

Abstract
Biophysical studies have shown that each molecule of calsequestrin 1 (CASQ1) can bind about 70–80 Ca2+ ions. However, the nature of Ca2+-binding sites has not yet been fully characterized. In this study, we employed in-silico approaches to identify the Ca2+ binding sites and to understand the molecular basis of CASQ1-Ca2+ recognition. We built the protein model by extracting the atomic coordinates for the back-to-back dimeric unit from the recently solved hexameric CASQ1 structure (PDB id: 3UOM) and adding the missing C-terminal residues (aa350–364). Using this model we performed extensive 30 ns molecular dynamics simulations exposed to wide range of Ca2+ concentrations ([Ca2+]). Our results show that the Ca2+-binding sites on CASQ1 differ both in affinity and geometry. The high affinity Ca2+-binding sites share a similar geometry and interestingly, majority of them were found to be induced by increased [Ca2+]. We also found that the system undergoes maximal Ca2+-binding to the CAS (consecutive aspartate stretch at the C-terminus) before the rest of the CASQ1 surface becomes saturated. Simulated data shows that the CASQ1 back-to-back stacking is progressively stabilized by emergence of an increasing number of hydrophobic interactions with increasing [Ca2+]. Further, this study shows that the CAS domain assumes a compact structure with increase in Ca2+ binding, which suggests that the CAS domain might function as a Ca2+-sensor that may be a novel structural motif to sense metal. We propose the term “Dn-motif” for the CAS domain.
Keywords: calcium binding protein, molecular dynamics, entropy, electrostatic interaction, structural motif
Introduction
CASQ1 is the major Ca2+ buffering protein present in the sarcoendoplasmic reticulum (SR) of the striated muscles1–5 where it also regulates Ca2+ release by its interaction with Ryanodine receptor (RyR) complex1. Crystallographic studies showed that CASQ1 monomer consists of three α-β globular domains formed by five β-strands arranged into a sheet sandwiched by four α-helices, which is structurally similar to ubiquitous redox protein “thioredoxin”6–7. Beyond the structural domain III, CASQ1 possesses a unique C-terminus that is composed of repeating aspartic acid residues. The length of this consecutive aspartate stretch (CAS) varies between species and typically among mammals it is of 9–14 residues. Biophysico-chemical studies suggest that CASQ1 binds large amount of Ca2+ ions (~70–80 Ca2+/molecule) with low affinity3, 8–11. However, CASQ1 lacks any known Ca2+ binding motif like EF-hand6, 12–14 or double-clamp15 and the nature of Ca2+-binding sites are not fully characterized. It has been proposed that Ca2+ binding to CASQ1 is purely a surface phenomenon governed by the highly negatively charged molecular surface7, 11.
Based on biochemical and structural observations it has been proposed that CASQ1 forms polymeric structure in a Ca2+ concentration ([Ca2+]) dependent manner6–7, 9. At first, two monomers interact to form front-to-front dimer by insertion of 24 N-terminal residues into the partner monomer, resulting in a very stable dimer7, 11, 16. Then, two such front-to-front dimers stack by back-to-back interactions leading to formation of a tetramer and subsequently resulting in the formation of linear polymer in a Ca2+-dependent manner. However, detailed structural information about the interactions at the back-to-back interface in the CASQ1 polymer has been speculative and it is currently unclear if the back-to-back stacking is dependent on [Ca2+]. Further, the CAS residues are located at the back-to-back interface and can significantly influence with the stacking interactions17. It is also unclear if CAS domain plays a role in facilitating back-to-back stacking and Ca2+-binding, given that the structure of the CAS residues has not been resolved in any of the currently available CASQ1 structures.
The recently solved CASQ1 structure (PDB Id: 3UOM12) in the hexameric state provides the opportunity to apply computational approaches to define the physico-chemical properties of the Ca2+-binding sites and the role of CAS domain in [Ca2+]-dependent protein-protein interaction. The hexamer consists of three repeating dimeric units formed by front-to-front dimerization, which on the other hand are held by back-to-back interaction. In the light of this scenario, the goals of this study are 1) identify and characterize the Ca2+-binding sites on CASQ1; 2) better understand how the surface of CASQ1 react to Ca2+-binding and 3) gain insight into the functional role of CAS domain in CASQ1-Ca2+ interaction. To accomplish these goals, we built a model of back-to-back dimeric unit from the hexameric CASQ1 structure, added the missing CAS residues (aa350–364) and performed extensive molecular dynamics (MD) simulations under a wide range of [Ca2+].
Results and Discussions
[Ca2+]-dependent charge distribution on CASQ1 surface
Before starting a detailed structural investigation, we first analysed the stability of CASQ1 dimeric unit during MD simulation by calculating the backbone RMSD and entropy of the system. The stability of the dimeric system increases at higher [Ca2+] as reflected both from decrease of RMSD and entropy values (Supplemental Table 1). Then, we determined the surface charge of CASQ1 after MD simulation in all the systems exposed at different [Ca2+]. As shown in Figure 1, the surface of CASQ1 in a Ca2+ free system is highly acidic, as also previously reported11, 18. Then, as expected, a gradual surface neutralization, mediated by Ca2+ binding, was observed increasing [Ca2+]. However, up to the presence of 40 Ca2+ ions, the surface was significantly negative and only some regions were found to be neutralized by Ca2+-binding. The CASQ1 surface for the system having 60 Ca2+ ions seems to be further neutralized and becomes fully neutral in the presence of 80 Ca2+. Interestingly, the electrostatic surface charge calculation shows highly electropositive surface for the CASQ1 in the system having 120 Ca2+ ions.
