

Abstract
c-Src and Lyn are the only Src family kinases (SFKs) with established activity in osteoclasts (OCs). c-Src promotes function via cytoskeletal organization of the mature resorptive cell while Lyn is a negative regulator of osteoclastogenesis. We establish that Fyn, another SFK, also impacts the OC, but in a manner distinctly different than c-Src and Lyn. Fyn deficiency principally alters cells throughout the osteoclastogenic process, resulting in diminished numbers of resorptive polykaryons. Arrested OC formation in the face of insufficient Fyn reflects reduced proliferation of precursors, in response to M-CSF and retarded RANK ligand (RANKL)-induced differentiation, attended by suppressed activation of the osteoclastogenic signaling molecules, c-Jun and NF-κB. The anti-apoptotic properties of RANKL are also compromised in cells deleted of Fyn, an event mediated by increased Bim expression and failed activation of Akt. The defective osteoclastogenesis of Fyn-/- OCs dampens bone resorption, in vitro. Finally, while Fyn deficiency does not regulate basal osteoclastogenesis, in vivo, it reduces that stimulated by RANKL by approximately 2/3. Thus, Fyn is a pro-resorptive SFK, which exerts its effects by prompting proliferation and differentiation while attenuating apoptosis of OC lineage cells.
Keywords: Osteoclasts, Fyn, Src family kinase (SFK), M-CSF, RANK ligand
OCs are multinucleated, hematopoietic cells with the unique capacity to degrade bone. They are generated under the aegis of M-CSF and RANKL, the latter binding to its receptor, RANK, on OC precursors [Teitelbaum, 2007]. Occupied RANK, in turn, recruits TRAF6 followed by activation of NF-κB and AP-1transcription factors [Karin et al., 2002; Tanaka et al., 2005; Teitelbaum and Ross, 2003]. Once differentiated, OCs undergo rapid apoptosis but their survival is regulated by hormones and cytokines, including M-CSF and RANKL. For example, M-CSF withdrawal enhances expression of the Bcl-2 family member, Bim, which is proapoptotic in the OC [Akiyama et al., 2003]. RANKL, on the other hand, likely promotes OC survival by activating PI3K/Akt signaling [Wong et al., 1999].
SFKs regulate proliferation, differentiation, and cytoskeletal organization of virtually all cells [Lowell and Soriano, 1996]. In 1991, Soriano, et al, noted that c-Src-deficient mice develop severe osteopetrosis despite increased numbers of dysfunctional OCs [Soriano et al., 1991]. Until recently, c-Src was the only SFK established as operative in the OC [Soriano et al., 1991] but Lyn has proven to be a negative regulator of osteoclastogenesis [Kim et al., 2009].
Fyn, another SFK, is particularly important in T-cell receptor- and adhesion-mediated signaling, mast cell degranulation and microtubule formation [Beggs et al., 1997; Kojima et al., 1997; Manie et al., 1997; Martin-Cofreces et al., 2006; Nishida et al., 2005; Parravicini et al., 2002; Stein et al., 1992; Umemori et al., 1994]. We find that Fyn, which is expressed constitutively in OCs and their precursors, regulates the cell’s resorptive activity. Unlike c-Src, which exerts its bone-degrading effects exclusively by organizing the OC cytoskeleton [Boyce et al., 1992] or Lyn which negatively impacts OC recruitment [Kim et al., 2009], Fyn stimulates osteoclastogenesis by facilitating proliferation and differentiation of OC precursors and retarding apoptosis of those committed to the resorptive phenotype. These events appear related to RANKL-activation of NF-κB, c-Jun and Akt and to inhibited Bim expression. Because Fyn deficiency, in vivo, suppresses activated but not physiological bone resorption, its therapeutic blockade may inhibit osteoporosis while sparing normal skeletal remodeling.
MATERIALS AND METHODS
MICE
Fyn-deficient mice were obtained from Jackson Laboratories. All mice were 6-10 weeks old and maintained at the Animal Facility of Washington University School of Medicine. Experiments were approved by the Animal Ethics Committee of Washington University.
OSTEOCLAST CULTURES
Whole bone marrow cells were isolated from 6-10 week-old mice as described [Kim et al., 2006] and cultured in the presence of RANKL (100 ng/ml) and M-CSF (10 ng/ml) in α-MEM containing 10% FBS. Media were changed every 2 days. Osteoclasts were identified as TRAP expressing multinucleated giant cells. To assess actin ring formation, BMMs were cultured on bone with RANKL (100 ng/ml) and M-CSF (10 ng/ml). After 5 days, cells were fixed and stained with FITC-phalloidin. Alternatively, the same cells were washed with cytokine-free cold medium to dissociate actin rings. The cells were then incubated with RANKL for 1 and 3 hrs and the percentage of osteoclasts which had re-formed actin rings, determined [Kim et al., 2006].
