

Abstract
Bioassay-guided phytochemical investigation of Zygogynum calothyrsum using the human colon carcinoma cell lines COLO205 and KM12 led to the isolation of three new drimane-type sesquiterpenoids, 1β-p-coumaroyloxydrimeninol (1), 1β-coumaroyloxy-5α-hydroxydrimeninol (2) and methyl ether of 1β-coumaroyloxy-12α-methoxydrimeninol (3), the known 1β-p-coumaroyloxypolygodial (4) together with two new tetralones, 3'-deoxyisozygolone (5) and calothyrlone A (9), three known tetralones, isozygolone A (6), zygolone A (7) and 4'-O-methylzygolone A (8) and a known cinnamolide (10). Compounds 1, 7 and 8 demonstrated higher cytotoxicity against COLO205 (GI50 = 18, 17 and 11 μM, respectively) and KM12 (GI50 = 14, 14 and 17 μM, respectively) than the other compounds.
Keywords: Winteraceae, Zygogynum calothyrsum, tetralone, drimane, colon cancer
Colon cancer is the second and third most prevalent cancer among men and women, respectively, in the United States.1 Mortality resulting from this disease, as in many solid tumors, is largely due to metastatic disease, not the primary tumor.2 Despite the use of aggressive surgical resection and chemotherapy, nearly 50% of patients with colorectal carcinoma develop recurrent disease, highlighting the need for improved therapies.3 Extracts of plants and specific compounds derived from plants have exhibited in vivo and in vitro colon cancer activity,4,5 thus, the search for new natural compounds which display specific activity against colon cancer is of great interest. To that end, we have analyzed data from the NCI 60 cell screening program to identify samples with selective activity against the colon cancer cell line panel. We identified a number of plant extracts which showed such activity, and among them, an organic solvent extract of Zygogynum calothyrsum (Winteraceae) was selected for bioassay guided fractionation using the COLO205 and KM12 colon cancer cell lines.
The Winteraceae are a family of flowering plants predominantly distributed in South-East Asia, Australia, New Zealand, Madagascar, Mexico and South America.6,7 The family includes around 120 species of trees and shrubs in 9 genera. The genus Zygogynum consist of about 41 species mainly distributed in Australia, Guinea and Caledonia, of which about 18 species are endemic to New Caledonia.8,9 In recent years, bioactive compounds have been reported from the genus Zygogynum,10,11 however, no phytochemical investigation has so far been reported on Z. calothyrsum. Bioassay-guided fractionation and isolation of compounds from Z. calothyrsum using the colon cancer cell lines COLO205 and KM12 yielded five new (1–3, 5 and 9) and five known (4, 6–8 and 10) compounds. Of these, compounds 1, 7 and 8 exhibited the most potent cell growth inhibition.
RESULTS AND DISCUSSION
Chromatography using successive diol, Sephadex LH-20 and C18 HPLC separation of colon cancer active fractions of Z. calothyrsum led to the isolation of five new (1–3, 5 and 9) and five known (4, 6–8 and 10) compounds (Fig. 1).
Figure 1.
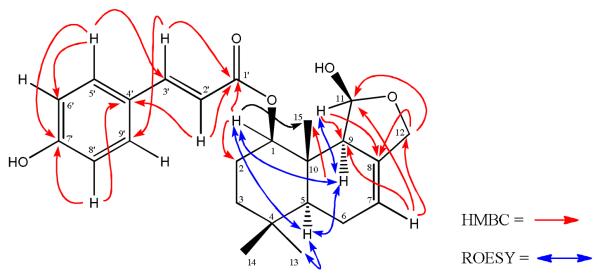
Key HMBC and ROESY correlations in compound 1.