Figure 1. Changes in the surface charge pattern of CASQ1 with increasing Ca2+ concentrations.
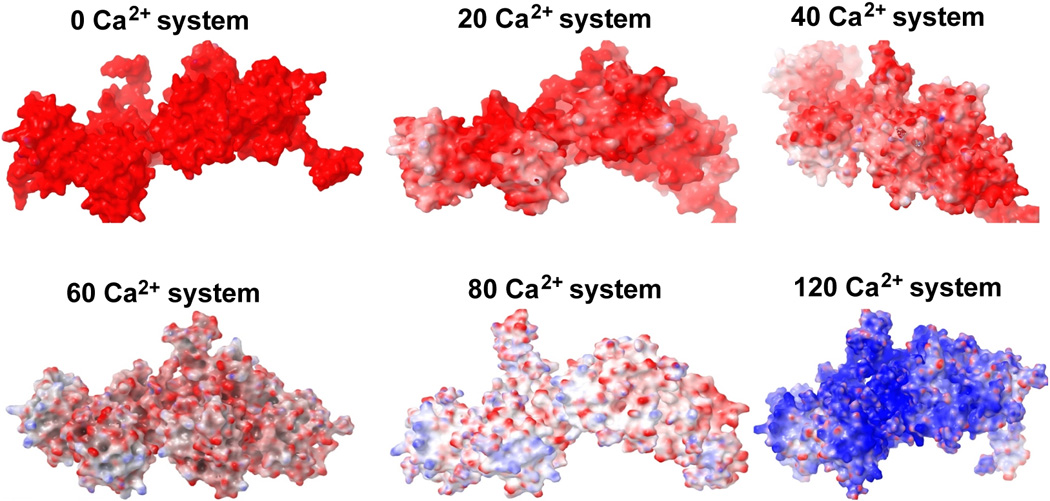
Surface charge was calculated for the average structure for each system indicated in the figure. Red, blue and white indicate negative, positive and neutral charge on the molecular surface. The color scale used to represent the Poisson Boltzmann electrostatic potential isosurface range is from −4 to +4 KT/e (where K - Boltzmann constant, T - temperature, and e - electron charge. CASQ1 gains electro-positivity on the surface in the 120 Ca2+-system, even if the surface negative charge is almost neutralized already in the 80 Ca2+-system.
Differential Ca2+-binding to CAS and rest of CASQ1
Next, we calculated the number of Ca2+ ions that interact with the CASQ1 protein after the MD simulation. Interestingly, we found that the CAS residues get saturated with Ca2+ before the rest of the protein surface. As shown in Figure 2A, the CAS residues (350–364) of chain B and C, bind to an increasing number of Ca2+ ions as [Ca2+] increases. However, in both the chains, CAS gets saturated when [Ca2+] reached 80 ions in the system, and after this threshold no further increase in number of bound Ca2+ was observed. This indicates that the maximum Ca2+ binding capacity of the CAS of chain B and C is ~6 and ~8 Ca2+ ions respectively, and the CAS cannot account for more than 10% of the Ca2+ binding of each CASQ1 monomer (bind ~70–80 Ca2+). On the other hand, the surface of rest of the protein (residues 1–350) binds progressively more number of Ca2+ ions as the [Ca2+] was increased. From the slope of the Ca2+-binding curve (Figure 2B), it appears that the CASQ1 surface has not reached saturation even in the system with 120 Ca2+ ions, where each chain has bound ~55 Ca2+ ions. These findings, and the observation that CAS may hinder back-to-back stacking due to charge-charge repulsion, indicate that the CAS domain undergoes complete charge neutralization before rest of the CASQ1 surface gets saturated with Ca2+. Furthermore, these observations suggest that the affinity of different Ca2+-binding sites on CASQ1 must be different, in such a way to allow systematic neutralization of electronegative surface for increasing [Ca2+]. The negative potential neutralization process would then lead to the formation of globular CASQ1 conformation that is ready to undergo polymerization.
Figure 2. Number of bound Ca2+ in the presence of increasing [Ca2+].
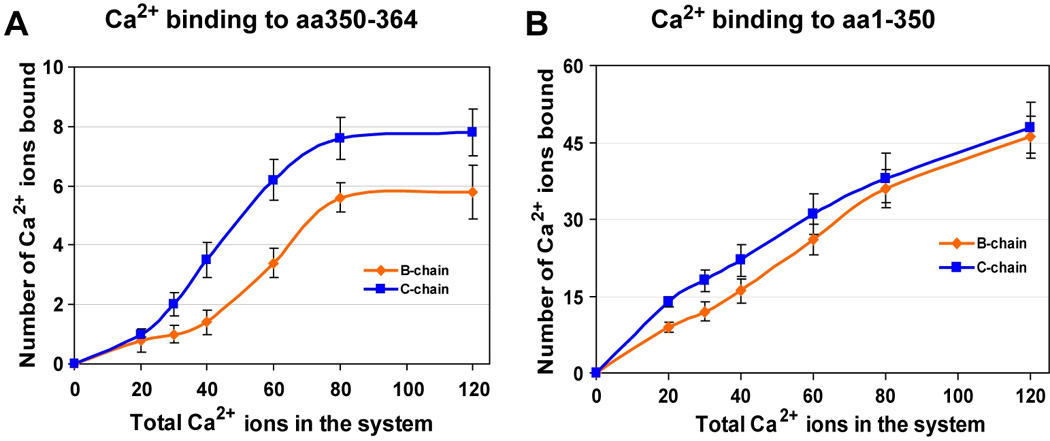
(A) Ca2+ bound to the Dn-motif (from amino acid 350 to 364 on the C-terminus) of each monomer. The Dn-motif at the dimeric interface binds less Ca2+ than the free Dn-motif. (B) Total number of Ca2+ ions bound to the rest of the protein (amino acids 1–350) surface of each monomer. Both the monomers bind almost similar number of Ca2+ ions although at intermediate [Ca2+] the monomer “chain B” binds more Ca2+ ions than “chain C”. Observing the slope of the curves it seems that both the monomers still possess the ability to bind even a higher number of Ca2+ ions, if provided.