RT-PCR
Total RNA (1μg) extracted from cultured cells was used as a template for cDNA synthesis. Primers were synthesized on the basis of the reported mouse cDNA sequence. The following primers were used: for TRAP, 5’-ACAGCCCCCCACTCCCACCCT-3’ and 3’-TCAGGGTCTGGGTCTCCTTGG-5’; for MMP-9, 5’-CCTGTGTGTTCCCGTTCATCT-3’ and 3’-CGCTGGAATGATCTAAGCCCA-5’; for cathepsin K, 5’-GGAAGAAGACTCACCAGAAGC-3’ and 3’-GCTATATAGCCGCCTCCACAG-5’; for CTR, 5′-CATTCCTGTTACTTGGTTGGC-3′ and 3′-AGCAATCGACAAGGAGTGAC; for GAPDH, 5’-ACTTTGTCAAGCTCATTTCC-3’ and 3’-TGCAGCGAACTTTATTGATG-5’. Amplification was conducted for 22-30 cycles, each of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. Ten microliters of each reaction mixture was analyzed by 1.5% agarose gel electrophoresis.
PROLIFERATION ASSAY AND CELL DEATH DETECTION ELISA
BMMs plated in 96-well plates were grown for 3 days and labeled with BrdU for the last 4 hrs of culture. The BrdU ELISA was performed using the cell proliferation Biotrak ELISA system (Amersham Biosciences) Cell death assay was performed using cell death detection ELISAPLUS kit (Roche Applied Science), which detects cytoplasmic histone-associated DNA fragmentation.
WESTERN BLOTTING AND ANTIBODIES
BMMs starved in serum-free α-MEM for 16 hrs or pre-OCs starved for 3 hrs were stimulated with RANKL (100 ng/ml) or M-CSF (50 ng/ml). Cells were washed with cold PBS and harvested with RIPA buffer containing protease and phosphatase inhibitors. Immunoblots were generated using monoclonal antibodies to phospho-JNK, phospho-Akt, and phospho-ERK (Cell Signaling) or polyclonal antibodies to JNK, Akt and ERK (Cell Signaling). Polyclonal antibody to phospho-p38 and monoclonal antibody to phospho-IκBα were obtained from Cell Signaling. Polyclonal antibody to Bim was from BD Pharmigen. Lyn mAb and phospho-Lyn polyclonal antibodies were purchased from Cell Signaling and Pierce respectively. mAbs to Fyn and phospho-SFK were purchased from Novus Biologicals and Cell Signaling, respectively. Total and phosphorylated Vav3 were analyzed as described [Faccio et al., 2005].
SERUM TRAP ASSAY
RANKL (100 μg per mouse per day) was injected subcutaneously daily for 7 days. One day after the final injection, serum was collected. Serum TRAP 5b was measured by ELISA (Immuno Diagnostic Systems).
STATISTICS
All data are presented as mean +/- S.D. Statistical significance was determined by 2-tailed Student’s t test.
RESULTS
FYN REGULATES OSTEOCLASTOGENESIS
c-Src is a non-receptor tyrosine kinase whose expression increases as BMMs commit to the OC phenotype, in vitro, whereas Lyn is constitutively expressed throughout osteoclastogenesis [Abu-Amer et al., 1997; Kim et al., 2009]. To determine Fyn expression, we cultured WT BMMs in M-CSF and RANKL and temporally immunoblotted equal amounts of lysate protein for Fyn and c-Src. Expectedly, c-Src is not detectable in BMMs but increases with osteoclast differentiation [Abu-Amer et al., 1997] (Fig. 1A). Fyn, like Lyn, is constitutively expressed by BMMs and its abundance is unaltered by exposure to osteoclastogenic cytokines. M-CSF and RANKL, however, both activate Fyn (Fig. 1B).
Figure 1.
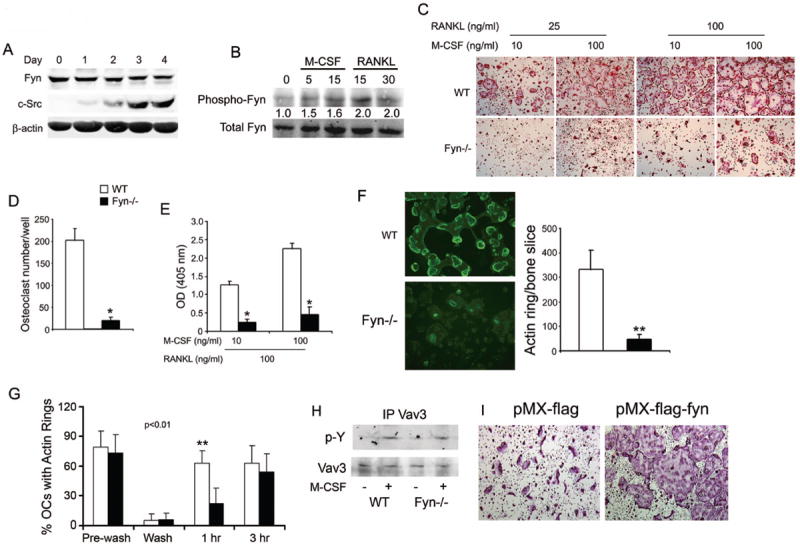
Fyn deficiency impairs osteoclast formation and spreading. (A) WT BMMs were cultured in M-CSF and RANKL. Lysates were collected daily for 4 days and immunoblot performed for Fyn and c-Src content. β-actin serves as loading control. B) WT BMMs plated in M-CSF were cytokine- and serum-starved overnight and then exposed to M-CSF or RANKL with time dictated by established signaling kinetics. Fyn immunoprecipitates were immunoblotted for total and phosphorylated Fyn. Blot intensity was densitometrically quantitated and the phospho-Fyn/total Fyn ratio normalized to time 0. (C-E) WT and Fyn-/- BMMs were cultured with M-CSF and RANKL. (C) Cells were stained for TRAP activity at day 5. (D) Statistical analysis of the number of WT and Fyn-/- TRAP-expressing multinucleated giant cells/well at day 5. (E) TRAP activity determined by ELISA at day 4. (F) WT and Fyn-/- BMMs were cultured on bone with M-CSF and RANKL. (left panel) After 5 days, the cells were fixed and stained with FITC-phalloidin to visualize actin rings. (right panel) Statistical analysis of the number of WT and Fyn-/- actin rings/bone slice at day 5. (G) Mature WT and Fyn-/- osteoclasts cultured on bone slices, were washed with cytokine-free cold medium to dissociate actin rings. The cells were then incubated with RANKL for 1 or 3 hrs and the percentage of osteoclasts, which had re-formed actin rings, determined. (H) WT and Fyn-/- BMMs were cytokine starved for 6 hrs prior to a 5 min exposure to M-CSF or carrier. Lysates were immunoprecipitated with anti-Vav3 mAb and the product immunoblotted for phosphotyrosine and Vav3 content. (I) Fyn-/- BMMs were retrovirally transfected with vectors bearing FLAG or FLAG- Fyn. OCs were generated by 5 days in RANKL and M-CSF. (*p< 0.01, **p< 0.005).
