

Abstract
Objective:
To evaluate the utility of targeted exome sequencing for the molecular diagnosis of mitochondrial disorders, which exhibit marked phenotypic and genetic heterogeneity.
Methods:
We considered a diverse set of 102 patients with suspected mitochondrial disorders based on clinical, biochemical, and/or molecular findings, and whose disease ranged from mild to severe, with varying age at onset. We sequenced the mitochondrial genome (mtDNA) and the exons of 1,598 nuclear-encoded genes implicated in mitochondrial biology, mitochondrial disease, or monogenic disorders with phenotypic overlap. We prioritized variants likely to underlie disease and established molecular diagnoses in accordance with current clinical genetic guidelines.
Results:
Targeted exome sequencing yielded molecular diagnoses in established disease loci in 22% of cases, including 17 of 18 (94%) with prior molecular diagnoses and 5 of 84 (6%) without. The 5 new diagnoses implicated 2 genes associated with canonical mitochondrial disorders (NDUFV1, POLG2), and 3 genes known to underlie other neurologic disorders (DPYD, KARS, WFS1), underscoring the phenotypic and biochemical overlap with other inborn errors. We prioritized variants in an additional 26 patients, including recessive, X-linked, and mtDNA variants that were enriched 2-fold over background and await further support of pathogenicity. In one case, we modeled patient mutations in yeast to provide evidence that recessive mutations in ATP5A1 can underlie combined respiratory chain deficiency.
Conclusion:
The results demonstrate that targeted exome sequencing is an effective alternative to the sequential testing of mtDNA and individual nuclear genes as part of the investigation of mitochondrial disease. Our study underscores the ongoing challenge of variant interpretation in the clinical setting.
Mitochondrial disorders are a heterogeneous collection of rare disorders caused by mitochondrial dysfunction.1 Multiple organ systems can be affected, with features that can include myopathy, encephalopathy, seizures, lactic acidosis, sensorineural deafness, optic atrophy, diabetes mellitus, liver failure, and ataxia.2 Mitochondrial disorders can be caused by mutations in the nuclear or mitochondrial genomes, and more than 100 genetic loci have been identified to date.3,4 Whereas these disorders may be inherited in a maternal, recessive, X-linked, or dominant manner, many patients have no obvious family history of the disorder.5–7 Because of phenotypic and locus heterogeneity, traditional sequential genetic tests result in molecular diagnoses for only a fraction of patients.8 Limitations in traditional genetic testing have motivated our group and others to develop “next-generation sequencing” (NGS) approaches for molecular diagnosis.9–12
Previously, we developed a targeted “MitoExome” sequencing approach for the molecular diagnosis of mitochondrial disease and benchmarked its performance in a cohort of 42 severe, infantile cases with biochemically proven mitochondrial respiratory chain disease.13 MitoExome sequencing enabled new molecular diagnoses in 24% of cases and, in an additional 29% of cases, identified recessive candidate disease genes, 3 of which have since been independently validated.14–16 Here, we evaluate a similar approach in a collection of 102 patients with suspected mitochondrial disease, spanning a broad range of age and phenotypic severity.
METHODS
Subjects.
We considered patients that were referred to the Massachusetts General Hospital (MGH) Mitochondrial Disorders Clinic for evaluation of possible mitochondrial disease and enrolled in the Partners DNA and Tissue Repository for Molecular Studies in Disorders of Energy Metabolism during 2004 to 2011. Of the 159 unrelated patients for whom high-quality DNA was available, we selected the 102 subjects with the highest clinical suspicion of mitochondrial disease, based on chart review and review of biochemical testing results. The selected cases included as positive controls all 18 patients with prior molecular diagnoses acquired through traditional genetic testing (13 mitochondrial DNA [mtDNA] and 5 nuclear mutations), 11 of which were made before referral to MGH. The remaining 84 patients had no prior molecular diagnosis, despite varying degrees of genetic testing in 74 cases. Of the 102 selected cases, 75% had histologic studies, 63% had blood metabolite studies, 33% had neuroimaging studies, and 68% had enzyme analysis of the electron transport chain (ETC) from muscle biopsy. Muscle biopsies were not typically performed in patients with prior molecular diagnoses.
Standard protocol approvals, registrations, and patient consents.
Study protocols were approved by the Partners Human Research Committee. All samples were obtained with informed consent for sequence analysis and data deposition into public databases.