[Ca2+] independent high affinity Ca2+-binding sites
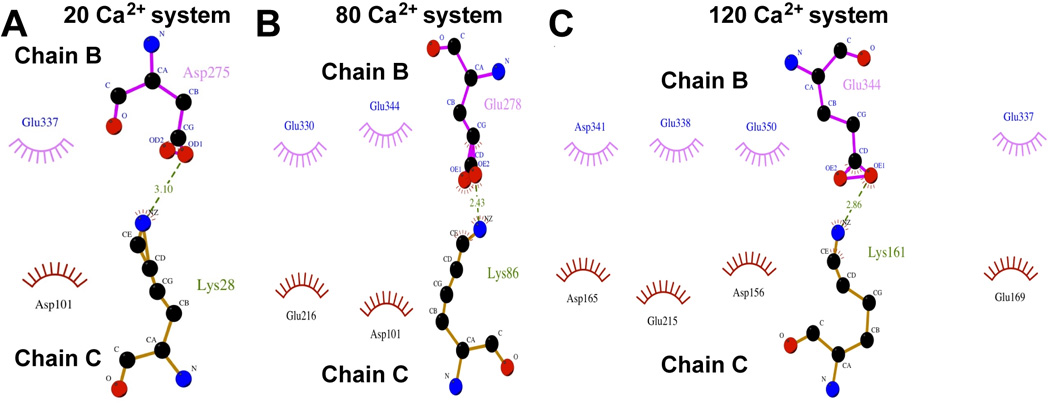
The recently solved crystal structure (PDB Id: 3UOM) provided information of about only 15 Ca2+-binding sites12, while our simulated data shows that each monomer (chain) binds about 55 Ca2+ in the system with 120 Ca2+ (Figure 2), indicating the existence of more Ca2+-binding sites. Therefore, we performed detailed investigation of the CASQ1 structures in the systems exposed to different [Ca2+], to identify and characterize other Ca2+ binding sites, in particular those not revealed by the crytallographic structure. Interestingly, the Ca2+-binding sites identified by the crystallographic study were also confirmed by MD simulation. The Ca2+-binding strength was analysed for a set of Ca2+-binding sites by determining the non-bonded (Van der Waals + Electrostatic) energy of interaction between the residues of the site and the Ca2+ ion. Based on the Ca2+-binding strength (summarized in Table 1) we further refined the classification of both high and low affinity sites into independent or dependent of [Ca2+]. The high affinity sites (site 1, 2, 3 and 4) are strictly independent of [Ca2+] and bind Ca2+ with high affinity as indicated by the high interaction energy (<−400 kcal/mol) in all the protein systems. We further found that the [Ca2+]-independent high affinity sites bind Ca2+ in a very favorable geometry in most of the systems at different [Ca2+] (Figure 3). However, only 13 [Ca2+]-independent high affinity sites on CASQ1 were found.
Table 1.
| Number of Ca2+ in the system |
Concentration independent high affinity sites (kcal/mol) |
Concentration dependent high affinity sites (kcal/mol) |
Concentration dependent intermediate affinity sites (kcal/mol) |
Concentration independent low affinity sites (kcal/mol) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(E39, E54 and D93) |
2(E102 and E169) |
3(E194 and D196) |
4(D356 and D357) |
5(E39, P40 and E42) |
6(D259 and D261) |
7(D328, E331 and D332) |
8(D306 and S308) |
9(E326 and E158) |
10(D319, E350 and E354) |
11(E66) | 12(E199 and T229) |
|
| 120 | −495 | −458 | −371 | −506 | −481 | −389 | −905 | X | −279 | −313 | X | −256 |
| 80 | −560 | −533 | −386 | −590 | −355 | −291 | −785 | −221 | −274 | −818 | −198 | X |
| 40 | −471 | −483 | −375 | −370 | −158 | −214 | −312 | −230 | −545 | −599 | X | −207 |
| 20 | −479 | −551 | −393 | −530 | X | X | X | X | −480 | X | −195 | −276 |
Figure 3. Identification of high affinity Ca2+-binding sites independent of [Ca2+].

Site 1 and site 3 are representative examples of high affinity Ca2+ binding sites on CASQ1 that are independent of [Ca2+]. Site 1 (A-B) and site 3 (C-D) bind Ca2+ with very similar geometry and affinity (see Table 1). System with 20 Ca2+ ions is referred to as “Low [Ca2+]”, whereas System with 80 Ca2+ ions is referred to as “High [Ca2+]”. The protein residues are represented in ball-stick representation, Ca2+ ion in magneta and water molecules that are present within 3.0 Å of Ca2+ ion in light blue.