c-Src deficiency arrests the bone resorptive function of mature OCs but does not modify differentiation of their precursors [Boyce et al., 1992]. Alternatively, absence of Lyn increases OC number [Kim et al., 2009]. We asked if Fyn also impacts the bone-resorbing polykaryon by culturing WT and Fyn-/- BMMs in low- and high-dose M-CSF and/or RANKL for 5 days, after which the cells were stained for the OC marker, tartrate resistant acid phosphatase (TRAP). Regardless of cytokine dose, absence of Fyn inhibits osteoclastogenesis, in vitro, as manifest by decreased numbers of TRAP-expressing multinuclear mature OCs (Fig. 1C,D) and dampened TRAP expression (Fig. 1E). Similar to c-Src-deficient OCs [Lakkakorpi et al., 2001], those lacking Fyn are capable of forming actin rings on bone, although the total number of these structures per bone slice is reduced due to impaired osteoclastogenesis (Fig. 1F). The rate at which Fyn-deficient osteoclasts generate actin rings is, however, retarded (Fig. 1G). This transient delay in sealing zone formation does not likely reflect failed activation of canonical cytoskeleton-organizing molecules as M-CSF-induced phosphorylation of its distal effector, Vav3, is unaltered by absence of Fyn [Reeve et al., 2009; Zou et al., 2008](Fig. 1H). Confirming the atypical phenotype of Fyn-/- OCs reflects absence of the SFK, it is normalized by retroviral reconstitution of the WT protein (Fig. 1I). Furthermore, the pro-osteoclastogenic properties of Fyn deficiency cannot be explained by enhanced RANK expression or that of its key signaling molecule, TRAF6 (Fig. S1). On the other hand, absence of Fyn activates Lyn in M-CSF-, but not RANKL-exposed osteoclasts (Fig. 2).
Figure 2.
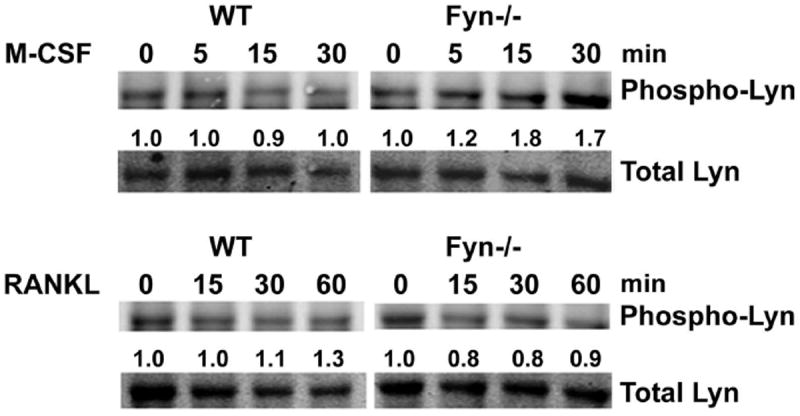
Fyn deficiency promotes M-CSF-stimulated Lyn phosphorylation. WT and Fyn-/- BMMs cultured in M-CSF were cytokine- and serum-starved overnight. The cells were exposed to M-CSF (top) or RANKL (bottom) with time dictated by established signaling kinetics. Lyn immunoprecipitates were immunoblotted for total and phosphorylated Lyn. Blot intensity was densitometrically quantitated and the phospho-Lyn/total Lyn ratio normalized to time 0.
FYN DEFICIENCY ARRESTS BONE RESORPTION IN VITRO
We assessed the impact of Fyn deficiency on skeletal resorption, in vitro, by generating WT and mutant OCs on cortical bone slices. Five days after initiation of RANKL and M-CSF exposure, resorption pits were visualized (Fig. 3A). Confirming suppressed resorption, collagen type 1 fragments (CTx) mobilized into Fyn-/- culture medium, are substantially less than WT (Fig. 3B). Thus, Fyn deficiency dampens bone degradation in vitro, likely representing diminished osteoclastogenesis.
Figure 3.