MitoExome sequencing and variant detection.
We expanded our previously reported MitoExome sequencing protocol to include additional genes of interest (appendix e‐1 on the Neurology® Web site at www.neurology.org). Specifically, we targeted 3.1 Mb of DNA including the mtDNA and coding exons of 1,598 nuclear-encoded genes (table e-1): 113 genes known to cause mitochondrial disease, 945 high-confidence mitochondrial genes from MitoCarta,17 327 genes predicted to associate with mitochondrial function, and 213 genes underlying monogenic disorders in the differential diagnosis, i.e., metabolic and neurologic disorders with phenotypic similarity to mitochondrial disease. DNA was extracted from whole blood or Epstein-Barr virus–immortalized lymphoblastoid cell lines. Targeted DNA regions were captured18 and sequenced (paired-end 76 base pair reads, Illumina GA-IIx or Hi-Seq)18,19 at the MGH Next-Generation Sequencing Core. Sequence alignment, variant detection, and annotation were performed as described in appendix e-1. mtDNA variants with ≥1% heteroplasmy based on read count were identified, and confirmatory quantitative PCR assays were performed at m.3243, m.8344, and m.8993 (Baylor College of Medicine, test 3006).
Variant prioritization.
We prioritized the following mtDNA variants: i) confirmed pathogenic variants in MITOMAP,20 with allele frequency (AF) <0.5% in mtDB,21 ii) mtDNA deletions/rearrangements detected through de novo mtDNA assembly,13 and iii) loss-of-function (i.e., nonsense or frameshift) mutations with AF <0.3% in mtDB21 and present in <10% of 102 cases. Variants with heteroplasmy <20% were confirmed in an independent DNA sample or were considered prioritized variants of unknown significance.
We prioritized the following nuclear variants: i) known pathogenic variants (Human Gene Mutation Database22 version 2010.3) with AF <1% in 1000 Genomes Project23 (low coverage release 20110511), and ii) variants with AF <0.5% that were likely deleterious, including nonsense, splice site, coding indel, and missense variants at sites conserved in ≥10 of 44 aligned vertebrate species.13 For established autosomal recessive disease genes, we required presence of homozygous or 2 heterozygous variants. For X-linked recessive disease genes, we required 2 variants in females or a hemizygous variant in males. Nuclear variants in candidate genes not previously linked to disease were prioritized as above but considered to be prioritized variants of unknown significance. For dominant disease genes (autosomal or X-linked), we required AF <0.1%. All prioritized variants were manually reviewed to remove likely sequencing artifacts and to phase potential compound heterozygous variants (appendix e-1). Prioritized variants are listed in table e-2.
Estimating the background frequency of variants.
mtDNA was analyzed in 284 healthy individuals of European descent with whole-genome data from the 1000 Genomes Project. Nuclear DNA was analyzed in 371 healthy individuals of European descent with whole-exome data from the National Institute of Mental Health control sample repository. Background frequency comparisons were limited to sequence regions well covered in all individuals, as previously described.13 Enrichment was calculated as the percent of cases vs controls containing at least 1 qualifying variant, with significance assessed by a 1-tailed binomial exact test.
Modeling the ATP5A1 mutation in yeast.
The Saccharomyces cerevisiae atp1 null mutant was created in an ade2− background and transformed with the pRS305 plasmid expressing wild-type or mutant alleles of ATP1 (appendix e-1). Mitochondrial membrane potential, phosphate/oxygen ratio, respiratory control rate, and adenosine triphosphatase activity were measured as described.24 Petite frequency (percent white colonies, where lack of red pigment in ade2− strains indicates mtDNA loss/deletion) was assessed in single haploid colonies grown, plated, and incubated in YPD (yeast extract, peptone, and glucose), as previously described.24 Loss of mtDNA in petite colonies was confirmed via mating with a ρ0 strain.
Data availability.
Sequence and phenotype data are available in dbGaP, accession phs000339.
RESULTS
MitoExome sequencing in 102 patients.
MitoExome sequencing was applied to 102 MGH patients with a suspected mitochondrial disorder. The cohort represented a broad spectrum of phenotypic severity ranging from lethal infantile to mild adult-onset disease (figure 1). Likelihood of mitochondrial disease was assessed as “definite,” “probable,” or “possible” based on the Bernier and Morava criteria.25,26 Fifty-one percent of patients had a biochemical ETC defect in muscle, defined as ≤30% of normal activity of any complex after normalization to citrate synthase activity. Eleven percent had a positive family history, defined as presence of a first-degree relative with a highly similar phenotype. Age at diagnosis, defined as the date of initial biochemical or genetic testing for a mitochondrial disorder, ranged from newborn to 64 years, with median 27 years. Sixty-seven percent of patients were female and 87% were of European descent.