Compound 1 was obtained as a colorless solid. The IR spectrum of 1 showed absorptions at 3369, 1732, 1604 and 1514 cm−1 for hydroxyl, carbonyl, olefinic and aromatic bonds, respectively.12 The HREIMS of 1 supported a molecular composition of C24H30O5, representing 10 degrees of unsaturation. In the 1H and 13C NMR spectra of 1 (Table 1), three methyl signals resonating at δH 0.89, 0.95 and 0.98 and δC 32.6, 21.1 and 9.6 were assigned to C-13, C-14 and C-15, respectively, consistent with a drimane skeleton.13 The relative downfield shift of C-11 (δC 99.7) and upfield shift of C-15 in the 13C NMR spectrum are characteristic for a drimane derivative with adjacent hemiacetal and cinnamate moieties.11 An oxymethine group at δH 5.50 (d, J11,9 = 3.7 Hz, H-11) and an isolated asymmetrical oxymethylene group resonating at δH 4.10 and 4.38 (d, J = 11.1 Hz, H-12) suggested the presence of a tetrahydrofuran-2-ol ring.11 The gem-couplings of H-12 required a quaternary carbon at C-8, which was supported by the vinyl proton of H-7 appearing at δH 5.52 as a broad singlet. The tetrahydrofuran-2-ol moiety in 1 was supported by HMBC correlations (Figure 1) in which H-12 showed correlation to C-7 (δC 116.9), C-8 (δC 136.7), C-9 (δC 60.9) and C-11 (δC 99.7); H-11 correlated to C-8, C-9, C-10 (δC 60.9), and C-12 (δC 68.7); and H-7 correlated with C-6 (δC 23.5), C-8, C-9 and C-12. The p-hydroxy-E-cinnamoyl moiety in 1 was observed by the characteristic aromatic doublet signals of H-2', H-3', H-5' and H-6' in its 1H NMR spectrum and the signals of C-1' to C-7' in its 13C NMR spectrum (Table 1).14 In the HMBC spectrum, correlations observed from the proton at δH 4.76 (1H, dd, J = 4.3, 11.6 Hz, H-1) to C-2 (δC 24.6), C-3 (δC 40.0), C-9, C-10, C-15 (δC 9.6), and C-1' (δC 167.0), suggested that the p-hydroxy-E-cinnamoyl moiety was located at C-1 (Figure 1). The relative configurations of the asymmetric carbons were established by analysis of coupling constants and the ROESY spectrum. In the ROESY spectrum (Figure 1), the correlation of H-5 (δH 1.39) with H-1, H-13, and H-9 (δH 2.33) indicated that they were located on the same face of the molecule. Furthermore, a negative [α]D value has been observed for compounds having a β-oriented H-9, whereas a positive value (+ 42.3° for 1) has generally been observed for compounds having an α-oriented H-9.11 Based on these observations, the structure of 1 was established as 1β-p-hydroxy-E-cinnamoyldrimeninol.
Table 1.
1H and 13C NMR data (600 MHz and 150 MHz, CDCl3) for compounds 1–3
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| δH (m, J Hz) | δ C | δH (m, J Hz) | δ C | δH (m, J Hz) | δ C | |
| 1 | 4.76 (dd, 4.3, 11.6) | 81.5 | 5.25 (dd, 4.2, 11.8) | 77.3 | 4.70 (dd, 4.2, 11.6) | 81.4 |
| 2 | 1.68, 1.78 (m) | 24.6 | 1.69, 1.82 (m) | 24.6 | 1.63, 1.77 (m) | 24.5 |
| 3 | 1.50, 1.46 (m) | 40.0 | 2.00. 2.50 (m) | 31.6 | 1.47. 1.48 (m) | 39.8 |
| 4 | - | 32.9 | - | 42.6 | - | 32.7 |
| 5 | 1.39 (dd, 5.4, 11.4) | 49.3 | - | 78.5 | 1.41 (dd, 5.3, 11.5) | 49.1 |
| 6 | 1.99, 2.50 (m) | 23.5 | 1.23, 2.02 (m) | 34.6 | 2.01, 2.20 (m) | 23.4 |
| 7 | 5.52 (brs) | 116.9 | 5.42 (brs) | 114.0 | 5.78 (brs) | 120.9 |
| 8 | - | 136.7 | - | 138.0 | - | 137.5 |
| 9 | 2.33 (brs) | 60.9 | 2.92 (brs) | 55.9 | 2.57 (brs) | 57.1 |
| 10 | - | 38.0 | - | 38.1 | - | 37.6 |
| 11 | 5.50 (d, 3.7) | 99.7 | 5.53 (d, 3.9) | 100.1 | 5.10 (d, 3.9) | 107.3 |