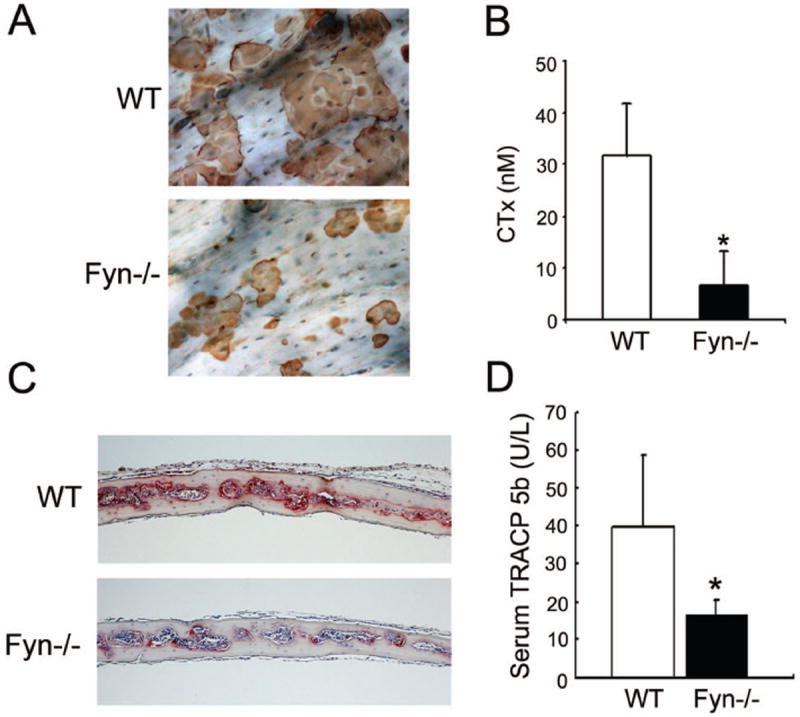
Absence of Fyn decreases bone resorption in vitro and in vivo. WT and Fyn-/- BMMs were cultured on bone with M-CSF and RANKL for 5 days. (A) Resorption lacuna formation was examined at day 5. (B) Resorptive activity was determined by collagen type I fragment (CTx) ELISA of culture media at day 5. (C, D) WT and Fyn-/- mice were injected, supracalvarially, with RANKL daily, for 7 days. (C) TRAP-stained (red reaction product) histological sections of calvariae. (D) Serum TRACP 5b levels at sacrifice were determined by ELISA. (*p< 0.05).
FYN ENHANCES RANKL-STIMULATED OSTEOCLAST DEVELOPMENT IN VIVO
Despite, evidence of increased bone formation in naïve Fyn-/- mice, as manifest by enhanced circulating osteocalcin (WT 27.4+/-2.31 vs Fyn-/- 70.0+/-12.6 ng/ml; p<.002), skeletal mass, following ovariectomy, is indistinguishable from WT (Fig. S2). On the other hand, OC generation, in vitro, probably represents an activated state [Novack et al., 2003] and therefore we asked if cytokine-stimulated osteoclastogenesis, in vivo, is impacted by Fyn deficiency. OC formation, in response to a 7 day course of daily supracalvarial RANKL injections, is blunted in Fyn-/- mice, as manifest by an approximate 2/3 reduction in circulating TRACP 5B relative to their WT counterparts (Fig. 3C, D). Thus, Fyn is not required for basal osteoclastogenesis but is a positive modulator of RANKL-stimulated OC development, in vivo.
FYN DEFICIENCY RETARDS PROLIFERATION AND DIFFERENTIATION AND ACCELERATES APOPTOSIS OF OC LINEAGE CELLS
OC number is dictated by the rate of precursor proliferation, differentiation and apoptosis of lineage cells. To assess proliferation of early OC precursors, we cultured Fyn-/- and WT BMMS, in increasing amounts of M-CSF, for 3 days and measured BrdU incorporation during the last 4 hrs. As expected, proliferation increases as a function of M-CSF, but at all concentrations of the cytokine, Fyn-/- BMMs incorporate less BrdU than WT (Fig. 4A). We next asked if the same holds in the context of progenitor cells committed to the OC phenotype. To this end, we cultured BMMs for 3 days in the same concentrations of M-CSF, but with100ng/ml RANKL added. Once again, proliferation increases as a function of M-CSF with significant differences between cells expressing Fyn and those lacking the SFK (Fig. 4B). Thus, Fyn deficiency blunts proliferation of early and committed OC precursors.
Figure 4.
