

Abstract
Whole genome amplification and sequencing of single microbial cells enables genomic characterization without the need of cultivation 1-3. Viruses, which are ubiquitous and the most numerous entities on our planet 4 and important in all environments 5, have yet to be revealed via similar approaches. Here we describe an approach for isolating and characterizing the genomes of single virions called 'Single Virus Genomics' (SVG). SVG utilizes flow cytometry to isolate individual viruses and whole genome amplification to obtain high molecular weight genomic DNA (gDNA) that can be used in subsequent sequencing reactions.
Keywords: Genetics, Issue 75, Microbiology, Immunology, Virology, Molecular Biology, Environmental Sciences, Genomics, environmental genomics, Single virus, single virus genomics, SVG, whole genome amplification, flow cytometry, viral ecology, virion, genome analysis, DNA, PCR, sequencing
Protocol
1. Preparation of Viral Suspensions
Before isolation of single virions via flow cytometry, prepare unfixed viral suspensions.
Isolate viral particles using previously established protocols making sure final viral suspension is contained in 0.1 μm-filtered TE (Tris-EDTA, pH 8). We recommend using tangential flow filtration (TFF) approaches 6 although other methods have been developed that use chemical flocculation 7.
Enumerate viral particles using established protocols, e.g. using epifluorescence 8-10 or flow cytometry 11,12.
Aliquot viral suspension into two Eppendorf tubes (final volume is recommended to be 1,000 μl).
2. Flow Cytometry
To sort viruses using the BD FACSAria II Flow Cytometer equipped with a custom Forward Scatter PMT (FSC PMT), dilute viral particles in 0.1 μm-filtered TE, pH 8.0 to an appropriate titer for an event rate of 200 events sec-1.
Set thresholds to maximize signal-to-noise rations, e.g. for a mixed assemblage containing T4 and lambda phage particles the following are recommended: FSC PMT at 1,000 and SSC at 200.
Run 100 μl of "blank" samples containing only 1) 0.1 μm-filtered TE and 2) 0.1 μm-filtered TE and SybrGreen1 at 1e-4 dilution of stock (10,000X).
Run a small aliquot (100 μl is recommended) of unstained viral suspension to assess background, followed by the stained viral suspension (SybrGreen1 at 1e-4 dilution of stock), viral particles to a total of 5,000 events each. This is to check concentration and adjust sorting gates, if necessary, prior to sort. We recommend a tight gate in the center of viral particles. Also, for generating gates we use the SYBR Green and FSC PMT plot.
Add 5 μl of 1% low melting point (LMP) agarose (in TBE buffer) cooled to 37 °C to each well on a polytetrafluoroethylene (PTFE) microscope slide (Electron Microscopy Sciences).
Load the PTFE microscope slide into the flow cytometer to capture sorted viral particles.
Begin sort of SYBR green stained viral particles directly onto PTFE slide with LMP agarose. We recommend sorting 10 or more events (viral particles) in some wells to aid in the detection of the depth of viruses during confocal laser scanning microscopy (CSLM).
When sorting is complete, remove slide from the flow cytometer and add 5 μl more LMP agarose cooled to 37 °C to each well, thus embedding the viral particle(s).
3. Visualizing Single Virions using Confocal Microscopy
If a single virion is desired, then determining if each well contains an individual virus is imperative and CLSM is necessary to visualize the embedded virion(s) in 3D to verify that only a single particle is present and that multiple viruses are not stacked on top of each other.
Set the microscope laser to the wavelength 488 nm excitation.
Image the embedded viral particles using a 63X long working distance objective.
4. Whole Genome Amplification
Once wells with single viruses are identified with CLSM, perform whole genome amplification using multiple displacement amplification (MDA) within the agarose (in situ).
To decrease the likelihood of introducing DNA contamination, remove the desired agarose 'beads' from the slide and placed in a sterile PCR tube. Use a pair of sterile tweezers and/or razor blade for this step.
Lyse the viral particle in situ using heat (94 °C) for 3 min in the heat block of a thermocycler.