| 12 | 4.10, 4.38 (d, 11.1) | 68.7 | 4.14, 4.46 (d, 11.1) | 68.6 | 5.08 (s) | 104.0 |
| 13 | 0.89 (s) | 32.6 | 0.99 (s) | 27.3 | 0.88 (s) | 32.5 |
| 14 | 0.95 (s) | 21.1 | 1.14 (s) | 25.4 | 0.93 (s) | 21.6 |
| 15 | 0.98 (s) | 9.6 | 1.07 (s) | 12.9 | 0.95 (s) | 9.6 |
| 1' | - | 167.0 | - | 167.2 | - | 166.1 |
| 2' | 6.25 (d, 15.9) | 116.3 | 6.20 (d, 15.9) | 116.2 | 6.26 (d, 15.9) | 116.8 |
| 3' | 7.54 (d, 15.9) | 144.2 | 7.49 (d, 15.9) | 144.3 | 7.57 (d, 15.9) | 143.6 |
| 4' | - | 127.2 | - | 127.1 | - | 127.6 |
| 5' & 9' | 7.35 (d, 8.4) | 130.1 | 7.27 (d, 8.4) | 130.1 | 7.41 (d, 8.5) | 130.0 |
| 6' & 8' | 6.79 (d, 8.4) | 116.1 | 6.75 (d, 8.4) | 116.0 | 6.81 (d, 8.5) | 116.1 |
| 7' | - | 158.0 | - | 158.1 | - | 157.7 |
| OMe-11 | - | - | - | - | 3.21 (s) | 56.4 |
| OMe-12 | - | - | - | - | 3.33 (s) | 54.2 |
Compound 2 was obtained as a colorless solid. The IR spectrum of 2 showed absorptions at 3386, 1734, 1605 and 1515 cm−1 for hydroxyl, carbonyl, olefinic and aromatic bonds, respectively.12 The HREIMS of 2 supported a molecular composition of C24H30O6, representing 10 degrees of unsaturation. The molecular formula of 2 showed one more oxygen than 1, indicating the presence of an additional hydroxyl group in 2. This was supported by changes in the 13C NMR shift values of C-5 (δC 78.5), C-6 (δC 34.6), C-13 (δC 27.3), C-14 (δC 25.4) and C-15 (δC 12.9) in 2 from those observed in 1 (Table 1), and loss of the H-5 resonance indicating the presence of an hydroxyl group at the C-5 position. This was further supported by HMBC correlations of δH 1.23 and 2.02 (H-6), δH 0.99 (H-13), δH 1.14 (H-14) and δH 1.07 (H-15) with δC 78.5 (C-5). Other NMR data for 2 are similar to those of 1 (Table 1). Based on these observations, the structure of 2 was established as 1β-hydroxy-E-cinnamoyl-5α-hydroxydrimeninol.
Compound 3 was obtained as a colorless solid. The IR spectrum of 3 showed absorptions at 3347, 1728, 1604 and 1515 cm−1 for hydroxyl, carbonyl, olefinic and aromatic bonds, respectively.12 The HREIMS of 3 suggested a molecular composition of C26H34O6 representing 10 degrees of unsaturation. The 1H and 13C NMR spectra of 3 (Table 1) were similar to those for 1 except for the presence of two methoxy groups. The two methoxy groups resonating at δH 3.21 and 3.33 were placed at the C-11 and C-12 positions, respectively. Both hydrogens geminal to the methoxy groups appeared downfield at δH 5.10 (d, J11,9 = 3.9 Hz, H-11) and 5.08 (s, H-12) with carbon shifts of δC 107.3 and 104.0, respectively. In the HMBC spectrum, two methoxy proton signals were correlated with C-11 and C-12, respectively. The sharp singlet of H-12 indicated a quaternary carbon at C-8. In the ROESY spectrum, the correlation of H-5 (δH 1.41) with H-1 (δH 4.70), H-13 (δH 0.88), and H-9 (δH 2.57) and H-9 with H-11 indicated that they were on the same face of the molecule. Similarly, H-11 and H-12 were assigned to opposite faces due to the lack of ROESY correlation between them. All other NMR data of 3 were consistent with 1 (Table 1). Although compound 3 might seem to be an artifact of compound 4 during the course of extraction and purification using methanol, Ferreto et. al. repeated the experimental steps in absence of methanol and obtained this type of compound, which argues for a natural origin.13 Based on these observation, the structure of 3 was established as methyl ether of 1β-hydroxy-E-cinnamoyl-12α-methoxydrimeninol.