Existence of [Ca2+]-induced Ca2+-binding sites
One of the most interesting findings of this study is that some Ca2+-binding sites are solely dependent on [Ca2+]. We found that some sites, for instance site 5, 6 and 7, bind Ca2+ with increasingly high affinity, reaching the highest value (correspoding to energy of <−400 kcal/mol) in the system with 120 Ca2+ ions (Table 1). These sites are referred as “[Ca2+]-dependent high affinity sites” and do not bind Ca2+ when the system has only 20 Ca2+ ions. Their interaction energy gradually increases as [Ca2+] increases, reaching a maximum value (<−400 kcal/mol) for the system containing 120 Ca2+ ions. Moreover, sites 5, 6 and 7 show a more favorable geometry of Ca2+-binding when [Ca2+] increases, demonstrating that these sites are induced upon Ca2+-binding (Figure 4). Our data suggest that the [Ca2+]-independent high affinity sites gets occupied first and then the binding events initiates structural alterations, bringing the residues involved in the [Ca2+]-dependent high affinity sites in a favorable geometry for subsequent Ca2+-binding.
Figure 4. Identification of [Ca2+] dependent high affinity Ca2+-binding sites.

Representative examples of Ca2+ binding sites on CASQ1 that are induced by an increase in [Ca2+] in the system. Site 5 (A-B) and site 7 (C-D) show progressively a more favorable Ca2+-binding geometry and better binding affinity increasing Ca2+ in the system (see Table 1).
Interestingly, some inducible sites (site 8, 9 and 10) were observed to bind Ca2+ with high affinity only at intermediate [Ca2+] (Table 1), thus we refer to them as “[Ca2+]-dependent intermediate affinity sites”. The geometry of site 10 in systems with different [Ca2+] is shown in Figure 5. As can be seen in Figure 5, the residues in site 10 are not in a proper orientation to support Ca2+ recognition in 20 Ca2+ ions. system. Thereafter, up to the system having 80 Ca2+, these residues gradually reorient to a correct geometry for ion Ca2+-binding, with all three acidic residues involved in bidentate interaction with the Ca2+ ion. However, further increase in [Ca2+] in the system disrupted the optimal geometry of the site 10, thus leading to a sharp decline of the interaction energy (Figure 5 and Table 1). Distance probability distributions also showed that in the system having 80 Ca2+ ions the distance between the residues and Ca2+ is the least value (Supplemental Figure 1). As a whole, we have found 25 inducible Ca2+-binding sites in CASQ1. Additionally, we also found that CASQ1 has many low affinity sites (for instance site 11 and 12) that are formed by either an acidic residue (Asp or Glu) alone or in combination with any other supporting residue (Supplemental Figure 1D). These sites bind Ca2+ with low affinity, with an interaction energy higher than −250 kcal/mol independent of [Ca2+] in the system.
Figure 5. [Ca2+] dependent intermediate affinity Ca2+ binding sites on CASQ1.

Some Ca2+ binding sites on CASQ1, like site 10, are induced to bind Ca2+ with high affinity only at intermediate [Ca2+]. (A) Site 10 is constituted by residues D319, E350 and E354, that show no sensitivity to Ca2+ when only 20 Ca2+ ions are present in the system. (B) Site 10 binds Ca2+ when 40 Ca2+ ions are present in the system. (C) Site 10 recognizes Ca2+ and binds with very high affinity and an optimal geometry when [Ca2+] increases to 80 ions. (D) With further increase in [Ca2+] to 120 ions, we note disruption of the optimal binding topology of site 10 and thus the interaction energy drastically reduces.
[Ca2+]-mediated dimer stabilization by hydrophobic interactions
We investigated the structure of the back-to-back dimeric interface in all the protein systems. Importantly, we found only one hydrogen bond at the interface, while interestingly the number of hydrophobic interactions increased with the rise in [Ca2+] (Figure 6). Then, on increasing the number of Ca2+ ions, we observed a gradual increase of hydrophobic interactions, and maximum of four hydrophobic interactions were found in the system with 120 Ca2+ ions. However, we did not observe any committed residue-to-residue contact, rather a set of interfacial residues that promiscuously established hydrophobic contacts as [Ca2+] increases. Nevertheless, all the hydrophobic interactions were found between acidic (glutamic and aspartic) residues. This suggests that the Ca2+-binding mediated charge neutralization of these interfacial acidic residues provide them the ability to establish hydrophobic interactions. We also observed that the number of bound water molecules in the interfacial region and to the CAS of chain B is unchanged in all the protein systems indicating that water plays only a minor role in back-to-back stacking. On the other hand, the free CAS of chain C binds more water with increasing [Ca2+] in the system (Supplemental Figure 2).
Figure 6. Characterization of the inter-molecular interaction at the back-to-back dimeric interface of CASQ1.
(A-C) Back-to-back stacking is stabilized by promiscuous hydrophobic interactions that increase with the increase in [Ca2+]. Hydrogen bonding (shown in green dotted lines, with distance in Å) does not play a major role in the back-to-back interaction.