- Perform the MDA reaction following manufacturer's recommendations (GenomiPhi HY kit, GE Healthcare) with the following modifications
- Reduce the volume of sample and reaction buffers to 11.25 μl and incubate at 30 °C for a maximum of 2 hr to reduce nonspecific amplification.
Purify the amplified genomic material by transferring genomic DNA (gDNA) to a 1.7 ml Eppendorf tube and add 1/10 volume 3 M sodium acetate (NaOAc) in TBE buffer (same buffer used in LMP agarose preparation).
Place sample(s) at 55 °C for 10 min to dissolve agarose.
Move tube(s) to 42 °C to reduce temperature prior to adding enzyme.
Add 2 units β-agarase to each tube and incubate for 2 hr.
Add 1 volume of a buffer-saturated phenol solution to the tube, mix vigorously and centrifuge at 3,500 x g for 15 min. Transfer supernatant containing DNA to a new 1.7 ml Eppendorf tube.
Add one volume of 100% isopropanol and 1μl of Glycoblue. Mix well by inverting tube.
Centrifuge at 28,000 x g at 4 °C for 60 min. Decant isopropanol making sure not to disturb the pellet. Add 150 μl of 70% ethanol to each tube. Spin at 28,000 x g and RT for 10 min. Decant the ethanol, air dry the DNA pellet and resuspend the DNA in an appropriate volume of TE buffer.
We recommend repeating steps 4.4-4.9 with the following modifications. Set up 3-5 replicate MDA reactions and reduce incubation at 30 °C to 1 hr. Pool samples following purification and precipitate using 95-100% ethanol to obtain desired concentration.
5. Representative Results
Figure 1 summarizes the SVG process. These methods use flow cytometry to sort viral particles onto PTFE microscope slides containing agarose to isolate single virions from a mixed assemblage followed by MDA to obtain quantities of gDNA that are sufficient for sequencing.
As a proof-of-concept, bacteriophage lambda and T4 were mixed and subjected to the SVG process 11. Once virions were captured and embedded in agarose, CLSM was used to visualize and confirm that a single virion was obtained; results are shown in Figure 2. Once wells with single virions were identified, they were selected for MDA of gDNA. We then used multiplex PCR specific for T4 and lambda to identify the virion isolated, Figure 3. An isolated phage lambda was selected for genome sequencing.
Genome sequencing was performed using 454-Titanium technology and sequencing reads were subjected to reference mapping to identify the level of coverage obtained using SVG. Figure 4 shows that almost the entire lambda genome was recovered (with the exception of the first 5 bp).
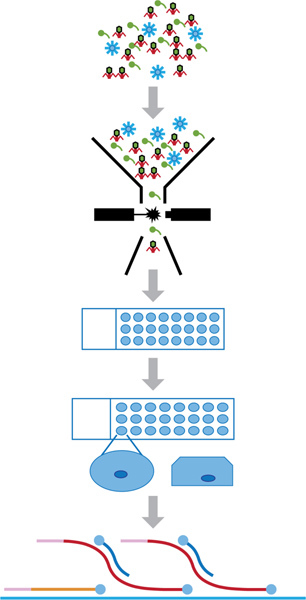 Figure 1. SVG methodology. Viral suspensions containing a mixed assemblage are reduced to single virus particles via flow cytometry that are then sorted onto individual agarose "beads". The virus is embedded within the agarose bead by overlaying with an additional layer of agarose post flow cytometric sorting. Lastly, whole genome amplification is performed in situ.
Figure 1. SVG methodology. Viral suspensions containing a mixed assemblage are reduced to single virus particles via flow cytometry that are then sorted onto individual agarose "beads". The virus is embedded within the agarose bead by overlaying with an additional layer of agarose post flow cytometric sorting. Lastly, whole genome amplification is performed in situ.