Compound 5, obtained as a yellow oil, possessed a molecular formula of C21H20O4 supported by HREIMS. The IR spectral data of 5 suggested the presence of an aromatic ring (1515 and 1455 cm−1), an hydroxyl group (3393 cm−1) and a hydrogen bonded conjugated carbonyl (1641 cm−1).12 The 1H and 13C NMR spectra (Table 2) of 5 showed characteristic signals of the rare 3-tetralone series, and were similar to those of the known tetralone 6 except the signals for aromatic ring B.10 The 1H NMR spectrum showed signals for aromatic ring B with a characteristic AA'BB'system of para-disubstitution at δH 7.12 (2H, d, J = 8.4 Hz, H-2' and H-6') and δH 6.79 (2H, d, J = 8.4 Hz, H-3' and H-5'). The HMBC correlation of H-3 with C-1' (δC 135.6) showed that ring B was connected to C-3 of the tetralone. The absolute configuration at C-3 is assigned on the basis of positive optical rotation value of 5 which is consistent to the reported data.10 All of these assignments led to the structure of 5 as 8-hydroxy-3-(4'-hydroxyphenyl)-(2”-propenyl-2”-3”-dihydrofuran) [4”,5”:6,7]-tetralone, trivially named 3'-deoxyisozygolone.
Table 2.
1H and 13C NMR data (600 MHz and 150 MHz, CDCl3) for compounds 5 and 9
| 5 | 9 | |||
|---|---|---|---|---|
|
|
||||
| δH (m, J Hz) | δ C | δH(m, J Hz) | δ C | |
| 1 | - | 202.6 | - | 202.5 |
| 2a | 2.73 (dd, 17.0, 12.6) | 45.8 | 2.72 (dd, 17.0, 12.3) | 45.7 |
| 2b | 2.81 (dd, 17.1, 3.4) | - | 2.81 (dd, 17.1, 3.4) | - |
| 3 | 3.30 (m) | 40.2 | 3.24 (m) | 40.1 |
| 4 | 3.01 (m) | 38.7 | 3.00 (m) | 38.1 |
| 5 | 6.23 (s) | 101.8 | 6.25 (brs) | 107.2 |
| 6 | - | 167.2 | - | 166.3 |
| 7 | - | 111.8 | 6.28 (d, 2.1) | 99.1 |
| 8 | - | 160.8 | - | 165.9 |
| 9 | - | 110.9 | - | 111.3 |
| 10 | - | 147.5 | - | 146.3 |
| 1' | - | 135.6 | - | 136.5 |
| 2' | 7.12 (d, 8.4) | 128.8 | 6.77 (d, 1.7) | 114.0 |
| 3' | 6.79 (d, 8.4) | 115.7 | - | 146.3 |
| 4' | - | 154.6 | - | 142.5 |
| 5' | 6.79 (d, 8.4) | 115.7 | 6.81 (d, 8.1) | 115.8 |
| 6' | 7.12 (d, 8.4) | 128.8 | 6.66 (dd, 8.1, 1.7) | 119.2 |
| 2” | 5.26 (t, 8.2) | 88.1 | - | - |
| 3”a | 2.94 (dd, 15.4, 7.6) | 30.9 | - | - |
| 3”b | 3.28 (dd, 15.6, 10.0) | - | - | - |
| 1”'a | 4.90 (brs) | 112.8 | - | - |
| 1”'b | 5.05 (brs) | - | - | - |
| 2”' | - | 143.4 | - | - |
| 3”' | 1.74 (s) | 17.2 | - | - |
| OMe | - | - | 3.81 (s) | 55.7 |
Compound 9 was obtained as a yellow oil with the molecular formula of C17H16O5 supported by HREIMS. The IR spectral data of 9 suggested the presence of an aromatic ring (1515 and 1455 cm−1), an hydroxyl group (3393 cm−1) and a conjugated hydrogen bonded carbonyl group (1630 cm−1).12 The 1H and 13C NMR spectra (Table 2) of 9 showed characteristic signals for the tetralone skeleton,10 having some structural modifications differentiating it from compound 5. The substituted dihydrofuran ring that was attached in ring A of 5 was absent in 9. A proton resonating at δH 6.28 (1H, d, J = 2.1 Hz, H-7) meta-coupled with H-5, showed HMBC correlations with C-5 (δC 107.2), C-6 (δC 166.3) and C-8 (δC 165.9). The 3H signal at δH 3.81 showed an HMBC correlation with C-6, indicating the attachment of the methoxy group at this carbon. These data, together with other 1H and 13C NMR data of 9 (Table 2) indicated that ring A was tetra-substituted. The 1H NMR spectrum also showed signals for aromatic ring B with a characteristic AMX system of one meta-coupled proton at δH 6.77 (1H, d, J = 1.7 Hz, H-2'), one ortho-coupled proton at δH 6.81 (1H, d, J = 8.1 Hz, H-5') and one ortho- and meta-coupled proton at δH 6.66 (1H, d, J = 8.1, 1.7 Hz, H-5') suggesting the presence of a 1,3,4-trisubstituted asymmetric aromatic ring. The HMBC correlation of H-3 (δH = 3.24) with C-1' (δC 136.5) showed that ring B was connected to C-3 of the tetralone. The absolute configuration at C-3 is assigned on the basis of positive optical rotation value of 9 which is consistent to the reported data.10 All of these assignments led to the structure of 9 as 8-hydroxy-6-methoxy-3-(3',4'-dihydroxyphenyl)-tetralone, trivially named calothyrlone A.