Figure 1. Characteristics of 102 Massachusetts General Hospital patients with suspected mitochondrial disease.

ETC = electron transport chain; GI = gastrointestinal; mito. = mitochondrial.
The approach yielded extremely high sequence coverage with mean 226X at targeted nuclear sites and 12,680X at mtDNA sites (table e-3). Ninety-five percent of targeted bases were covered with at least 10 reads, enabling confident detection of variants at the majority of sites. The deep coverage of mtDNA enabled sensitive detection of known heteroplasmic variants, including 5 of 6 variants present at <10% heteroplasmy (appendix e-2).
We reasoned that high-confidence “molecular diagnoses” could only be established within genes proven to cause disease consistent with the patient's phenotype. We therefore first examined variants in established disease loci, adopting guidelines from the American College of Medical Genetics to make new molecular diagnoses.27 Prioritized variants not consistent with previously reported phenotypes, or prioritized variants in genes not previously implicated with disease, were designated “prioritized variants of unknown significance” (pVUS).
Diagnoses in mtDNA.
Within mtDNA, we prioritized previously reported pathogenic variants, large deletions or rearrangements, and rare loss-of-function variants (Methods). Other rare missense and tRNA variants were not prioritized because their collective frequency could not be distinguished from that observed in healthy individuals (figure e-1).
Our diagnostic procedure correctly recovered molecular diagnoses for 12 of 13 patients with previously identified pathogenic mtDNA mutations (table e-4). The exception was a patient with m.3243A>G present at 3% heteroplasmy based on quantitative PCR in the sequenced lymphoblastoid cell line sample, underscoring the importance of tissue selection in studies of mitochondrial disease.28
No confident mtDNA diagnoses were established in the remaining individuals. However, we identified 6 patients with pVUS in mtDNA (appendix e-2, table e-5), representing a 2.5-fold excess over the background rate in healthy controls (p < 0.05).
Diagnoses in previously established nuclear disease genes.
We next prioritized nuclear variants within 113 genes known to underlie mitochondrial disease and 213 genes underlying monogenic disorders in the differential diagnosis. Our diagnostic procedure recovered the prior molecular diagnoses for 5 of 5 patients with pathogenic nuclear mutations (table e-6), and established new molecular diagnoses in 5 patients (table 1).
Table 1.
Individuals with new molecular diagnoses from MitoExome sequencing
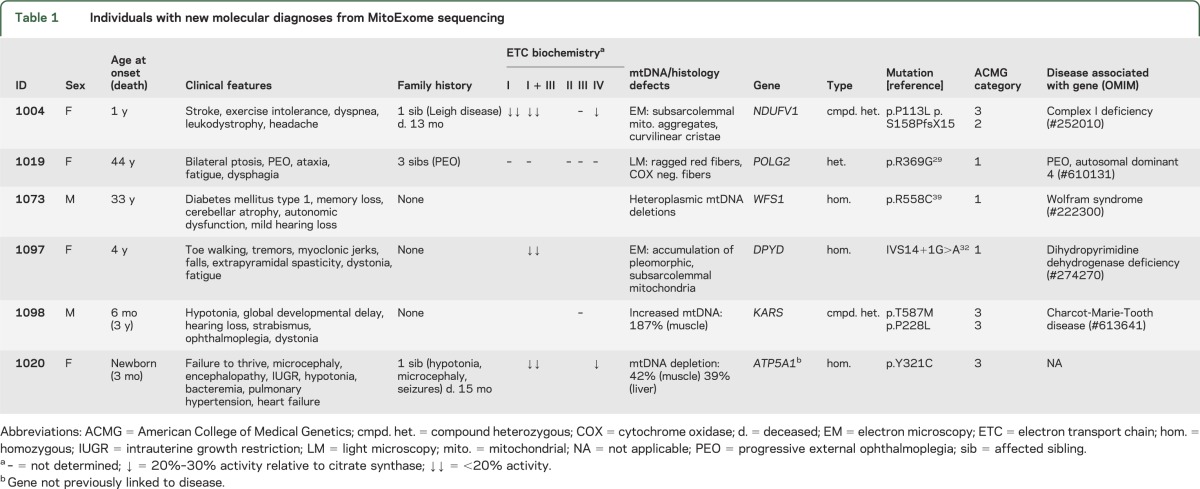
Two of the 5 new molecular diagnoses implicated genes underlying primary mitochondrial disorders: a heterozygous POLG2 mutation known to be pathogenic29,30 was detected in an individual with progressive external ophthalmoplegia, and compound heterozygous mutations in NDUFV131 were detected in an individual with complex I deficiency in whom prior NDUFV1 sequencing had detected only 1 variant.