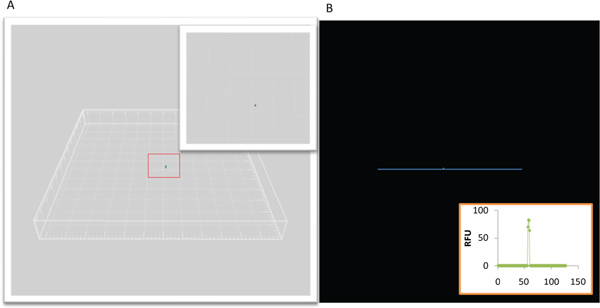 Figure 2. Confocal laser scanning micrograph of a sorted viral particle embedded in an agarose bead.A) Three dimensional reconstruction of a Sybr Green I-stained viral particle within an agarose bead. Inset: higher magnification of the isolated viral particle. B) Profile plot of relative fluorescence of the stained single viral particle within an agarose bead.
Figure 2. Confocal laser scanning micrograph of a sorted viral particle embedded in an agarose bead.A) Three dimensional reconstruction of a Sybr Green I-stained viral particle within an agarose bead. Inset: higher magnification of the isolated viral particle. B) Profile plot of relative fluorescence of the stained single viral particle within an agarose bead.
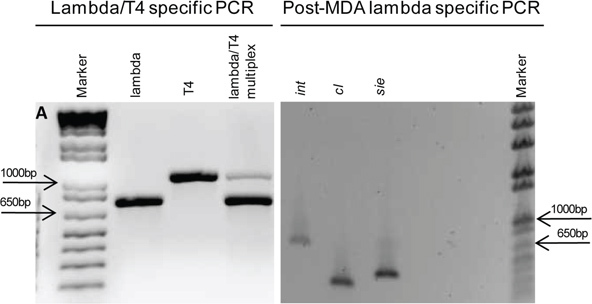 Figure 3. Bacteriophage identification using PCR.A) Multiplex PCR using T4 and lambda-specific primers to genotype, Lanes: 1. TrackIt 1 kb plus ladder (Invitrogen), 2. lambda integrase (750 bp), 3. T4 major capsid protein (1,050 bp), 4. Mix of lambda integrase and T4 major capsid protein. B) Subsequent lambda specific PCR with additional loci to further confirm phage genome isolation, Lanes: 1. Lambda integrase (750 bp), 2. Lambda repressor (356 bp) 3. Lambda sie (superinfection exclusion) (456 bp) 4. TrackIt 1 kb plus ladder (Invitrogen).
Figure 3. Bacteriophage identification using PCR.A) Multiplex PCR using T4 and lambda-specific primers to genotype, Lanes: 1. TrackIt 1 kb plus ladder (Invitrogen), 2. lambda integrase (750 bp), 3. T4 major capsid protein (1,050 bp), 4. Mix of lambda integrase and T4 major capsid protein. B) Subsequent lambda specific PCR with additional loci to further confirm phage genome isolation, Lanes: 1. Lambda integrase (750 bp), 2. Lambda repressor (356 bp) 3. Lambda sie (superinfection exclusion) (456 bp) 4. TrackIt 1 kb plus ladder (Invitrogen).
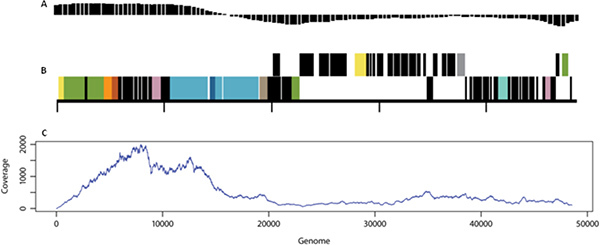 Figure 4. Lambda genome attributes and coverage.A) GC plot, B) Genome map of lambda (adapted from http://img.jgi.doe.gov), and C) Mapping of SVG reads to the reference bacteriophage lambda, x-axis is genome position, y-axis is % coverage.
Figure 4. Lambda genome attributes and coverage.A) GC plot, B) Genome map of lambda (adapted from http://img.jgi.doe.gov), and C) Mapping of SVG reads to the reference bacteriophage lambda, x-axis is genome position, y-axis is % coverage.