Together with five new compounds, five known compounds were also isolated, characterized and identified based on comparison of their spectroscopic data with the literature. The known compounds were identified as 1β-p-hydroxy-E-cinnamoylpolygodial (4),13,14 isozygolone A (6),10 zygolone A (7),10 4'-O-methyl zygolone A (8),10 and cinnamolide (10).15
All compounds were evaluated in vitro against two human colon cancer cell lines COLO205 and KM12 (Table 3). In the drimane series, compound 1, which possesses a β-hydroxyl group on the epoxide ring, was found to more potently inhibit cell growth (COLO205, GI50 = 18 μM and KM12, GI50 = 14 μM) than compound 2 which had an additional α-hydroxyl group at C-5 (COLO205, GI50 = 29 μM and KM12, GI50 = 25 μM). Interestingly, compound 3 bearing methoxy moieties in ring C was found to be less active. Similarly, the zygolones 7 and 8 were found to be more potent (COLO205, GI50 = 17 and 11 μM, respectively and KM12, GI50 = 14 and 17 μM, respectively) than the isozygolones 5 and 6 (COLO205, GI50 = 35 and 32 μM, respectively and KM12, GI50 = 29 and 32 μM, respectively).
Table 3.
Cytotoxicity of compounds 1–10 in colon tumor cells COLO205 and KM12
| Compound | GI50 (μM)a |
|
|---|---|---|
| COLO205 | KM12 | |
| 1 | 18 | 14 |
| 2 | 29 | 25 |
| 3 | >45 | >45 |
| 4 | 29 | 25 |
| 5 | 35 | 29 |
| 6 | 32 | 32 |
| 7 | 17 | 14 |
| 8 | 11 | 17 |
| 9 | >67 | >67 |
| 10 | >85 | >85 |
The GI50 values are the concentration corresponding to 50% growth inhibition. Data are an average of at least two tests.
NCI 60 cell testing of compounds 1, 2, 4, 6, 7 and 8 was also conducted. In initial one-dose testing at 10−5 M, compounds 1 and 4 showed selectivity towards colon cancer cell lines (see SI), while the other compounds did not. Compounds 1, 4, 6, 7, and 8 were selected and tested in the full five-dose screen, however, none of them showed colon cancer cell line selectivity in this assay (see SI). The failure to isolate compounds that clearly reproduce the observed selectivity of the extract (see SI for NCI 60 extract data) may perhaps be due to the instability of dialdehyde compounds such as 4 in biological matrices, or in pure form.
The cytotoxicity of drimanes from Zygogynum baillonii has been previously studied by the CNRS group, who found similarly modest activity against KB cells.16 Other species of Zygogynum studied by the same group had more potent activity against three cancer cell lines.11 Cytotoxicity of tetralones from Zygogynum has also been reported by the same group.10
EXPERIMENTAL SECTION
General experimental procedures
Optical rotations ([α]D) were measured on a Perkin-Elmer 241 polarimeter in a 100 × 2 mm cell (units 10−1 deg cm2g−1). UV absorption spectra were obtained using a Varian Cary 50 Bio UV-visible spectrophotometer. IR spectra were measured using a Perkin Elmer FT-IR Spectrometer, Spectrum 2000. LCMS data were obtained using a Hewlett Packard Series 1100 MSD, whereas HRMS data were acquired on an Agilent 6520 Accurate Mass Q-TOF instrument with internal reference masses calibrated at 121.05087 and 922.00979, both within 5 ppm. The NMR experiments were performed on a Bruker 600 MHz NMR spectrometer. 1H and 13C spectra were referenced to deuterated solvent peaks. The diol DIO Spe-ed SPE cartridge and Sephadex LH-20 columns attached to a model UA-6 UV detector and Foxy 200 fraction collector (Teledyne Isco) were used for fractionation of the extract, whereas purification of the compounds was performed using a Varian ProStar 210/215 solvent delivery module HPLC equipped with a Varian ProStar 325 UV-Vis detector, operating under Star 6.41 chromatography workstation software. All solvents and chemicals were of analytical grade.