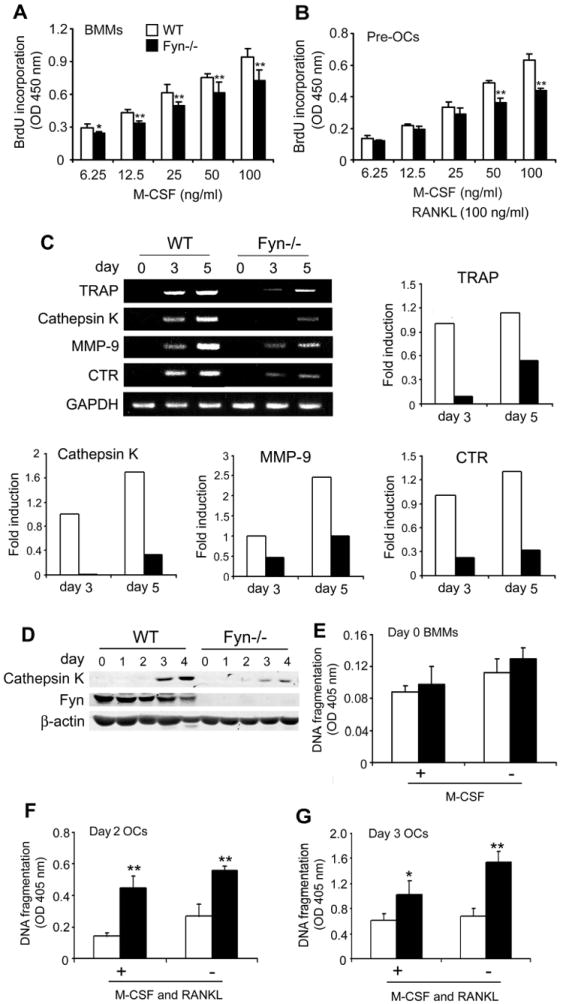
Fyn regulates proliferation, differentiation and survival of osteoclast lineage cells. Equal numbers of WT and Fyn-/- BMMs were cultured with the indicated concentrations of M-CSF (A) without or (B) with RANKL for 3 days. Cells were exposed to BrdU for the last 4 hrs and incorporation determined. (C) WT and Fyn-/- BMMs were exposed to M-CSF and RANKL, with time. Cathepsin K, MMP-9, and CTR mRNA was assessed by RT-PCR. GAPDH serves as loading control. The gels were quantitated densitometrically and fold induction of the various mRNAs on day 3 and 5 of osteoclastogenesis was expressed relative to day 3 WT cells. (D) Lysates of WT and Fyn-/- BMMs, maintained as above in M-CSF and RANKL, were immunoblotted daily for Cathepsin K and Fyn content. β-actin serves as loading control. (E) WT and Fyn-/- BMMs were incubated for 4 hrs in the presence (+) or absence (-) of M-CSF. WT and Fyn-/- BMMs were incubated with RANKL and M-CSF for (F) 2 or (G) 3 days. The cytokines were maintained (+) or withdrawn (-) during the last 4 hrs of culture. The magnitude of apoptosis was measured by ELISA. (* p < 0.05, ** p < 0.01).
We determined the impact of Fyn deletion on OC differentiation by measuring temporal gene expression of osteoclastogenic markers by mutant and WT BMMs in RANKL and M-CSF. Confirming arrested OC differentiation, mRNA levels of TRAP, Cathepsin K, MMP-9, and calcitonin receptor (CTR) are reduced in day 3 and day 5 Fyn-/- cells (Fig. 4C). Mirroring its mRNA, Cathepsin K protein is diminished in the absence of Fyn (Fig. 4D).
Turning to apoptosis, we measured DNA fragmentation by ELISA in cytokine-starved BMMs previously exposed only to M-CSF or M-CSF and RANKL. While absence of Fyn does not impact survival of naïve BMMs, in the presence or absence of M-CSF (Fig. 4E), Fyn-/- cells committed to the OC lineage by RANKL treatment for 2 or 3 days undergo accelerated apoptosis (Fig. 4F and G).
FYN DEFICIENCY IMPAIRS RANKL-INDUCED AKT, IκBα AND C-JUN PHOSPHORYLATION
The data presented suggest that Fyn deficiency dampens osteoclastogenesis by retarding proliferation and differentiation and promoting apoptosis of lineage cells. We, therefore, examined the role of the two key cytokines, M-CSF and RANKL, in Fyn-deficient OC suppression. M-CSF recruits OCs by promoting precursor proliferation and prolonging longevity. In keeping with our observation that it does not accelerate BMM apoptosis, Fyn deficiency fails to impact M-CSF activation of Akt, a principal mechanism by which the cytokine prevents cell death (Fig. 5A).
Figure 5.
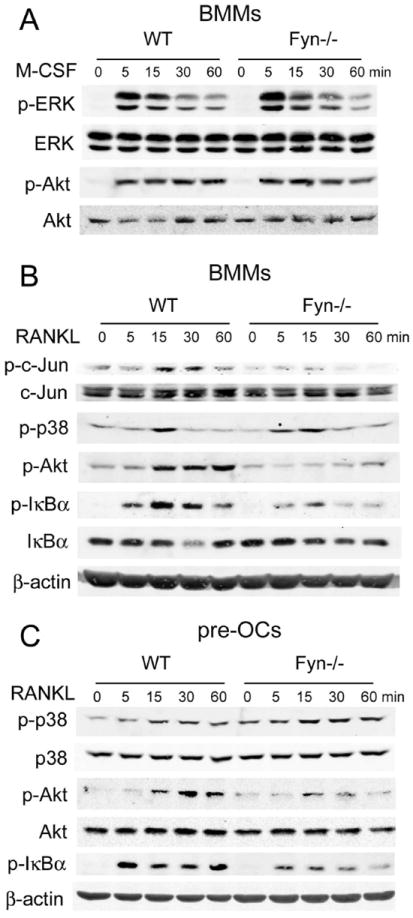
RANKL-induced Akt and IκBα phosphorylation is impaired in Fyn-/- osteoclast lineage cells. (A) Cytokine- and serum-starved WT and Fyn-/- BMMs were exposed to M-CSF with time and phosphorylation of Akt and ERK was determined by immunoblot. Total ERK and Akt levels serve as loading control. (B) WT and Fyn-/- BMMs or (C) those committed to the OC phenotype by 3 days of M-CSF and RANKL exposure, were serum- and cytokine-starved. They were then treated with RANKL with time and total and phosphorylated signaling molecules measured by immunoblot. β-actin serves as loading control.