The other 3 new molecular diagnoses, described below, implicated autosomal recessive variants in genes known to cause monogenic disorders in the differential diagnosis: dihydropyrimidine dehydrogenase (DPD) deficiency (DPYD),32 Wolfram syndrome (WFS1),33 and a Charcot-Marie-Tooth–like axonal neuropathy (KARS).34 Importantly, the phenotypes of these patients were not clearly discernible even in retrospect from the remainder of the cohort, underscoring the phenotypic and biochemical overlap between canonical mitochondrial disease and other monogenic disorders.
Patient 1097, of Indian descent, developed multiple neurologic symptoms starting at age 4 years, including toe walking, tremors, and dystonia. Muscle tissue showed reduced complex I + III activity and electron microscopy showed accumulation of pleomorphic subsarcolemmal mitochondria, some with complex cristae. Sequencing revealed a homozygous splice mutation in DPYD previously associated with recessive DPD deficiency and 5-fluorouracil toxicity.32,35 The diagnosis of DPD deficiency was confirmed by subsequent urine tests that showed highly elevated uracil (395 mmol/moL creatinine; reference range <28) and thymine (300 mmoL/moL creatinine; reference range <6).
Patient 1073, of Ashkenazi ancestry, presented with type 1 diabetes at age 33 years and developed autonomic dysfunction, cerebellar atrophy, and severe memory loss. Muscle biopsy showed heteroplasmic mtDNA deletions. MitoExome sequencing revealed a homozygous missense mutation in WFS1 underlying Wolfram syndrome, which was not detected by initial clinical gene sequencing, as previously described.36
Patient 1098, of European descent, presented at 6 months of age with developmental delay, hypotonia, ophthalmoplegia, and other neurologic manifestations (table 1), and died by age 4 years. EEG and MRI were unremarkable; however, brainstem auditory-evoked potential study at 10 months was markedly abnormal, suggesting severe peripheral conduction defects in the auditory pathways bilaterally. Metabolic studies showed elevated plasma alanine (657 μmol/L; reference range 142–421) and CSF lactate (2.4 mmol/L; reference range 0.5–2.2); increased mtDNA levels were found in muscle (187% of controls). Sequencing revealed compound heterozygous missense mutations in KARS, which encodes both the cytoplasmic and mitochondrial lysyl-tRNA synthetase (figure e-2). Although formal experimental proof is needed, given the predicted severity of the mutations at highly conserved residues and the previous report of recessive KARS mutations in patients with overlapping symptoms, the observed mutations are likely to underlie our patient's phenotype, as discussed in appendix e-2.
In addition to 5 new molecular diagnoses, 11 patients harbored pVUS in established nuclear disease loci (appendix e-2): 7 in recessive-acting genes (4-fold enrichment over background frequency, p < 0.05), and 4 in dominant-acting genes (no enrichment over background) (Methods).
Altogether, analysis of >300 established disease loci in the mitochondrial and nuclear genomes recovered the diagnoses of 17 of 18 patients with prior molecular diagnoses and led to new molecular diagnoses in 5 of 84 previously unsolved cases (figure 2).
Figure 2. MitoExome sequencing in 102 patients with suspected mitochondrial disease.