Ca2+-induced CAS folding
Next, we investigated the alteration in structure of CAS upon Ca2+-binding throughout the MD trajectory in all the systems. One of the most exciting finding of this study is that the CAS acquires a compact 3-dimensional structure upon Ca2+-binding. Analysis of MD simulation trajectory suggest that CAS domain can exist in three distinct conformations; linear, intermediate and compact, as shown in Figure 7. The linear conformation of CAS forms 2–3 hydrogen bonds, the intermediate conformation 4–5 hydrogen bonds, and the compact conformation is stabilized with 6–8 hydrogen bonds. Then, we calculated the energy of the different CAS conformational populations, which is pertinent with the number of hydrogen bonds. Furthermore, we calculated the percentage of occurrence of the different CAS conformational populations at various [Ca2+]. At [Ca2+]=0, CAS domain remains primarily in the linear conformation. We noted that the ratio of compact to intermediate CAS conformations increased gradually with [Ca2+] increase in the system. At highest investigated [Ca2+] (120 Ca2+ ions), the compact CAS conformation definitely predominates (see Supplemental movie). This finding suggests that the free unstructured CAS can undergo reorganization in a Ca2+ dependent manner to form a 3-dimensional structure and might function as a novel structural motif for sensing metal ions. In an earlier study, Evans et al using dihedral angle based Monte Carlo calculation under solvation condition, suggested that the C-terminal tail of CASQ1 may acquire a supercoiled conformation 19. Based on these observations, we propose the term “Dn-motif” for the CAS and suggest that the Dn-motif might play a key role either in CASQ1 polymerization-depolymerization kinetics and/or interaction with other partner proteins of the RyR1 complex in a [Ca2+]-dependent manner.
Figure 7. The Dn-motif of CASQ1 assumes compact conformation upon binding of Ca2+ ions.
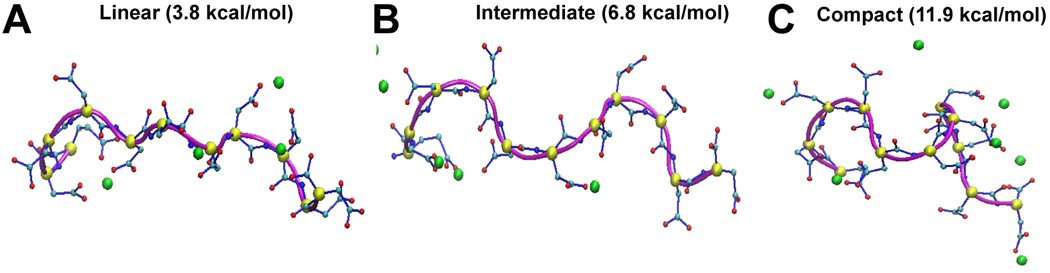
(A-C) The CAS or the Dn-motif is observed to exist in three main conformations. The linear conformation of CAS is characterized by the presence of the least number of Ca2+-bound as well as by the energetically least stable state. Additional Ca2+-binding leads to to an intermediate conformer, which was found to be energetically more stable. Finally, the Dn-motif saturation by Ca2+ resulted in the most energetically stable and a very compact structure.
Biological Implication
Although the alteration in the currents generated by coordinated flow of metal atoms is known to cause many diseases, including muscle dysfunction, present knowledge about structural recognition of metal concentrations by regulatory proteins is extremely limited20–23. CASQ1 proteins belong to a family of nonconventional metal-interacting proteins, expressed only in striated muscles17, 24–26. CASQ1 possess two very unique structural properties; 1) Ca2+ selectivity, despite lacking known Ca2+-binding motifs, 2) presence of C-terminus composed solely of aspartic acid residues (more than 10). The present study provide structural information towards better understanding of both these properties.
Biophysical studies have shown that CASQ1 binds a large number of Ca2+ (≥70 ions/monomer)11. However, the structural details of Ca2+-CASQ1 interaction at different ionic concentrations are missing. The detailed analysis presented here revealed the uniqueness of binding geometry of the Ca2+-CASQ1 interactions, and provides a firsthand insight into the overall quantitative as well as qualitative features of the interaction. The presence of Ca2+-binding sites of different affinities enable CASQ1 to buffer Ca2+ ions under a wide range of [Ca2+] (Figure 8). The CAS domain is neutralized before the complete saturation of the rest of CASQ1 protein, suggest that CASQ1 polymerization might occur at a low [Ca2+] that is needed just to neutralise the CAS located at the interface of back-to-back dimer. The rest of CASQ1 protein contains many Ca2+-binding sites of different affinities, suggesting that the CASQ1 polymer can sequester and deliver Ca2+ ions without need to depolymerize, under the normal physiological range of [Ca2+] oscillation in the skeletal muscles. Further, our data showed that promiscuous interfacial hydrophobic interactions stabilize the back-to-back dimer in a [Ca2+] dependent manner, which suggests that the back-to-back stacking is not extremely stable. Such a labile, but energetically stable back-to-back stacking could support the dynamic depolymerization of CASQ1 if required at extreme Ca2+ demand (Figure 8). Dynamic depolymerization of CASQ2 isoform has been suggested recently27–28, and such an interpretation is also experimentally supported by the recent dynamic measurement of the calcium buffering properties of the sarcoplasmic reticulum in the mouse skeletal muscle by Manno et. al29. Our current study shows that MD simulations of aqueous solution of metals (at various concentrations) can be successfully applied to gain a structural and functional insight into protein-metal interaction of metalloproteins.
Figure 8. Proposed model for Ca2+-CASQ1 interaction and polymerization.