Activation of MAP kinases, particularly ERKs, is a key event in the cytokine’s ability to stimulate BMM proliferation. Curiously, in spite of their blunted proliferative response to M-CSF, the cytokine does not alter ERK activation in these cells (Fig. 5A).
Our next efforts focused on RANKL. To this end, we serum- and cytokine-starved BMMs previously exposed only to M-CSF or committed to the OC linage by 3 days with M-CSF and RANKL (pre-fusion OCs). Both were then RANKL-treated for up to one hour and canonical RANK signaling assessed by immunoblot. Upon binding to RANK, RANKL activates JNK, which phosphorylates c-Jun. Activated c-Jun, in turn, is an essential component of the AP-1 osteoclastogenic transcription complex. In keeping with their arrested differentiation into OCs, RANKL-induced phosphorylation of c-Jun is reduced in Fyn-/- BMMs. In contrast, RANKL activation of p38, another pro-osteoclastogenic MAP kinase, is not impaired. (Fig. 5B,C).
Unlike their unaltered response to M-CSF, Fyn-/- osteoclastic cells fail to effectively phosphorylate Akt when exposed to RANKL (Fig. 5B,C). Similarly, NF-κB activation, a necessary event in OC differentiation, is retarded in BMMs or committed pre-fusion OCs exposed to the cytokine, as manifest by inhibited phosphorylation of IκBα (Fig. 5C). Thus, Fyn modulates RANK signaling, thereby affecting c-Jun, Akt and NF-κB and enhancing survival and differentiation of OC lineage cells.
FYN DEFICIENCY ENHANCES BIM EXPRESSION
OC programmed cell death, in other circumstances, is accompanied by abundant Bim, a pro-apoptotic BH3-only, Bcl-2 family member [Akiyama et al., 2003]. Hence, we asked if Fyn deficiency increases Bim in osteoclastic cells. Fyn-/- and WT pre-OCs were maintained in apoptosis-promoting, cytokine-free conditions. Consonant with induction of programmed cell death in OCs, Bim is increased in Fyn-/- cells at the time of RANKL/M-CSF withdrawal and after 3 hrs (Fig. 6). Alternatively, Bid, another anti-apoptotic Bcl family protein, which mediates other states of OC apoptosis [Vaira et al., 2008], is unchanged by Fyn deficiency.
Figure 6. Fyn suppresses BIM expression.
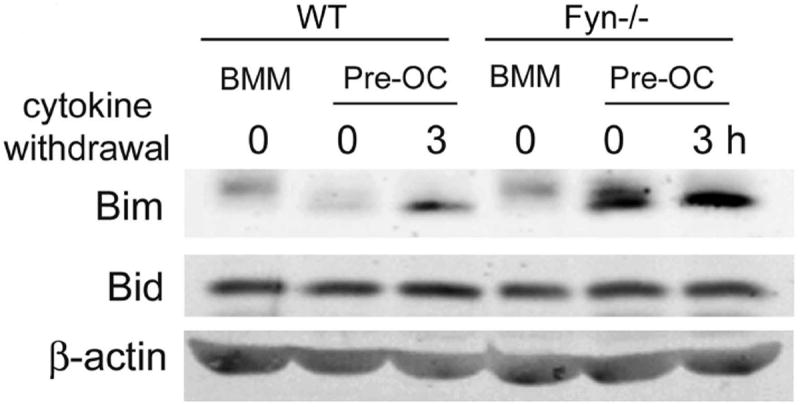
Lysates were obtained from naïve WT and FYN-/- BMMs or pre- fusion osteoclasts generated by 3 days of culture with RANKL and M-CSF and maintained in the presence or absence of cytokines for the last 3 hrs. Bim and Bid content was determined by immunoblot. β-actin serves as loading control.
DISCUSSION
The discovery in 1991 that c-Src-deficient mice are osteopetrotic heralded the importance of SFKs in the bone resorptive process [Soriano et al., 1991]. While c-Src remained the only family member with established function in OCs for virtually two decades, the role of Lyn was recently elucidated in the bone resorptive cell [Kim et al., 2009]. In the present study, we demonstrate a third SFK, Fyn, also regulates the OC but does so in a manner distinctly different than c-Src or Lyn. Specifically, c-Src directly encourages bone resorption by organizing the cytoskeleton of mature OCs, whereas Lyn exclusively suppresses osteoclastogenesis. Fyn, while modestly accelerating actin ring formation, primarily promotes osteoclast accumulation. Interestingly, Lyn phosphorylation is enhanced in Fyn-/- OC lineage cells raising the possibility its negative influence contributes to the compromised number of Fyn-deficient, TRAP-expressing polykaryons.
OC abundance reflects rates of proliferation, differentiation and apoptosis of lineage cells. The mature resorptive polykaryon is non-proliferative and terminally differentiated but its BMM precursors expand under the aegis of M-CSF interacting with its receptor, c-Fms. In keeping with osteoclast number, Fyn-/- BMMs and pre-OCs are less proliferative in response to M-CSF than are their WT counterparts. While the classical model holds that the Ras/MAP kinase pathway, particularly by activating ERK, is central to M-CSF-stimulated BMM proliferation [Takeshita et al., 2007] we find reduced propagation of Fyn-/- BMMs in response to M-CSF is unattended by changes phosphorylated ERK.