Discussion
A number of important factors must be taken into consideration when applying SVG approaches. Genotyping, as was performed during the proof-of-concept experiment 13, is not a valid option for environmental or unknown isolates as conserved primers are not available across all viral groups. Also, background DNA synthesis or nonspecific amplification is commonly reported during amplification using the MDA reaction 14. Nonspecific amplification has been attributed to contaminating DNA emerging from reaction reagents and/or through a mechanism that enables amplification from the random hexamers within the reaction mixture. When working with viral assemblages, there is perhaps a higher likelihood of nonspecific DNAs preferentially amplified due to the lower quantity of template viral DNA as opposed to single bacterial cells as a result of the significant difference in particle (cell) size and genomic DNA content (25-100 nm; ~1.5 fg for viruses, as opposed to 0.2-1.5 um; ~14 fg for bacteria). The following methods are recommended to reduce the level of nonspecific amplification: treatment of viral assemblages with DNase I, examination of virus containing agarose beads using CLSM, reduction of MDA incubation time and increased sequencing depth (i.e. as is capable with next-generation sequencing technologies like 454-Titanium and Illumina).
When performing SVG on environmental samples, optimized de novo assembly is imperative to obtain complete or near-complete genome sequences. We have found that reducing the redundant reads prior to assembly is necessary to obtain greater coverage 13.
Disclosures
No conflicts of interest declared.
Acknowledgments
We would like to thank Ken Nealson for his insight and advice throughout the manuscript preparation process.
References
- Raghunathan A, et al. Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 2005;71:3342–337. doi: 10.1128/AEM.71.6.3342-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken RS. Single-cell genomic sequencing using Multiple Displacement Amplification. Curr. Opin. Microbiol. 2007;10:510–516. doi: 10.1016/j.mib.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Ishoey T, Woyke T, Stepanauskas R, Novotny M, Lasken RS. Genomic sequencing of single microbial cells from environmental samples. Curr. Opin. Microbiol. 2008;11:198–204. doi: 10.1016/j.mib.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Rohwer F. Viral metagenomics. Nature Reviews Microbiology. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Prangishvili D, Lindell D. Roles of viruses in the environment. Environ. Microbiol. 2009;11:2771–2774. doi: 10.1111/j.1462-2920.2009.02101.x. [DOI] [PubMed] [Google Scholar]
- Williamson SJ, et al. The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE. 2008;3:e1456. doi: 10.1371/journal.pone.0001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SG, et al. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011;3:195–202. doi: 10.1111/j.1758-2229.2010.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble R, Fuhrman J. Use of SYBR Green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat Microb Ecol. 1998;14:113–118. [Google Scholar]
- Hara S, Terauchi K, Koike I. Abundance of viruses in marine waters: assessment by epifluorescence and transmission electron microscopy. Appl. Environ. Microbiol. 1991;57:2731–2734. doi: 10.1128/aem.57.9.2731-2734.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennes KP, Suttle CA. Direct counts of viruses in natural waters and laboratory cultures by epifluorescence microscopy. Limnol. Oceanogr. 1995;40:1050–1055. [Google Scholar]
- Marie D, Brussaard CPD, Thyrhaug R, Bratbak G, Vaulot D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl. Environ. Microbiol. 1999;65 doi: 10.1128/aem.65.1.45-52.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussaard CP. Enumeration of bacteriophages using flow cytometry. Methods Mol. Biol. 2009;501:97–111. doi: 10.1007/978-1-60327-164-6_11. [DOI] [PubMed] [Google Scholar]
- Allen LZ, et al. Single Virus Genomics: A New Tool for Virus Discovery. Plos One. 2011;6 doi: 10.1371/journal.pone.0017722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA, Smith HO, Pfannkoch C, Venter JC. Cell-free cloning using phi 29 DNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17332–17336. doi: 10.1073/pnas.0508809102. [DOI] [PMC free article] [PubMed] [Google Scholar]