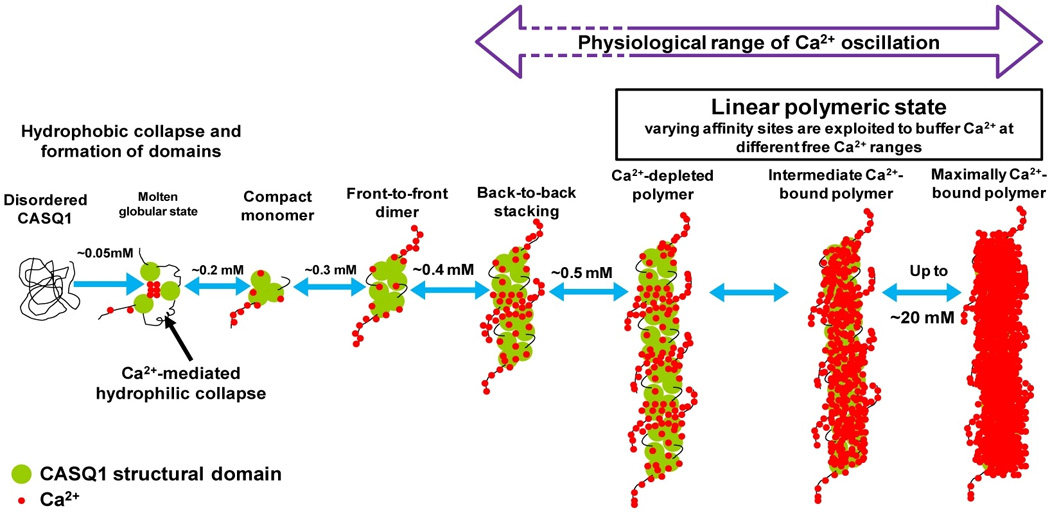
The centre of each domain of the CASQ1 molecule is hydrophobic and must undergo hydrophobic collapse to form the domains. The core region of CASQ1 at the interface, between the three domains is acidic, which is characteristic feature of the CASQ-protein family. The inter-domain charge neutralization by Ca2+ is necessary for the three domains to come to close proximity to form a compact monomer. The Dn-motif of CASQ1 is composed of 13 aspartic acids that can not bind more than 8 Ca2+ ions, whereas CASQ1 monomer has capacity to bind 70–80 Ca2+ ions. Therefore, the CASQ1 structural domains can bind more than 50 Ca2+. Existence of sites that can switch to an high affinity sites at varying [Ca2+] allows CASQ1 to release Ca2+ without the necessity of undergoing depolymerization. This observation holds in most part of the “physiological range of Ca2+ variations”. Dotted lines indicate the extreme Ca2+ concentrations that might be important only during higher physiological Calcium demand.
We would like to emphasize that the presence of more than 10 consecutive aspartic acid residues (Dn-motif) in a protein sequence is a very rare feature that is found only in about 20 proteins in the human genome, out of more than 30,000 predicted proteins. An evident common property shared among these proteins is metal binding capability, which indicates that the Dn-motif might be important as metal concentration sensors or for the metal sensing and binding, and to best of our knowledge, has not been investigated so far. For instance, these 20 proteins include; zinc finger protein castor homolog 1 (genebank accession id: NP_001073312), Histidine-rich calcium-binding protein (AAI12356), ring finger protein 34 (EAW98263), E3 ubiquitin-protein ligase RNF34 isoform 3 (NP_001243787), and asporin (CAI16698). Searches in PDB indicate that the structure of Dn-motif has also not been determined through experimental methods for any protein and functional role of Dn-motif in metalloproteins is currently unknown. Our result showed that Dn-motif of CASQ1 undergoes folding at increasing [Ca2+], which suggests that this motif might have a metal-sensing function.
Materials and method
CASQ1 Model Building with CAS motif
The recent crystal structure of the hexameric model of skeletal calsequestrin (PDB Id: 3UOM30), was adopted as a base model for our MD studies. The protein preparation wizard of the Schrodinger suite was used to optimize the model structure for it chemical correctness.31 In general, before simulations, the crystal structures requires fixing the common issues related to the experimental adopted methodology, that is missing hydrogen atoms, missing side chains, bond order assignments, charge states, and conformational orientations of symmetrical groups. During the structure refinement the orientation of hydroxyl (or thiol) groups, the terminal amide groups in asparagine (Asn) and glutamine (Gln), and the ring of histidine (His), those not already determined in the X-ray structure, were corrected. Flipping the terminal amide groups and the histidine ring can improve charge-charge interactions with neighboring groups as well as improving hydrogen bonding. The 180° flips preserve the heavy-atom placement deduced from the X-ray electron density. In addition, the protonation state of histidine is varied to optimize hydrogen bonding and charge interactions. The optimization was done using exhaustive sampling, which allowed increasing the number of iteration to an adequate level. The protein was further minimized using protein preparation wizard using OPLS2005 force field with the maximum convergence of 0.3 Å of heavy atoms. As shown in Supplemental Figure 3A, the CASQ1 monomers (labeled A, B, C, D, E and F) are arranged in a repetitive manner. AB, CD and EF are the front-to-front dimers and back-to-back interactions occur between B and C chains. In this study we are interested to understand the dynamics of back-to-back stacking and C-terminus-to-Ca2+ interaction, thus we chose to consider chains B and C only. The missing C-terminal residues were added using prime and builder module of Schrodinger to the chain B and C, without disturbing the structure of the remaining regions of the chains.32 After C-terminus addition, the residues were corrected by extended serial loop sampling method, using loop refinement module of Prime (Supplemental Figure 3B).33 The dimeric structure with the CAS region was subjected to global minimization through the Macromodel34 module of the Schrodinger suite using the OPLS 2005 force field with a GB/SA continuum water solvation model. This was followed by a Polak-Ribiere Conjugate Gradient energy minimization stopped after 5000 steps or when the energy difference between two subsequent structures was inferior to 0.05 kJ/mol. Finally, varying number of Ca2+ ions were added randomly to the system to build up the final CASQ1-Ca2+ complexes using System Building Module of the Desmond suite to perform the following MD simulations.35