Plant material
The twigs of Z. calothyrsum (Diels) Vink (Winteraceae) (400 g dry weight, Q66O7253-U) were collected on March 12, 1989 by Wayne Takeuchi (Takeuchi 4509) from Morobe Province, Biaru area of Papua New Guinea (1956 m elevation) in mountainous terrain between the Ekuti and Kuper ranges, mixed Castanopsis - podocarp forest; emerging from Nastus thicket. The plant was identified by M. Van Balgooy. A voucher specimen No. 2031934 collected from the same individual September 22, 1988 was deposited in the Field Museum of Natural History. Samples of roots, stem bark and wood, and leaves were also obtained; extracts from these plant parts lacked selectivity in the NCI 60 cell screen.
Extraction and Isolation
400 g of dried twigs of Z. calothyrsum was ground and extracted with CH2Cl2--MeOH (1:1 v/v) by the standard NCI method17 to yield 18.14 g of organic extract. 1.62 g of this extract was used for the colon cancer cell based bioassay-guided extraction and isolation of compounds. The extract was loaded on twelve diol DIO Spe-ed SPE cartridges (135.0 mg in each cartridge) and eluted with 6.0 mL each of hexane/CH2Cl2 (9:1), CH2Cl2/EtOAc (20:1), EtOAc, EtOAc/MeOH (5:1) and MeOH to give five fractions A-E of 474.2 mg, 392.6 mg, 120.6 mg, 210.4 mg and 295.1 mg, respectively. The colon cancer activity of fractions A, B and C was found to be higher than other fractions were selected for further fractionation and isolation of compounds.
Fraction A (470.0 mg) was chromatographed on a 40 × 2.5 cm i.d., Sephadex LH-20 column and eluted with hexane/CH2Cl2/MeOH (2:5:1, v/v), with 250 drop fractions collected in each tube. Based upon the UV-absorption (254 nm), eleven fractions A1-K1 were collected. The fractions A1-K1 were between the tubes 5–7, 8–12, 13–14, 15–16, 17–20, 21–23, 26–30, 31–33, 37–41, 42–47 and 48–56, respectively. Among the eleven fractions, fraction F1 showed activity. Similarly, fraction B (390.0 mg) was also fractionated by using a similar procedure as with fraction A, by which total of fourteen fractions A2-N2 were collected. Among the fourteen fractions, fraction E2-H2 and J2-N2 were found to show activity. In the same way, fraction C (120 mg) was chromatographed on a 40 × 1.5 cm i.d., Sephadex LH-20 column and eluted with CH2Cl2/MeOH (1:1, v/v), with 250 drop fractions collected in each tube. Based upon the UV-absorption (254 nm), nine fractions A3-I3 were collected. Among them, fractions D3 and E3 were found to have activity.