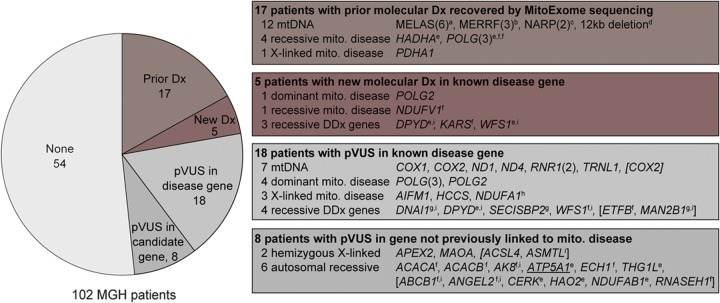
Number of patients with molecular diagnoses or prioritized variants of unknown significance (pVUS), with the prioritized loci listed at right. Patients who harbored multiple variants were annotated based on their highest priority variant, with all additional pVUS listed in brackets. Parentheses indicate number of patients. Underline indicates experimental support of pathogenicity. a m.3243A>G; b m.8344A>G; c m.8993T>G/C; d m.3643_15569del; e homozygous; f compound heterozygous (phased); g potential compound heterozygous (unphased); h heterozygous in female; i not in MitoCarta. DDx = differential diagnosis; mito. = mitochondrial; mtDNA = mitochondrial DNA.
Parental sequencing.
To assess whether de novo mutations could be a major contributor to sporadic disease, we performed MitoExome sequencing on parental DNA from 5 probands without family history of disease. No confirmed de novo mutations were identified.
Prioritized recessive and X-linked variants in genes not previously linked to disease.
We next explored whether disease-causing mutations may lie in 1,283 nuclear-encoded candidate genes associated with the mitochondrion but not previously linked to disease.
If candidate genes truly underlie disease, we might expect an increased number of predicted deleterious variants in patients compared with healthy individuals (figure e-3). We observed modest 2-fold enrichment for likely recessive variants (p < 0.05) and 7-fold enrichment for hemizygous X-linked variants in male patients (p < 0.01). No significant enrichment was observed for prioritized heterozygous variants, X-linked variants in females, or synonymous variants.
Altogether, 17 candidate genes not previously linked to mitochondrial disease harbored autosomal recessive or hemizygous X-linked variants (figure 2, table e-5). We emphasize that because of the modest enrichment over background, only a subset is expected to be causal. Further genetic and experimental studies are needed to establish pathogenicity.
Pathogenicity of ATP5A1 mutations in a yeast model.
In one such case, we investigated prioritized variants detected in an infant with combined oxidative phosphorylation (OXPHOS) deficiency and mtDNA depletion (figure 3). Patient 1020, born to consanguineous first-cousin parents of Moroccan descent, presented at birth with microcephaly, pulmonary hypertension, and heart failure requiring extracorporeal membrane oxygenation, hypotonia, hyperalaninemia, and bacteremia. She died at the age of 3 months. Muscle tissue showed decreased ETC activity (22% complex I + III and 30% complex IV activity) and mtDNA depletion (42% of controls in muscle, 39% in liver). The patient had 1 healthy sister and 1 sister who died at 15 months with a similar clinical presentation and combined OXPHOS deficiency in muscle, consistent with a recessive disorder.
Figure 3. Modeling ATP5A1 mutations in yeast.

(A) Family pedigree and genotype at ATP5A1:c.962A>G. (B) Electron transport chain (ETC) biochemistry and mitochondrial DNA (mtDNA) quantitation in muscle. (C) Schematic of ATP5A1 protein, with sequence alignment shown in inset. Vertical bars indicate patient mutation (red) and yeast mitochondrial genome integrity (mgi) mutations (black). (D) Yeast atp1 deletion strains show normal (red) and petite (white) colonies. (E) Mitochondrial physiology of yeast deletion strains report mean values ± SD across 3 replicates. *p < 0.01, 1-tailed t test. ↓ = 20%–30% activity relative to citrate synthase;↓↓ = <20%; ATPase = adenosine triphosphatase; d. = deceased; na = not available; nd = not determined; P/O = phosphate/oxygen; RCR = respiratory control rate.
MitoExome sequencing revealed a homozygous ATP5A1:c.962A>G (p.Y321C) variant, which was not present in 1000 Genomes or in the Exome Variant Server, and introduced a missense mutation at a residue evolutionary conserved to bacteria (figure 3B). The variant was homozygous in the affected sister and heterozygous in their mother. DNA from the father and unaffected sister was unavailable. The only other homozygous variant prioritized in the proband (HAO2:p.D259E) did not segregate with disease in the family.
