

Abstract
Objective:
We examined the clinical and pathologic phenotypes of GRN mutation carriers with the pathogenic A9D (g.26C>A) missense mutation.
Methods:
Three patients with GRN A9D mutations were evaluated clinically and came to autopsy with subsequent neuropathologic examination.
Results:
The clinical diagnoses of patients with GRN A9D mutations were amyotrophic lateral sclerosis, atypical extrapyramidal disorder, and behavioral variant frontotemporal dementia. Immunohistochemistry for TAR DNA-binding protein 43 (TDP-43) revealed variability in morphology and distribution of pathology. One patient had notable involvement of motor neurons in the spinal cord as well as type B TDP-43, whereas 2 other patients had type A TDP-43.
Conclusions:
The clinical presentation of the GRN A9D missense mutation is not restricted to behavioral variant frontotemporal dementia and may include aphasia, extrapyramidal features, and, notably, amyotrophic lateral sclerosis.
Mutations in the progranulin (GRN) gene are a common cause of frontotemporal lobar degeneration (FTLD), with mutations identified in up to 10% of patients with FTLD.1–3 Behavioral variant frontotemporal dementia (bvFTD) and primary progressive aphasia are the most frequent clinical presentations, but parkinsonism, corticobasal syndrome, and amnestic syndrome suggesting Alzheimer disease have also been reported.4 In contrast, pathology associated with GRN mutations is relatively homogeneous, and cortical atrophy tends to be most severe in frontal lobes.5,6 Atrophy of the parietal lobe and neuronal loss in the substantia nigra occur more often than in FTLD without GRN mutations. TAR DNA-binding protein 43 (TDP-43) is the major pathologic protein in inclusions of FTLD,7 and GRN mutation carriers have a highly consistent pattern of TDP-43 histopathology.5,6 The combined experience of investigators at a number of centers where FTLD is regularly seen has led to a harmonized classification system for TDP-43 pathology, with GRN mutation carriers usually exhibiting type A pathology.8
The majority of GRN mutations promote premature termination of the coding sequence, which results in haploinsufficiency due to nonsense-mediated RNA decay.1,2 An exception is the A9D (g.26C>A) missense mutation, which results in haploinsufficiency due to loss of GRN secretion.9 Additional missense, silent, and intronic mutations have unknown pathogenicity or have been identified in controls and are not considered pathogenic (www.molgen.ua.ac.be/FTDMutations). A few missense mutations of unknown pathogenicity have been reported in patients with amyotrophic lateral sclerosis (ALS) and Alzheimer disease,10,11 suggesting that GRN variants may increase risk for diseases other than FTLD. Herein, we report that the GRN A9D mutation causes clinical and pathologic heterogeneity, which includes features of ALS.
METHODS
Standard protocol approvals, registrations, and patient consents.
The clinical and neuropathologic studies in this report were approved by the Mayo Clinic institutional review board, and informed consent was obtained from all participants, including authorization to disclose video recording. Autopsies were performed after informed consent by the legal next of kin.
Genetic screening.
GRN sequencing was performed as previously described.1 All cases were positive for the A9D (g.26C>A) mutation in GRN. Screening for C9ORF72 hexanucleotide repeats was performed as previously described.12
Pathologic analysis.
Tissue from frontal (including precentral gyrus), temporal, parietal, and occipital neocortex, in addition to hippocampus, basal ganglia, thalamus, midbrain, pons, medulla, and cerebellum were reviewed. If available, spinal cord and skeletal muscle were also studied. Histologic methods used to characterize the cases included hematoxylin & eosin stain, Gomori trichrome stain, Bielschowsky silver stain, and thioflavin-S fluorescent microscopy. Immunohistochemistry for ubiquitin (Ubi-1, 1:60,000; Millipore, Billerica, MA) and phospho-TDP-43 (ps409/410, 1:5,000; Cosmobio Co., Tokyo, Japan) was used to detect pathologic inclusions. Antibodies detecting ionized calcium-binding adaptor molecule 1 (IBA1, 1:3,000; Wako Chemicals, Richmond, VA), glial fibrillary acidic protein (GFAP, 1:5,000; Biogenex, Femont, CA), fused in sarcoma (1:250; Sigma, St. Louis, MO), superoxide dismutase-1 (1:10,000; a gift from David Borchelt, University of Florida), and α-synuclein (NACP, 1:3,000)13 were used in a subset of sections.
