrahedron 2011, 67, 9283. [Google Scholar]; k Xarnod C.; Huang W.; Ren R.-G.; Liu R.-C.; Wei B.-G. Tetrahedron 2012, 68, 6688. [Google Scholar]; l Devi T. J.; Saikia B.; Barua N. C. Tetrahedron 2013, 69, 3817. [Google Scholar]
- For recent syntheses of (+)-spisulosine, see:; a Yun J. M.; Sim T. B.; Hahm H. S.; Lee W. K. J. Org. Chem. 2003, 68, 7675. [DOI] [PubMed] [Google Scholar]; b Allepuz A. C.; Badorrey R.; Díaz-de-Villegas M. D.; Gálvez J. A. Eur. J. Org. Chem. 2009, 6172. [Google Scholar]; c Amarante G. W.; Cavallaro M.; Coelho F. Tetrahedron Lett. 2010, 51, 2597. [Google Scholar]; d Ghosal P.; Shaw A. K. Tetrahedron Lett. 2010, 51, 4140. [Google Scholar]; e Dinda S. K.; Das S. K.; Panda G. Tetrahedron 2010, 66, 9304. [Google Scholar]; f Chen B.-S.; Yang L.-H.; Ye J.-L.; Huang T.; Ruan Y.-P.; Fu J.; Huang P.-Q. Eur. J. Med. Chem. 2011, 46, 5480. [DOI] [PubMed] [Google Scholar]; g Malik G.; Estéoule A.; Retailleau P.; Dauban P. J. Org. Chem. 2011, 76, 7438. [DOI] [PubMed] [Google Scholar]; h Xu K.; Lai G.; Zha Z.; Pan S.; Chen H.; Wang Z. Chem.—Eur. J. 2012, 18, 12357. [DOI] [PubMed] [Google Scholar]
- Jamieson A. G.; Sutherland A. Org. Biomol. Chem. 2005, 3, 735. [DOI] [PubMed] [Google Scholar]
- a Jamieson A. G.; Sutherland A. Org. Biomol. Chem. 2006, 4, 2932. [DOI] [PubMed] [Google Scholar]; b Jamieson A. G.; Sutherland A. Tetrahedron 2007, 63, 2123. [Google Scholar]
- Fanning K. N.; Jamieson A. G.; Sutherland A. Org. Biomol. Chem. 2005, 3, 3749. [DOI] [PubMed] [Google Scholar]
- Jamieson A. G.; Sutherland A. Org. Lett. 2007, 9, 1609.17371038 [Google Scholar]
- Zaed A. M.; Sutherland A. Org. Biomol. Chem. 2010, 8, 4394. [DOI] [PubMed] [Google Scholar]
- Zaed A.; Sutherland A. Org. Biomol. Chem. 2011, 9, 8030. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; VanNieuwenhze M. S.; Sharpless K. B. Chem. Rev. 1994, 94, 2483. [Google Scholar]
- a Hubieki M. P.; Gandour R. D.; Ashendel C. L. J. Org. Chem. 1996, 61, 9379. [Google Scholar]; b Murakami T.; Hirono R.; Furusawa K. Tetrahedron 2005, 61, 9233. [Google Scholar]
- Blanchette M. A.; Choy W.; Davis J. T.; Essenfeld A. P.; Masamune S.; Roush W. R.; Sakai T. Tetrahedron Lett. 1984, 25, 2183. [Google Scholar]
- Overman L. E.; Carpenter N. E. In Organic Reactions; Overman L. E., Ed.; Wiley: Hoboken, NJ, 2005; Vol. 66, pp 1–107 and references therein. [Google Scholar]
- Watanabe M.; Harada N. J. Org. Chem. 1995, 60, 7372. [Google Scholar]
- Barrett A. G. M.; Lebold S. A. J. Org. Chem. 1990, 55, 3853. [Google Scholar]
- Bernet B.; Vasella A. Helv. Chim. Acta 1979, 62, 1990. [Google Scholar]
- a Lipshutz B. H.; Pollart D.; Monforte J.; Kotsuki H. Tetrahedron Lett. 1985, 26, 705. [Google Scholar]; b Swift M. D.; Sutherland A. Tetrahedron 2008, 64, 9521. [Google Scholar]
- Because the Pd(II)-catalyzed rearrangement of allylic trichloroacetimidate 25 failed to give the corresponding allylic trichloracetamide, the synthesis of a bis-MOM derivative of allylic alcohol 24 was investigated. Although the acetonide group could be removed from compound 23 (2 M HCl), subsequent MOM protection of the resulting diol under standard conditions (see synthesis of 7) gave only mono-MOM protection at the C-5 position. Further attempts using more forceful conditions (sealed tube, 80 °C) gave none of the bis-MOM product and led to substantial decomposition.
- The general effects of 1,3-allylic strain controlling the stereochemical outcome of reactions have been reviewed:Hoffmann R. W. Chem. Rev. 1989, 89, 1841. [Google Scholar]
- a Corey E. J.; Bakshi R. K.; Shibata S. J. Am. Chem. Soc. 1987, 109, 5551. [Google Scholar]; b Corey E. J.; Bakshi R. K.; Shibata S.; Chen C.-P.; Singh V. K. J. Am. Chem. Soc. 1987, 109, 7925. [Google Scholar]
- For a recent novel application of the Overman rearrangement under thermal conditions, see:Lee S. I.; Moon S. Y.; Hwang G.-S.; Ryu D. H. Org. Lett. 2010, 12, 3234. [DOI] [PubMed] [Google Scholar]
- The two-step approach for the deprotection of 32 was found to give d-ribo-phytosphingosine (3) more cleanly and in higher yield than a single-step deprotection of both protecting groups under acidic conditions at elevated temperatures (6 M HCl, 60 °C).
- Prabhakar P.; Rajaram S.; Reddy D. K.; Shekar V.; Venkateswarlu Y. Tetrahedron: Asymmetry 2010, 21, 216. [Google Scholar]
- Davies S. G.; Foster E. M.; Frost A. B.; Lee J. A.; Roberts P. M.; Thomson J. E. Org. Biomol. Chem. 2012, 10, 6186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.