Because cultured patient fibroblasts did not survive lentiviral transduction required for human complementation studies, we modeled the ATP5A1:p.Y321C mutation in yeast. This mutation lies within a highly conserved region of the protein associated with mitochondrial genomic integrity (mgi) in yeast; nearby mutations result in decreased coupling of the adenosine triphosphate (ATP) synthase and lead to a high rate of mtDNA loss (figure e-4).24 We expressed the analogous yeast variant (ATP1:p.Y315C) in an atp1 knockout strain and observed a 3-fold increase in petite frequency, reflective of the rate of mtDNA loss, and a 2-fold decrease of mitochondrial membrane potential (p < 0.01, t test) compared with expression of wild-type ATP1 (figure 3D), consistent with reported mgi mutants.24
Collectively, the familial segregation, severe amino acid change of a highly conserved residue, and increased rate of mtDNA loss in a yeast model consistent with the patient phenotype strongly support the pathogenicity of this ATP5A1 mutation.
Predictors of diagnostic efficacy.
We investigated whether any patient features correlated with the rate of diagnostic success, including family history, age of onset, and ETC deficiency. We observed significant enrichment of molecular diagnoses only in cases with clear family history (table e-7).
Cohort comparison.
Lastly, we compared the rate of molecular diagnosis from this study and our previous sequencing study of 42 infants with severe, biochemically proven OXPHOS disease.13 We limited the analysis to cases without prior molecular diagnoses, and only focused on the subset of 1,034 genes that were analyzed in both studies.13 Compared with the infantile cohort, the 84 MGH patients with suspected mitochondrial disorders contained markedly fewer new diagnoses and prioritized recessive variants in mitochondrial proteins not yet linked to disease (figure 4). Similar differences were observed when considering all patients, including those with prior molecular diagnoses (figure e-5).
Figure 4. New molecular diagnoses and prioritized recessive genes.
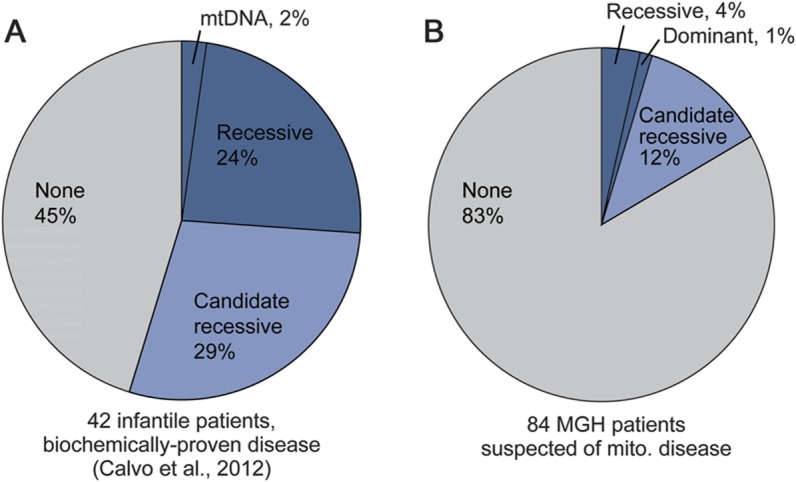
Comparison of MitoExome analysis from our (A) previous study13 and (B) current study, showing the fraction of patients with firm molecular diagnoses (dark blue) and autosomal recessive candidate genes (light blue). Comparison is restricted to the subset of patients who lacked molecular diagnosis before MitoExome sequencing and to the 1,034 genes analyzed in both studies (thereby excluding 212 differential diagnosis genes from the current study). MGH = Massachusetts General Hospital; mito. = mitochondrial; mtDNA = mitochondrial DNA.
DISCUSSION
We assessed the efficacy of targeted NGS for molecular diagnosis in 102 patients with a suspected mitochondrial disorder, providing new molecular insights into the genetic basis of mitochondrial disease and lessons for incorporating NGS in the clinic. Unlike our previous study of biochemically proven infantile cases, the current study includes many patients with late-onset presentations and uncertain clinical diagnoses. MitoExome sequencing recovered 94% of prior molecular diagnoses and yielded molecular diagnoses for 7% of patients refractory to traditional genetic testing, less than a third the diagnostic success rate (24%) observed in our previous study.13 Two major possibilities exist to explain the lower yield. First, milder cases may be attributable not to severe, recessive, coding mutations, but perhaps to variants with incomplete penetrance, regulatory variants, or genetic interaction of weaker alleles. Second, a subset of our MGH patients may not actually have primary mitochondrial disease, despite clinical and biochemical features used in standard diagnostic algorithms.