RESULTS
Clinical descriptions.
Case 1.
At age 67 years, this woman developed rapidly progressive walking difficulties and nearly continuous muscle twitching of her thigh muscles with a disease course of less than 2 years. She had initial difficulty with balance and control of her legs, and at age 68, she had a fall without head injury. She developed trouble expressing herself, with soft, dysarthric and repetitive speech that over the course of a year became unintelligible. She tended to repeat words and phrases over and over, and repeated nonsequitur words and phrases with literal paraphasic errors. Despite her expressive language difficulty, she appeared able to understand what was said to her. She lost facial expression, blink rate was reduced, and she developed dysphagia. She would keep food in her mouth without swallowing, and she had choking episodes. She lost 30 pounds over 3 months. She was unable to dress or bathe herself. She was continent of urine and stool.
Formal neurologic examination performed 10 days before her death was notable for repetitious speech with paraphasia, yet intact comprehension and preserved recognition of current personal and family information. She was easily distracted, tended to reach for nearby stimuli (stimulus-bound behavior), and exhibited perseveration (verbal and motor) and echopraxia. On the Mini-Mental State Examination, she scored 9 of 30 points. She scored below the first percentile on both the Boston Naming Test Short Form and the Token Test, but she had relatively intact recognition memory (9/10 correct with 0 false-positive errors) on the CERAD List Learning Test.
On neurologic examination, she had hypomimia, reduced blink rate, and hypophonia. Eye movements were reduced in all directions on voluntary gaze. She had slow saccades, but normal oculocephalic reflexes. Deep tendon reflexes, including jaw jerk, were hyperactive, and there was a left extensor plantar response. Her muscle tone had features of spasticity and rigidity. She had difficulty standing without assistance. She was able to take a few steps, but walking was difficult because of leg weakness and bilateral foot drop. Widespread fasciculations were observed (see video).
Nerve conduction studies showed mildly slow conduction velocities and reduced compound muscle action potential amplitudes, particularly in the lower extremities. Needle EMG examination demonstrated widespread fibrillation and fasciculation potentials and signs of chronic partial motor denervation and reinnervation. Brain MRI showed focal right temporal pole and subcortical volume loss (figure 1A). 18F-fluorodeoxyglucose PET revealed asymmetrical (right worse than left) focal cortical hypometabolism in temporal lobes (figure 1B). Genetic analysis detected the A9D missense mutation in GRN (figure 1C).
Figure 1. Case 1 brain imaging and genetic studies.
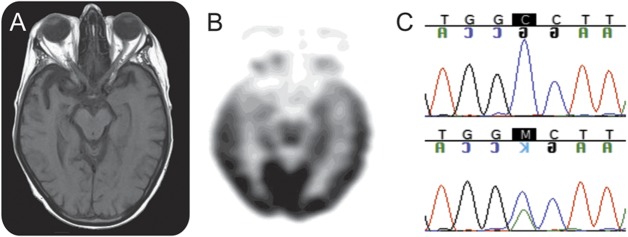
(A) T1-weighted axial MRI performed on 1.5 tesla shows focal volume loss in the right temporal lobe. (B) 18F-fluorodeoxyglucose PET shows focal cortical hypometabolism in the bilateral temporal lobes, predominantly left side. (C) Chromatogram of a normal control (top) and case 1 (bottom) displaying the A9D mutation.
Her antemortem clinical diagnosis was ALS with evidence of frontal executive dysfunction on neuropsychological studies. She died of peritonitis and acute renal failure due to embolism in the mesentery and renal arteries.
Cases 2 and 3.