Like proliferation, commitment of Fyn-/- BMMs to the OC phenotype is retarded, as manifest by expression of OC-specific markers with time of osteoclastogenesis, which requires RANKL activation of NF-κB. Therefore, arrested differentiation is in keeping with blunted RANKL-induced NF-κB activity in Fyn-deficient pre-OCs and their non-committed precursors.
RANKL is anti-apoptotic in the context of the OC [Fuller et al., 1998; Jimi et al., 1999; Lacey et al., 2000], providing survival signaling through the PI3K/Akt cascade. In addition to its cytoskeleton-organizing properties, c-Src participates in RANKL-stimulated OC survival, in vitro, as its absence accelerates apoptosis, an event associated with failure to activate AKT [Wong et al., 1999]. However, c-Src deletion in the intact mouse increases, rather than decreases, OC abundance, questioning the biological relevance of the SFK’s in vitro anti-apoptotic properties.
Lack of Fyn also promotes OC apoptosis, which is in keeping with its in vivo phenotype of reduced osteoclastogenesis in response to RANKL. Why absence of Fyn, but not c-Src, diminishes OC number in vivo is unclear, but may relate to the unaltered presence of Fyn throughout the osteoclastogenic process. Fyn deficiency, for example, impairs proliferation of BMMs and blunts the pro-survival effects of RANKL after only two days exposure to the cytokine. In both circumstances, c-Src is minimally expressed. Thus, c-Src may protect the survival of only mature OCs whereas Fyn exerts its effects over a more prolonged time course, thereby impacting cells at multiple stages of differentiation.
M-CSF and RANKL are both pro-survival, but the accelerated apoptosis of Fyn-/- OCs reflects impaired response to the latter cytokine. This conclusion is based Fyn deficiency’s inability to alter longevity of BMMs exposed only to M-CSF. Furthermore, failure to activate Akt, in the mutant cells, which occurs in tandem with induction of apoptosis, appears only in response to RANKL. While controversial [Sugatani and Hruska, 2005], active Akt is likely a principal mediator of RANKL-stimulated OC survival [Lee and Kim, 2003; Wong et al., 1999]. On the other hand, Akt also promotes OC differentiation, perhaps by activating NF-κB [Sugatani and Hruska, 2005], which is compromised in the absence of Fyn. Thus, Akt appears central to the means by which Fyn promotes both OC differentiation and longevity.
Bim is a BH3-only Bcl-2 family member expressed in hematopoietic cells [O’Reilly et al., 2000] whose expression mediates OC apoptosis [Akiyama et al., 2003]. Fyn deficiency increases Bim upon M-CSF and RANKL withdrawal. Bim in Fyn-/- OCs is enhanced, despite the presence of M-CSF and RANKL, reflecting the fact that absence of Fyn compromises sensitivity of OCs to a survival-promoting cytokine. Consistent with its failure to impact apoptosis in BMMs, Fyn deficiency does not increase Bim upon exposure of these cells to M-CSF. Furthermore, ERK phosphorylation, which mediates Bim expression in response to M-CSF deprivation, is unaltered in Fyn-/- cells. These observations suggest that the accelerated apoptosis, extant in Fyn-deficient OCs, reflects resistance to RANKL and not M-CSF.
Despite its dramatic effect on OC number, in vitro, absence of Fyn does not influence the unstimulated skeleton. Because OCs generated, in vitro, from NF-κB-inducing kinase (NIK) [Novack et al., 2003] - or FHL2 [Bai et al., 2005] -deficient mice represent the activated, but not basal state of their in vivo counterparts, we asked if the same is true regarding absence of Fyn. As in the case of NIK and FHL2 deficiency, RANKL stimulation, in vivo, yields functional confirmation of the pro-osteoclastogenic properties of Fyn.
Fyn, therefore, joins c-Src and Lyn as functionally relevant SFKs in the OC. It is interesting that despite similarities in domain structure, these enzymes exert discrete effects on the bone resorptive polykaryon. Thus, c-Src is key to organizing the cell’s cytoskeleton whereas Lyn and Fyn respectively inhibit and stimulate osteoclastogenesis. These distinct properties provide insight into the means by which the three kinases, in concert, regulate bone degradation but also underscore the necessity of precise targeting of individual SFKs in design of potential anti-resorptive drugs.
Supplementary Material
Acknowledgments
This research was supported by Shriners’ Hospitals for Children Grant Number 8590 (SLT); National Institutes of Health Grant Numbers AR032788, AR046523, AR054618, AR057037 (SLT); and AR046852 (FPR); Korea Health 21 R&D Project, Ministry of Health & Welfare, the Government of Korea Grant Number A010252 (HJK).