It is notable that 2 proteins implicated in disease localize outside of mitochondria, which may help delineate pathways of crosstalk between mitochondria and other cellular compartments. Mutations in WFS1, a protein localized to the endoplasmic reticulum, have been previously linked with mtDNA deletions.37 DPYD, a cytosolic enzyme in pyrimidine metabolism also responsible for catabolism of the chemotherapeutic drug 5-fluorouracil, has not been linked to mitochondrial function, but defects in 2 adjacent metabolic enzymes cause mtDNA depletion syndromes: TYMP (OMIM #603041) and TK2 (OMIM #609560). Further studies are needed to explore whether mitochondrial defects are typical in DPD deficiency.
Our study identified 26 patients with prioritized variants of unknown significance in known or candidate disease genes. In one case, we provide evidence that recessive mutations in ATP5A1, the α subunit of the F1–Fo ATP synthase, can underlie combined OXPHOS deficiency. This is the second nuclear-encoded complex V subunit linked to disease.38 Our study suggests that mutations in this complex may have a secondary impact on mtDNA homeostasis and the respiratory chain (appendix e-2).
A number of lessons have emerged from our study that may be useful as NGS is introduced into the clinic. First, targeted exome sequencing is a cost-effective and time-efficient alternative to traditional, sequential testing of the mtDNA and individual nuclear genes. For a subset of patients, a firm molecular diagnosis can be established quickly in a minimally invasive manner. Prospective studies will be required to determine the efficacy of such technology as a first-line diagnostic test. Second, our study underscores the phenotypic overlap between mitochondrial disorders and other genetic syndromes, and as costs decrease, argues for whole exome or whole genome sequencing. Third, when possible, we advocate familial sequencing to facilitate phasing of haplotypes, detection of de novo variants, and filtering of candidate variants. Fourth, our study underscores the need for guidelines for interpretation of NGS data for mitochondrial disorders. Larger databases of healthy and morbid genomes will aid in future interpretation, and to this end, our data have been deposited into dbGaP. Finally, DNA sequence must not be interpreted alone, but rather in the context of the broader clinical picture. Principled methods combining genomic, biochemical, and clinical data will be required to usher NGS into the clinical arena.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Winnie Xin, Rosemary Kiely, Mary Anne Anderson, and Tammy Gillis for technical assistance; Khalid Shakir, David Jaffe, Mark DePristo, and the Broad GATK team for assistance with data analysis; Irina Anselm for clinical assistance; John Walker, David Holtzman, Elaine Lim, and Scott Vafai for discussion; Mark Daly, Benjamin Neale, the National Institute of Mental Health (NIMH) Schizophrenia Genetics Initiative, and the NIMH Control Sample repository for control exome data. Finally, the authors thank the patients and their families for their participation.
GLOSSARY
- AF
allele frequency
- ATP
adenosine triphosphate
- DPD
dihydropyrimidine dehydrogenase
- ETC
electron transport chain
- MGH
Massachusetts General Hospital
- mgi
mitochondrial genome integrity
- mtDNA
mitochondrial DNA
- NGS
next-generation sequencing
- OMIM
Online Mendelian Inheritance in Man
- OXPHOS
oxidative phosphorylation
- pVUS
prioritized variant of unknown significance
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Study design: S.E.C., V.K.M. Sequence data generation: S.L., B.A.C., M.L.B. Sequence data analysis: D.S.L., S.G.H., N.B.G. Variant confirmation: D.S.L. Patient registry and clinical assessment: N.G.S., D.R.T., G.T.B., J.D.S., K.B.S. Biological experiments: K.S., D.M.M. Manuscript preparation: D.S.L., S.E.C., V.K.M.
STUDY FUNDING
This work was supported by an NSF Graduate Research Fellowship Program grant (D.S.L.), a grant from NIH R01GM66223 (D.M.M.), and American Recovery and Reinvestment Act (ARRA) funds through grant number RC2HG005556 (V.K.M.) from the National Human Genome Research Institute, NIH.