Two additional A9D mutation carriers are also described briefly. Case 2 was an older sister of case 1 and has been previously reported.14 She had an atypical extrapyramidal disorder characterized by progressive dysarthria, apractic eye movements, motor impersistence, bradykinesia, and buccal myorhythmia initially thought to be due to Whipple disease. She did not respond to carbidopa/levodopa. Her disease course was about 2 years. In the end, she had major cognitive and motor impairment, she was nearly mute, and she required assistance for instrumental activities of daily living. Autopsy showed striatal gliosis with neuronal inclusions with TDP-43 immunohistochemistry, as well as cortical involvement consistent with type A.14
Case 3 was a 56-year-old male accountant who presented with poor job performance that forced him to change jobs. His difficulties were attributed to memory difficulties, but he also had difficulty paying bills, handling his checkbook, as well as significant visuospatial problems, apathy, dressing apraxia, and akathisia. He had little insight, and he improvised knowledge of his family and current events. He had perseverative behavior. His neurologic examination was normal, but he had disorientation to place and time, frontal executive dysfunction, and mild memory impairment (recalling 2 of 3 items after delay). He also demonstrated aphasia, including word-finding difficulties with frequent perseveration, plus acalculia and construction apraxia. He died after 11 years of a slowly progressive illness. At autopsy, he had severe brain atrophy affecting frontal and parietal lobes, with less involvement of temporal lobes. There was also striatal atrophy. He had TDP-43 pathology consistent with type A.
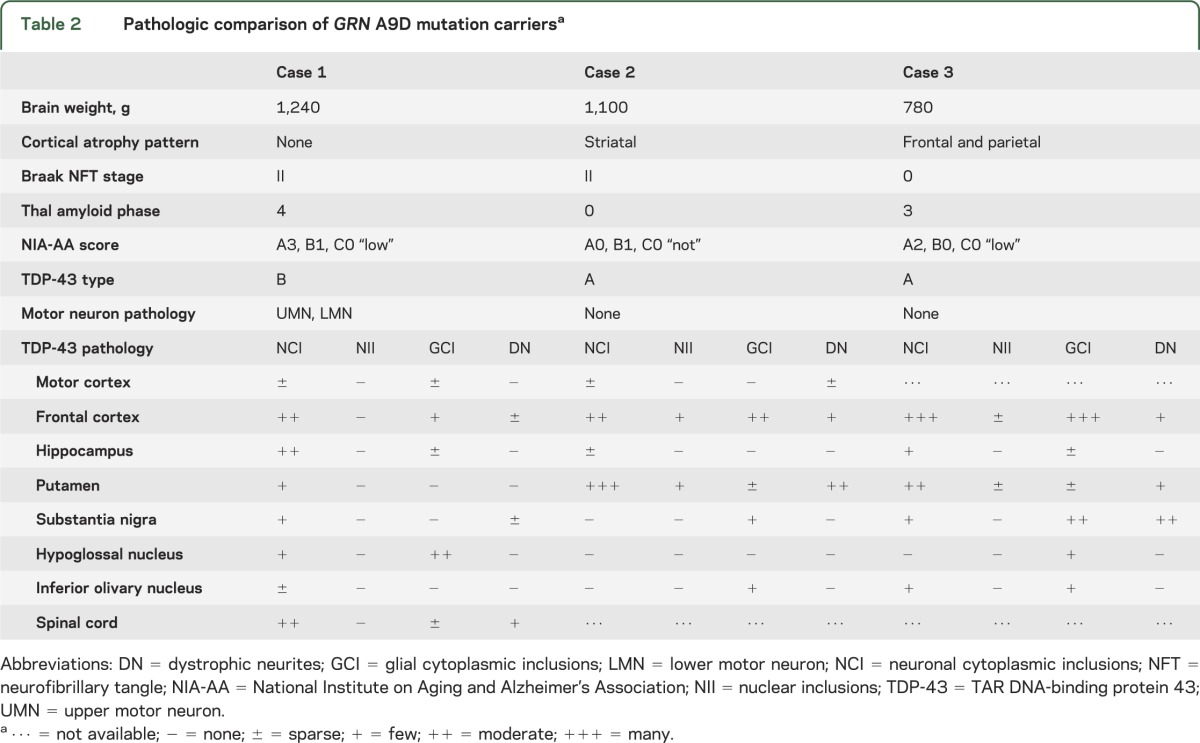
A comparison of clinical features of these 3 cases reveals significant heterogeneity (table 1). All cases had different initial symptoms and final clinical diagnoses. Furthermore, MAPT haplotype and ApoE genotypes do not correlate with clinical manifestations. All cases were negative for C9ORF72 hexanucleotide repeat expansion that has been associated with ALS and FTLD.