Molecular Dynamics Simulations
The parameters for the protein and ions were assigned using CHARMM27 force-field (C27-FF) parameters36. The protein-protein complex with different concentration of Ca2+ ions were alternatively inserted in water box, and additional counter-ions were added to neutralize the system, using the modules in VMD software37. We used the TIP3P parameters for water molecules38. Standard protonation states were assigned to all residues using the propKa software39. Each of the resulting molecular system was energy minimized and slowly heated to 300 K in steps of 30 K, with positional restraints of 50 kcal/(mol Å2) on C-alpha atoms for a simulation time of 0.2 ns. The positional restraints on the C-alpha atoms were then slowly released in steps of 10 kcal/(mol Å2) and after 0.3 ns of simulation all positional restraints on the C-alpha atoms were completely released. Then, equilibration of the molecular system was done for a simulation time of 3 ns. Subsequently, all production runs of 30 ns simulations were performed at 300 K and 1 atm pressure (NPT ensemble), using periodic boundary conditions. The initial dimension of the simulation box edges were [138, 110, 95] Å, for a total system of ~130.000 atoms. All bonds involving hydrogen atoms were constrained using SHAKE40, which allowed using an integration time step of 2 fs. The Long-range electrostatic interactions were evaluated using particle mesh Ewald with a [148 128 96] Å grid dimension41. We used a 12 Å cut-off radius for both Van der Waals and electrostatic interactions along with smooth particle mesh Ewald41. We used the NAMD software package42 to perform all-atom molecular dynamics (MD) simulations on 96 processors cluster.
Structure Analysis and Energy Calculations
To monitor the stability of protein structure during MD simulations, we calculated the root mean square deviation (RMSD) on heavy protein atoms using VMD software. The configurational entropy for CAS domain during MD simulations was calculated from covariance matrices of the heavy atom fluctuations at 200 ps time step, using CARMA software43–45. The interaction energy for the Ca2+ ion in the many sites we identified was done by evaluating non-bonded interactions (that is, Van der Waals and electrostatics) between the Ca2+ and the protein residues in the identified sites, using a cutoff of 12.0 Å. For the electrostatic interactions, we adopted the same scheme adopted for the whole MD simulations. The total number of Ca2+ ions around the CAS residues (residue id 350–364) and the rest of the protein (residue id 1–350) for the chains B, C was monitored using distance cutoff of 5.0 Å, at every 20 ps time step of each MD trajectory. Similarly, the number of water molecules around the CAS residues and the rest of the protein for the chains B and C was calculated using distance cutoff of 3.0 Å. The H-bond and hydrophobic interactions between the interface residues belonging to chain B, C were calculated using appropriate VMD scripts on average structure of the protein calculated at intervals of 2 ns. The two dimensional schematic representation for the interaction network were finally generated by using the software LIGPLOT46.
Supplementary Material
Acknowledgements
AS acknowledges for modelling software support from Department of Biotechnology (DBT), Delhi, through grant no BT/PR14237/MED/29/196/2010. AK thanks the computational facility at CRS4 (Polaris), Pula, Italy. HC supported by institute research fellowship from Birla Institute of Technology, Mesra. TB thanks CSIR, Delhi for providing Senior Research Fellowship. N.C.B. was supported by postdoctoral fellowship from the American Heart Association. This work was supported in part, by National Institutes of Health Grant R01 HL64014 to M. P.