DISCLOSURE
M. Borowsky is currently an employee at Novartis, although his contributions to this manuscript were made at the time he was a full-time employee at Massachusetts General Hospital. The remaining authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.DiMauro S, Hirano M, Schon EA. Mitochondrial Medicine. New York: Informa Healthcare; 2006 [Google Scholar]
- 2.Chinnery PF. Mitochondrial disorders overview. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle: University of Washington; 1993 [Google Scholar]
- 3.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 2012;366:1132–1141 [DOI] [PubMed] [Google Scholar]
- 4.Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature 2012;491:374–383 [DOI] [PubMed] [Google Scholar]
- 5.Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med 2005;37:222–232 [DOI] [PubMed] [Google Scholar]
- 6.Chinnery PF, Turnbull DM. Epidemiology and treatment of mitochondrial disorders. Am J Med Genet 2001;106:94–101 [DOI] [PubMed] [Google Scholar]
- 7.Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology 1999;52:1255–1264 [DOI] [PubMed] [Google Scholar]
- 8.Finsterer J, Jarius C, Eichberger H. Phenotype variability in 130 adult patients with respiratory chain disorders. J Inherit Metab Dis 2001;24:560–576 [DOI] [PubMed] [Google Scholar]
- 9.Vasta V, Ng S, Turner E, Shendure J, Hahn SH. Next generation sequence analysis for mitochondrial disorders. Genome Med 2009;1:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker EJ, Hershman SG, Kohrer C, et al. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab 2011;14:428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasta V, Merritt JL, II, Saneto RP, Hahn SH. Next-generation sequencing for mitochondrial diseases reveals wide diagnostic spectrum. Pediatr Int 2012;54:585–601 [DOI] [PubMed] [Google Scholar]
- 12.Calvo SE, Tucker EJ, Compton AG, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet 2010;42:851–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012;4:118ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayr JA, Haack TB, Graf E, et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am J Hum Genet 2012;90:314–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet 2012;49:277–283 [DOI] [PubMed] [Google Scholar]
- 16.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL' caused by EARS2 mutations. Brain 2012;135:1387–1394 [DOI] [PubMed] [Google Scholar]
- 17.Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008;134:112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009;27:182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008;456:53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruiz-Pesini E, Lott MT, Procaccio V, et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 2007;35:D823–D828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingman M, Gyllensten U. mtDB: human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res 2006;34:D749–D751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: 2008 update. Genome Med 2009;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The 1000 Genomes Project Consortium; Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Singh U, Mueller DM. Mitochondrial genome integrity mutations uncouple the yeast Saccharomyces cerevisiae ATP synthase. J Biol Chem 2007;282:8228–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology 2006;67:1823–1826 [DOI] [PubMed] [Google Scholar]
- 26.Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002;59:1406–1411 [DOI] [PubMed] [Google Scholar]
- 27.Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 2008;10:294–300 [DOI] [PubMed] [Google Scholar]
- 28.Shanske S, Pancrudo J, Kaufmann P, et al. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A 2004;130A:134–137 [DOI] [PubMed] [Google Scholar]
- 29.Craig K, Young MJ, Blakely EL, et al. A p.R369G POLG2 mutation associated with adPEO and multiple mtDNA deletions causes decreased affinity between polymerase gamma subunits. Mitochondrion 2012;12:313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young MJ, Longley MJ, Li FY, Kasiviswanathan R, Wong LJ, Copeland WC. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum Mol Genet 2011;20:3052–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuelke M, Smeitink J, Mariman E, et al. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet 1999;21:260–261 [DOI] [PubMed] [Google Scholar]
- 32.Al-Sanna'a NA, Van Kuilenburg AB, Atrak TM, Abdul-Jabbar MA, Van Gennip AH. Dihydropyrimidine dehydrogenase deficiency presenting at birth. J Inherit Metab Dis 2005;28:793–796 [DOI] [PubMed] [Google Scholar]
- 33.Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995;346:1458–1463 [DOI] [PubMed] [Google Scholar]
- 34.McLaughlin HM, Sakaguchi R, Liu C, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 2010;87:560–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maring JG, van Kuilenburg AB, Haasjes J, et al. Reduced 5-FU clearance in a patient with low DPD activity due to heterozygosity for a mutant allele of the DPYD gene. B J Cancer 2002;86:1028–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lieber DS, Vafai SB, Horton LC, et al. Atypical case of Wolfram syndrome revealed through targeted exome sequencing in a patient with suspected mitochondrial disease. BMC Med Genet 2012;13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rotig A, Cormier V, Chatelain P, et al. Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 1993;91:1095–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayr JA, Havlickova V, Zimmermann F, et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet 2010;19:3430–3439 [DOI] [PubMed] [Google Scholar]
- 39.Cano A, Rouzier C, Monnot S, et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A 2007;143A:1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence and phenotype data are available in dbGaP, accession phs000339.