Table 1.
Clinical and genetic comparison of GRN A9D mutation carriers
Neuropathologic findings of case 1.
The whole-brain weight, calculated from the fixed left hemibrain, was 1,240 g. Macroscopic examination of the cerebrum, brainstem, and cerebellum showed no atrophy or ventricular enlargement, and the demarcation between gray and white matter was sharp (figure e-1, A and B, on the Neurology® Web site at www.neurology.org). The only remarkable macroscopic finding was mild decreased pigmentation in the substantia nigra and locus ceruleus (figure e-1, C and D). Examination of spinal cord revealed diffuse atrophy and poor demarcation between gray and white matter.
Histologic analyses revealed the most significant abnormalities in the spinal cord and medulla, where there was neuronal loss affecting motor neurons as well as reactive astrocytosis and microgliosis (figure 2, A–D). Chromatolytic neurons and axonal spheroids were identified, as well as Bunina bodies and Lewy body–like hyaline inclusions (figure 2A, inset). Myelinated fiber loss and microgliosis were present in the anterior and lateral funiculi of the spinal cord and the pyramid of the medulla, consistent with corticospinal tract degeneration. Examination of the muscle demonstrated neurogenic atrophy with groups of small, acutely angulated fibers (figure 2, F and G). TDP-43 immunohistochemistry revealed Lewy body–like and skein-like neuronal cytoplasmic inclusions (NCI) in the anterior horn cells of the spinal cord (figure 2, H and I). Sparse TDP-43–positive NCI and glial cytoplasmic inclusions (GCIs) were also detected in the motor cortex (figure 2J and table 2), where there was mild depletion of Betz cells, scattered macrophages, and vacuolation of subjacent white matter. The predominance of NCI and paucity of dystrophic neurites was consistent with type B. The pattern of upper and lower motor neuronal loss with corticospinal tract degeneration and type B TDP-43 pathology was characteristic of ALS.
Figure 2. Pathologic assessment of case 1.

(A) Hematoxylin & eosin staining of anterior horn spinal cord tissue reveals Lewy body–like hyaline inclusions (inset). (B and C) Immunohistochemical staining of spinal cord tissue for GFAP (B) and IBA1 (C) shows gliosis. (D and E) Immunohistochemical staining of medulla for GFAP (D) and IBA1 (E) shows moderate gliosis. (F and G) Erector spinae muscle with trichrome stain demonstrates neurogenic atrophy of muscle fibers. (G) Higher magnification shows a cluster of angulated fibers. (H–J) Immunohistochemical staining for phosphorylated TDP-43 reveals Lewy body–like inclusions in anterior horn cell (H), skein-like inclusions in anterior horn cell (I), and neuronal cytoplasmic inclusions in Betz cells of motor cortex (J). Bar represents 100 μm. GFAP = glial fibrillary acidic protein; IBA1 = ionized calcium-binding adaptor molecule 1; TDP-43 = TAR DNA-binding protein 43.
Table 2.
Pathologic comparison of GRN A9D mutation carriersa
To exclude possible concomitant neurodegenerative disease processes, a battery of special stains was performed. Senile changes of Alzheimer type, as seen by thioflavin-S fluorescent microscopy, included nonneuritic (diffuse) amyloid deposits in corticolimbic regions (Thal phase 4) and mild medial temporal neurofibrillary degeneration (Braak NFT [neurofibrillary tangle] stage II; table 2 and figure e-2A). Furthermore, fused in sarcoma, superoxide dismutase-1, and α-synuclein immunostaining were negative (figure e-2, B–D), suggesting that TDP-43 was the major pathologic protein.