HPLC separation was performed on 3.0 mg of fraction F1 dissolved in 0.4 mL of MeOH and injected in 200 μL aliquots (approx. 1.5 mg/injection) onto a 2.5 × 1 cm i.d., Varian Dynamax Microsorb 60–8 C18 HPLC column. The detector wavelength was 225 nm and solvent elution condition was 4.0 mL/min. Elution begin with 80% MeOH/H2O, running isocratic for 3 minutes and switching to a linear gradient of 80% to 100% MeOH at 25 min. The column was flushed with 100% MeOH for 5 min (System A). System B began at 70% MeOH but was otherwise identical, while System C began at 60% MeOH. One major UV-absorbing peak was collected and evaporated in vacuo to yield 10 (0.9 mg) eluted at approx. 11.4 min (System A). A similar procedure was followed for fraction E2 (30.0 mg) using System B and yielded compound 3 (1.5 mg) eluting at 16.3 min. Employing System B on active fraction G2 (16.0 mg) yielded 3 (0.7 mg, 16.2 min) and 4 (1.1 mg, 12.5 min). System B applied to active fraction H2 (16.0 mg) yielded 5 (1.6 mg, 15.2 min). Similarly, chromatography of active fraction J2 (9.0 mg) yielded 1 (1.1 mg, 15.0 min) and fraction K2 (2.0 mg) produced 8 (1.3 mg, 14.3 min). The active fraction L2 (4.5 mg) using System B yielded 7 (1.4 mg, 17.2 min) and 6 (0.9 mg, 16.8 min), whereas fraction M2 (3.0 mg) yielded 2 (1.3 mg, 19.3 min). The fraction N2 (2.8 mg) with System B provided 9 (1.3 mg, 7.2 min). Using System C, active fraction D3 (18.0 mg) yielded 1 (1.5 mg) and 2 (0.6 mg) at elution of approx. 15.2 and 17.7 min, respectively. The last active fraction E3 (5.0 mg) using System C yielded 2 (0.8 mg, 14.7 min).
1β-p-hydroxy-E-cinnamoyldrimeninol (1), NSC#762012
Colorless solid; [α]25D = + 42.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 310 (3.9) nm; IR (CHCl3) νmax 3369, 1732, 1604 and 1514 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data, see Table 1; HREIMS [M+H]+ at m/z 399.2081 (calcd for C24H31O5 399.2074).
1β-hydroxy-E-cinnamoyl-5α-hydroxydrimeninol (2), NSC#762011
Colorless solid; [α]25D = + 52.7 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 310 (3.8), 224 (2.6) nm; IR (CHCl3) νmax 3386, 1734, 1605 and 1515 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data, see Table 1; HREIMS [M+H]+ at m/z 415.5022 (calcd for C24H31O6 415.5027).
Methyl ether of 1β-hydroxy-E-cinnamoyl-12α-methoxydrimeninol (3)
Colorless solid; [α]25D = + 33.4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 325 (2.9), 300 (3.1), 225 (2.4) nm; IR (CHCl3) νmax 3347, 1728, 1604 and 1515 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data, see Table 1; HREIMS [M+Na]+ at m/z 465.2234 (calcd for C26H34NaO6 465.2248).
3'-dehydroxyisozygolone (5)
Yellow oil; [α]25D = + 93.5 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 310 (4.2), 290 (3.9) nm; IR (CHCl3) νmax 3393, 1641, 1515and 1455 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data, see Table 2; HREIMS [M+H]+ at m/z 337.1437 (calcd for C21H21O4 337.1443).
Calythyrlone A (9)
Yellow oil; [α]25D = + 71.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 309 (4.1), 290 (3.9) nm; IR (CHCl3) νmax 3393, 1630, 1515and 1455 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data, see Table 2; HREIMS [M+H]+ at m/z 301.1078 (calcd. for C17H17O5 301.1071).
Cytotoxicity assay on colon cancer cells
The assay used for this study was a two-day, two cell line XTT bioassay, an in vitro antitumor colorimetric assay developed by the MTL Assay Development and Screening Section. Colon cancer cell lines used were COLO205 and KM12. Cells were maintained and passed weekly in RPMI-1640 medium with phenol red (GIBCO, Carlsbad, CA) and supplemented with 2mM l-glutamine (Quality Biologicals, Inc., Gaithersburg, MD) and 10% fetal bovine serum (Hyclone, Logan, UT). All work was done under sterile conditions using a laminar air-flow hood with external venting. Cells were placed in a humidified incubator with an atmosphere of 5% CO2, 95% air and a temperature of 37 °C. Cells were discarded after 25 passages to reduce the risk of “genetic drift” or cross contamination of cell lines. Cells used in the assay were harvested with RPMI-1640 medium, without phenol red (GIBCO, Carlsbad, CA) and supplemented with 2mM L-glutamine (Quality Biologicals, Inc., Gaithersburg, MD) and 10% fetal bovine serum without antibiotics. Harvested cells were counted using a Cellometer Auto T4 cell counter (Nexcelom Bioscience LLC, Lawrence MA) and plated in 384-well flat bottom polystyrene microtiter plates (Nunc - Nunc A/S, Denmark), at a density of 5,000 cells/well for COLO205 and 5,000 cells/well for KM12. The cells were incubated in a 5% CO2, 95% air and a temperature of 37 °C incubator for 24 hrs. After incubation, test samples were added to plates using a Biomek FX robotic liquid handling workstation (Beckman/Coulter, Fullerton, CA). The robot performed eight 2-fold serial dilutions of the sample and then transferred the sample from the source plate to the assay plate. The plates used were Costar 384-well round bottom plates, (Corning Inc., Corning, NY). Cells were further incubated with samples for (48) hours, at which time the XTT reagent was added.