The contents of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
contract grant sponsor: Shriners’ Hospitals for Children contract grant number: 8590
contract grant sponsor: National Institutes of Health contract grant number: AR032788
contract grant sponsor: National Institutes of Health contract grant number: AR046523
contract grant sponsor: National Institutes of Health contract grant number: AR054618
contract grant sponsor: National Institutes of Health contract grant number: AR057037
contract grant sponsor: National Institutes of Health contract grant number: AR046852
contract grant sponsor: Korea Health 21 R&D Project, Ministry of Health & Welfare, the Government of Korea contract grant number: A010252
References
- Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;100:1557–1565. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, Inaba T, Sanjay A, Baron R, Kawaguchi H, Oda H, Nakamura K, Strasser A, Tanaka S. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. EMBO J. 2003;22:6653–6664. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Kitaura H, Zhao H, Chen J, Muller JM, Schule R, Darnay B, Novack DV, Ross FP, Teitelbaum SL. FHL2 inhibits the activated osteoclast in a TRAF6-dependent manner. J Clin Invest. 2005;115:2742–2751. doi: 10.1172/JCI24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs HE, Baragona SC, Hemperly JJ, Maness PF. NCAM140 interacts with the focal adhesion kinase p125(fak) and the SRC-related tyrosine kinase p59(fyn) J Biol Chem. 1997;272:8310–8319. doi: 10.1074/jbc.272.13.8310. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccio R, Teitelbaum SL, Fujikawa K, Chappel J, Zallone A, Tybulewicz VL, Ross FP, Swat W. Vav3 regulates osteoclast function and bone mass. Nat Med. 2005;11:284–290. doi: 10.1038/nm1194. [DOI] [PubMed] [Google Scholar]
- Fuller K, Wong B, Fox S, Choi Y, Chambers TJ. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. 1998;188:997–1001. doi: 10.1084/jem.188.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimi E, Akiyama S, Tsurukai T, Okahashi N, Kobayashi K, Udagawa N, Nishihara T, Takahashi N, Suda T. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol. 1999;163:434–442. [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Zhang K, Zhang L, Ross FP, Teitelbaum SL, Faccio R. The Src family kinase, Lyn, suppresses osteoclastogenesis in vitro and in vivo. Proc Natl Acad Sci USA. 2009;106:2325–2330. doi: 10.1073/pnas.0806963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, Ross FP, Teitelbaum SL. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–2160. doi: 10.1172/JCI28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Tan HL, Lu J, Kaufman S, Van G, Qiu W, Rattan A, Scully S, Fletcher F, Juan T, Kelley M, Burgess TL, Boyle WJ, Polverino AJ. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol. 2000;157:435–448. doi: 10.1016/S0002-9440(10)64556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkakorpi P, Nakamura I, Young M, Lipfert L, Rodan G, Duong L. Abnormal localisation and hyperclustering of (alpha)(V)(beta)(3) integrins and associated proteins in Src-deficient or tyrphostin A9-treated osteoclasts. J Cell Sci. 2001;114:149–160. doi: 10.1242/jcs.114.1.149. [DOI] [PubMed] [Google Scholar]
- Lee ZH, Kim H-H. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun. 2003;305:211–214. doi: 10.1016/s0006-291x(03)00695-8. [DOI] [PubMed] [Google Scholar]
- Lowell CA, Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10:1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- Manie SN, Astier A, Haghayeghi N, Canty T, Druker BJ, Hirai H, Freedman AS. Regulation of integrin-mediated p130(Cas) tyrosine phosphorylation in human B cells. A role for p59(Fyn) and SHP2. J Biol Chem. 1997;272:15636–15641. doi: 10.1074/jbc.272.25.15636. [DOI] [PubMed] [Google Scholar]
- Martin-Cofreces NB, Sancho D, Fernandez E, Vicente-Manzanares M, Gordon-Alonso M, Montoya MC, Michel F, Acuto O, Alarcon B, Sanchez-Madrid F. Role of Fyn in the rearrangement of tubulin cytoskeleton induced through TCR. J Immunol. 2006;176:4201–4207. doi: 10.4049/jimmunol.176.7.4201. [DOI] [PubMed] [Google Scholar]
- Nishida K, Yamasaki S, Ito Y, Kabu K, Hattori K, Tezuka T, Nishizumi H, Kitamura D, Goitsuka R, Geha RS, Yamamoto T, Yagi T, Hirano T. FcεRI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J Cell Biol. 2005;170:115–126. doi: 10.1083/jcb.200501111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL. The IκB function of NF-kB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003;198:771–781. doi: 10.1084/jem.20030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–461. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez-Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O’Shea JJ, Rivera J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat Immunol. 2002;3:741–748. doi: 10.1038/ni817. [DOI] [PubMed] [Google Scholar]
- Reeve JL, Zou W, Liu Y, Maltzman JS, Ross FP, Teitelbaum SL. SLP-76 couples Syk to the osteoclast cytoskeleton. J Immunol. 2009;183:1804–1812. doi: 10.4049/jimmunol.0804206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src protooncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Stein PL, Lee HM, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y. [DOI] [PubMed] [Google Scholar]
- Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem. 2005;280:3583–3589. doi: 10.1074/jbc.M410480200. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Faccio R, Chappel J, Zheng L, Feng X, Weber JD, Teitelbaum SL, Ross FP. c-Fms tyrosine 559 is a major mediator of M-CSF-induced proliferation of primary macrophages. J Biol Chem. 2007;282:18980–18990. doi: 10.1074/jbc.M610938200. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakamura K, Takahasi N, Suda T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol Rev. 2005;208:30–49. doi: 10.1111/j.0105-2896.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170:427–435. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Umemori H, Sato S, Yagi T, Aizawa S, Yamamoto T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature. 1994;367:572–576. doi: 10.1038/367572a0. [DOI] [PubMed] [Google Scholar]
- Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118:2088–2097. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell. 2008;31:422–431. doi: 10.1016/j.molcel.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.