References
- 1.Beard NA, Laver DR, Dulhunty AF. Prog Biophys Mol Biol. 2004;85:33–69. doi: 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Caudwell B, Antoniw JF, Cohen P. Eur J Biochem. 1978;86:511–518. doi: 10.1111/j.1432-1033.1978.tb12334.x. [DOI] [PubMed] [Google Scholar]
- 3.Maurer A, Tanaka M, Ozawa T, Fleischer S. Proc Natl Acad Sci U S A. 1985;82:4036–4040. doi: 10.1073/pnas.82.12.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cala SE, Jones LR. J Biol Chem. 1983;258:11932–11936. [PubMed] [Google Scholar]
- 5.Franzini-Armstrong C, Kenney LJ, Varriano-Marston E. J Cell Biol. 1987;105:49–56. doi: 10.1083/jcb.105.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Nat Struct Biol. 1998;5:476–483. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- 7.Park H, Wu S, Dunker AK, Kang C. J Biol Chem. 2003;278:16176–16182. doi: 10.1074/jbc.M300120200. [DOI] [PubMed] [Google Scholar]
- 8.Cozens B, Reithmeier RA. J Biol Chem. 1984;259:6248–6252. [PubMed] [Google Scholar]
- 9.Aaron BM, Oikawa K, Reithmeier RA, Sykes BD. J Biol Chem. 1984;259:11876–11881. [PubMed] [Google Scholar]
- 10.Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- 11.Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C. J Biol Chem. 2004;279:18026–18033. doi: 10.1074/jbc.M311553200. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez EJ, Lewis KM, Danna BR, Kang C. J Biol Chem. 2012 doi: 10.1074/jbc.M111.335075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fliegel L, Ohnishi M, Carpenter MR, Khanna VK, Reithmeier RA, MacLennan DH. Proc Natl Acad Sci U S A. 1987;84:1167–1171. doi: 10.1073/pnas.84.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarain-Herzberg A, Fliegel L, MacLennan DH. J Biol Chem. 1988;263:4807–4812. [PubMed] [Google Scholar]
- 15.Mishra A, Suman SK, Srivastava SS, Sankaranarayanan R, Sharma Y. J Mol Biol. 2012;415:75–91. doi: 10.1016/j.jmb.2011.10.037. [DOI] [PubMed] [Google Scholar]
- 16.Kim E, Youn B, Kemper L, Campbell C, Milting H, Varsanyi M, Kang C. J Mol Biol. 2007;373:1047–1057. doi: 10.1016/j.jmb.2007.08.055. [DOI] [PubMed] [Google Scholar]
- 17.Gaburjakova M, Bal NC, Gaburjakova J, Periasamy M. Cell Mol Life Sci. 2012 doi: 10.1007/s00018-012-1199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bal NC, Kumar A, Chakravarty A, Balaraju T, Bal C, Jena N, Sharon A, Periasamy M. Biophysical Journal. 2013;104:173a. [Google Scholar]
- 19.Evans JS, Chan SI, Goddard WA., 3rd Protein Sci. 1995;4:2019–2031. doi: 10.1002/pro.5560041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priori SG, Chen SR. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg JM. Cold Spring Harb Symp Quant Biol. 1987;52:579–585. doi: 10.1101/sqb.1987.052.01.066. [DOI] [PubMed] [Google Scholar]
- 22.Ozawa T. Mol Med Rep. 2010;3:199–204. doi: 10.3892/mmr_00000240. [DOI] [PubMed] [Google Scholar]
- 23.Dulhunty AF, Beard NA, Hanna AD. J Gen Physiol. 2012;140:87–92. doi: 10.1085/jgp.201210862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikemoto N, Nagy B, Bhatnagar GM, Gergely J. J Biol Chem. 1974;249:2357–2365. [PubMed] [Google Scholar]
- 25.Bal NC, Jena N, Sopariwala D, Balaraju T, Shaikh S, Bal C, Sharon A, Gyorke S, Periasamy M. Biochem J. 2011;435:391–399. doi: 10.1042/BJ20101771. [DOI] [PubMed] [Google Scholar]
- 26.Scriven DR, Asghari P, Moore ED. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt025. [DOI] [PubMed] [Google Scholar]
- 27.Bal NC, Sharon A, Gupta SC, Jena N, Shaikh S, Gyorke S, Periasamy M. J Biol Chem. 2010;285:17188–17196. doi: 10.1074/jbc.M109.096354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KW, Maeng JS, Choi JY, Lee YR, Hwang CY, Park SS, Park HK, Chung BH, Lee SG, Kim YS, Jeon H, Eom SH, Kang C, Kim do H, Kwon KS. J Biol Chem. 2012;287:1679–1687. doi: 10.1074/jbc.M111.254045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manno C, Sztretye M, Figueroa L, Allen PD, Rios E. J Physiol. 2012 doi: 10.1113/jphysiol.2012.243444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez EJ, Lewis KM, Danna BR, Kang C. J Biol Chem. 2012;287:11592–11601. doi: 10.1074/jbc.M111.335075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park KW, Goo JH, Chung HS, Kim H, Kim DH, Park WJ. Gene. 1998;217:25–30. doi: 10.1016/s0378-1119(98)00372-2. [DOI] [PubMed] [Google Scholar]
- 32.Arai M, Alpert NR, Periasamy M. Gene. 1991;109:275–279. doi: 10.1016/0378-1119(91)90621-h. [DOI] [PubMed] [Google Scholar]
- 33.Fliegel L, Leberer E, Green NM, MacLennan DH. FEBS Lett. 1989;242:297–300. doi: 10.1016/0014-5793(89)80488-0. [DOI] [PubMed] [Google Scholar]
- 34.Damiani E, Volpe P, Margreth A. J Muscle Res Cell Motil. 1990;11:522–530. doi: 10.1007/BF01745219. [DOI] [PubMed] [Google Scholar]
- 35.Biral D, Volpe P, Damiani E, Margreth A. FEBS Lett. 1992;299:175–178. doi: 10.1016/0014-5793(92)80241-8. [DOI] [PubMed] [Google Scholar]
- 36.MacKerell AD, Jr, Banavali N, Foloppe N. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 37.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 39.Li H, Robertson AD, Jensen JH. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 40.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 41.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 42.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andricioaei I, Karplus M. J Chem Phys. 2001;115:6289–6292. [Google Scholar]
- 44.Glykos NM. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 45.Balaraju T, Kumar A, Bal C, Chattopadhyay D, Jena N, Bal NC, Sharon A. Structural Chemistry. 2012 In Press. [Google Scholar]
- 46.Wallace AC, Laskowski RA, Thornton JM. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.