Supplementary Material
Table 4.
Cytotoxicity of compounds in NCI 60-cell screen
| Compound | NSC# | Mean Percent inhibition at 10−5 M | Percent Range at 10−5 M | Mean GI50 Full Dose (μM) | Range at GI50 (log10 units) |
|---|---|---|---|---|---|
| 1 | 762012 | −2 | 131 | 16 | 0.55 |
| 2 | 762011 | 69 | 66 | NT | NT |
| 4 | 762013 | 83 | 78 | 9.1 | 0.99 |
| 6 | 762015 | 48 | 122 | 17 | 1.88 |
| 7 | 762014 | 42 | 120 | 6.3 | 0.93 |
| 8 | 756803 | 56 | 135 | >100 | 0.43 |
ACKNOWLEDGEMENTS
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. We thank Wayne Takeuchi for the plant collection, and Doel Soejarto and David Newman for help in documenting the collection, as well as the Natural Products Support Group at NCI-Frederick for extraction, and Sergey Tarasov and Marzena Dyba (Biophysics Resource Core, Structural Biophysics Laboratory, CCR) for assistance with mass spectrometry.
REFERENCES
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. CA Cancer J. Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Rajput A, Dominguez SM, I, Rose R, Beko A, Levea C, Sharratt E, Mazurchuk R, Hoffman RM, Brattain MG, Wang J. J. Surg. Res. 2008;147:276–281. doi: 10.1016/j.jss.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 3.Wils J, O'Dwyer P, Labianca R. Ann. Oncol. 2001;12:13–22. doi: 10.1023/a:1008357725209. [DOI] [PubMed] [Google Scholar]
- 4.Sandeep K, Shweta, Nisha S, Manjunath SM, Arti J. Drug Delivery Ther. 2012;2:97–105. [Google Scholar]
- 5.Park JJ, Seo SM, Kim EJ, Lee YJ, Ko YG, Ha J, Lee M. Biochem. Biophys. Res. Commun. 2012;426:461–467. doi: 10.1016/j.bbrc.2012.08.091. [DOI] [PubMed] [Google Scholar]
- 6.Doust AN. Ann. Missouri Bot. Garden. 2000;87:366–379. [Google Scholar]
- 7.Vink W. Taxon. 1988;37:691–698. [Google Scholar]
- 8.White F, Vink W. Flore de la Nouvella-Caledonie. Association de Botanique Tropicale, MNHN; Paris: 1993. Winteracees; pp. 90–171. [Google Scholar]
- 9.Marquinez X, Lohmann LG, Salatino ML, Salatino A, Gonzalez F. Mol. Phylogenet. Evol. 2009;53:435–449. doi: 10.1016/j.ympev.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Allouche N, Morleo B, Thoison O, Dumontet V, Nosjean O, Gueritte F, Sevenet T, Litaudon M. Phytochemistry. 2008;69:1750–1755. doi: 10.1016/j.phytochem.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 11.Allouche N, Apel C, Martin MT, Dumontet V, Gueritte F, Litaudon M. Phytochemistry. 2009;70:546–553. doi: 10.1016/j.phytochem.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Pavia DL, Lampman GM, Kriz GS. Introduction to Spectroscopy. Harcourt Brace College Publishers; 1996. [Google Scholar]
- 13.Ferreto L, Ciccio JF, Castro V, Andrade R. Spectros. Int. J. 1988;6:133–136. [Google Scholar]
- 14.Vichnewski W, Kulanthaivel P, Herz W. Phytochem. 1986;25:1476–1478. [Google Scholar]
- 15.Kioy D, Gray AI, Waterman PG. Phytochem. 1990;29:3535–3538. [Google Scholar]
- 16.Fotsop DF, Roussi F, Le Callonec C, Bousserouel H, Litaudon M, Gueritte F. Tetrahedron. 2008;64:2192–2197. [Google Scholar]
- 17.McCloud TG. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.