Because the neuropathology was unusual for a GRN mutation carrier, we compared cortical and brainstem TDP-43 pathology with the 2 other cases with the same A9D mutation (table 2 and figure 3), including the previously reported sister. The middle frontal cortex had some of the highest levels of TDP-43 pathology (figure 3). The frontal cortex of case 1 mainly consisted of “preinclusions” with diffuse granular cytoplasmic immunoreactivity, in addition to NCI and GCI, but neuronal intranuclear inclusions were absent (figure 3). This is consistent with type B TDP-43 pathology. In contrast, cases 2 and 3 exhibited NCI, intranuclear inclusions, GCI, and dystrophic neurites in the upper cortical layers (figure 3), consistent with type A TDP-43 pathology. Few inclusions were detected in the hippocampus of all cases, but case 1 had moderate numbers of neuronal preinclusions, particularly in the dentate fascia. Case 2 had the most inclusions in the putamen compared with all other regions examined and much higher than the other cases (figure 3). Moderate levels of TDP-43 pathology were detected in the substantia nigra of case 3, but less frequently in the other cases. In general, the medulla had low levels of TDP-43 pathology in all cases (figure 3). Unique to case 1 was the presence of TDP-43 pathology in the hypoglossal nucleus. Overall, both the TDP-43 distribution and morphology were heterogeneous in the GRN A9D mutation carriers, with a striking difference between the 2 siblings. Specifically, case 1 had motor neuron disease and case 2 had prominent striatal pathology. Of note, the National Institute on Aging and Alzheimer's Association composite score for Alzheimer pathology reveals a “low” score for cases 1 and 3, indicating low levels of Alzheimer pathology, and “none” for case 2 (table 2).
Figure 3. TDP-43 pathology comparison of GRN A9D mutation carriers.

Immunohistochemistry for phosphorylated TDP-43 in the middle frontal gyrus, putamen, and hypoglossal nucleus of case 1, case 2, and case 3. Bar represents 100 μm. TDP-43 = TAR DNA-binding protein 43.
DISCUSSION
We have demonstrated that a GRN mutation can present with clinical and pathologic features of ALS, and that the GRN A9D mutation has significant heterogeneity. In addition to ALS, case 1 also had neuropsychologic deficits consistent with aphasia, executive dysfunction, and mild parkinsonism. Clinical presentations of the other 2 cases with A9D mutations in progranulin included bvFTD and an atypical extrapyramidal syndrome. Neither of the other 2 had clinical or neuropathologic evidence of motor neuron disease, although an EMG examination was not performed. Other reports of GRN A9D mutation carriers have described patients with primary progressive aphasia,15 corticobasal syndrome,16 and hereditary dysphasic disinhibition dementia,17 but ALS features were never documented in any of these reports. The evidence to date suggests that although bvFTD is not a common clinical presentation of A9D mutation, it is the predominant phenotype of other GRN mutations. Furthermore, the A9D mutation is highly pleiotropic. Pleiotropy usually occurs when a single gene affects multiple traits; this often occurs with genes associated with ALS and FTLD (figure e-3), suggesting that genetic etiology does not necessarily predict clinical presentation. However, a single mutation affecting multiple traits like the GRN A9D mutation is unusual, but not without precedence. For example, MAPT mutations normally cause frontotemporal dementia with parkinsonism, but individuals with N279K and P301L mutations may also present with clinical features suggestive of progressive supranuclear palsy or corticobasal degeneration.18,19
Pathologic assessment of case 1 demonstrated that motor neurons in the brain and spinal cord were most severely affected by neuronal loss and TDP-43 inclusions. TDP-43 inclusions were also present in extramotor regions and were consistent with the type B subtype of TDP-43 pathology.8 In contrast, motor regions were relatively spared in cases 2 and 3 and represented type A TDP-43 pathology, consistent with prior reports of pathology associated with GRN mutations. Taken together, GRN mutations normally exhibit type A TDP-43 pathology; however, the A9D mutation does not necessarily conform to characteristic TDP-43 distribution and morphology.
The mechanism of this clinical and pathologic heterogeneity in A9D carriers is unknown. All other pathogenic GRN mutations cause a premature stop codon that results in nonsense-mediated mRNA decay. The A9D mutation is a pathogenic missense mutation that produces, and retains, its transcript; however, the resulting progranulin protein cannot be secreted to perform its normal functions.13 We can postulate that gene alterations or differences in the expression level of progranulin receptors such as sortilin 1 may influence progranulin function.20 An alternative hypothesis for A9D clinical and pathologic heterogeneity would be unidentified genetic modifiers, which could include known ALS-associated genes. Mutations in the most common ALS-associated genes, SOD1 and C9ORF72, were excluded in the current study.
ALS and FTLD are considered part of a clinicopathologic disease spectrum and have been genetically linked by C9ORF72 gene abnormalities. Our findings demonstrate that ALS must also be included in the range of phenotypes associated with mutations in GRN. Furthermore, this report illustrates the heterogeneity of GRN mutation carriers, specifically those with A9D mutations, which may be underrepresented.
Supplementary Material
ACKNOWLEDGMENT
The authors thank S.M. Traynor for assistance with logistics related to case 1 (Department of Neurology, Mayo Clinic, Jacksonville, FL; traynor.sharleen@mayo.edu).
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- bvFTD
behavioral variant frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- GCI
glial cytoplasmic inclusions
- GFAP
glial fibrillary acidic protein
- IBA1
ionized calcium-binding adaptor molecule 1
- NCI
neuronal cytoplasmic inclusions
- TDP-43
TAR DNA-binding protein 43
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
A. Cannon and S. Fujioka: drafting/revising the manuscript for content, analysis or interpretation of data. N.J. Rutherford: analysis or interpretation of data. T.J. Ferman: analysis or interpretation of data, drafting/revising the manuscript for content. D.F. Broderick: analysis or interpretation of data. K.B. Boylan: analysis or interpretation of data, drafting/revising the manuscript for content. N.R. Graff-Radford, R.J. Uitti, and R. Rademakers: analysis or interpretation of data. Z.K. Wszolek and D.W. Dickson: drafting/revising the manuscript for content, study concept or design.
STUDY FUNDING
Z.K.W., R.J.U., and D.W.D. are partially supported by the NIH/National Institute of Neurological Disorders and Stroke Morris K. Udall Center awarded to the Mayo Clinic Jacksonville (P50 NS07218703).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–919 [DOI] [PubMed] [Google Scholar]
- 2.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006;442:920–924 [DOI] [PubMed] [Google Scholar]
- 3.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15:2988–3001 [DOI] [PubMed] [Google Scholar]
- 4.van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 2008;7:965–974 [DOI] [PubMed] [Google Scholar]
- 5.Forman MS, Mackenzie IR, Cairns NJ, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 2006;65:571–581 [DOI] [PubMed] [Google Scholar]
- 6.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 2007;66:142–151 [DOI] [PubMed] [Google Scholar]
- 7.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133 [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shankaran SS, Capell A, Hruscha AT, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 2008;283:1744–1753 [DOI] [PubMed] [Google Scholar]
- 10.Brouwers N, Sleegers K, Engelborghs S, et al. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 2008;71:656–664 [DOI] [PubMed] [Google Scholar]
- 11.Sleegers K, Brouwers N, Maurer-Stroh S, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 2008;71:253–259 [DOI] [PubMed] [Google Scholar]
- 12.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dickson DW, Liu W, Hardy J, et al. Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol 1999;155:1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wider C, Uitti RJ, Wszolek ZK, et al. Progranulin gene mutation with an unusual clinical and neuropathologic presentation. Mov Disord 2008;23:1168–1173 [DOI] [PubMed] [Google Scholar]
- 15.Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging 2009;30:739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spina S, Murrell JR, Huey ED, et al. Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol 2007;66:892–900 [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 2006;60:314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wszolek ZK, Slowinski J, Golan M, Dickson DW. Frontotemporal dementia and parkinsonism linked to chromosome 17. Folia Neuropathol 2005;43:258–270 [PubMed] [Google Scholar]
- 19.Wszolek ZK, Tsuboi Y, Farrer M, Uitti RJ, Hutton ML. Hereditary tauopathies and parkinsonism. Adv Neurol 2003;91:153–163 [PubMed] [Google Scholar]
- 20.Hu F, Padukkavidana T, Vaegter CB, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 2010;68:654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.