

Abstract
Oncolytic measles viruses (MV) derived from the live attenuated vaccine strain have been engineered for increased antitumor activity, and are currently under investigation in clinical phase 1 trials. Approaches with other viral vectors have shown that insertion of immunomodulatory transgenes enhances the therapeutic potency. In this study, we engineered MV for expression of the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF). For the first time, therapeutic efficacy and adaptive immune response in the context of MV oncolysis could be evaluated in the previously established immunocompetent murine colon adenocarcinoma model MC38cea. MC38cea cells express the human carcinoembryonic antigen (CEA), allowing for infection with retargeted MV. Intratumoral application of MV-GMCSF significantly delayed tumor progression and prolonged median overall survival compared with control virus-treated mice. Importantly, more than one-third of mice treated with MV-GMCSF showed complete tumor remission and rejected successive tumor reengraftment, demonstrating robust long-term protection. An enhanced cell-mediated tumor-specific immune response could be detected by lactate dehydrogenase assay and interferon-γ enzyme-linked immunospot assay. Furthermore, MV-GMCSF treatment correlated with increased abundance of tumor-infiltrating CD3+ lymphocytes analyzed by quantitative microscopy of tumor sections. These findings underline the potential of oncolytic, GM-CSF-expressing MV as an effective therapeutic cancer vaccine actively recruiting adaptive immune responses for enhanced therapeutic impact and tumor elimination. Thus, the treatment benefit of this combined immunovirotherapy approach has direct implications for future clinical trials.
Grossardt and colleagues engineer measles viruses (MV) for expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) and evaluate its therapeutic efficacy and immune response in an established immunocompetent murine colon adenocarcinoma model. Intratumoral application of MV-GMCSF delayed tumor progression and significantly prolonged median overall survival, with more than one third of mice showing complete tumor remission as well as resistance to tumor re-engraftment.
Introduction
Oncolytic viruses (OVs) that selectively target tumor cells represent a promising approach for the treatment of cancer. Various oncolytic viral platforms have been tested preclinically for safety and therapeutic efficacy and are now being evaluated in clinical trials (Russell et al., 2012). Vaccine strain measles viruses (MVs) have substantial antitumor activity in multiple tumor entities while causing minimal damage to nonmalignant cells. At present, oncolytic MVs are under investigation in clinical trials for the treatment of ovarian cancer, glioma, and multiple myeloma, and no dose-limiting toxicity has been observed so far (Galanis et al., 2010; Msaouel et al., 2012).
Nevertheless, viral monotherapy may have limited efficacy, especially in advanced-stage cancer. Therefore, OVs have been armed with transgenes, combined with radiotherapy, chemotherapy, and—more recently—immunotherapy for enhanced therapeutic potential. However, the interaction of OV with the immune system is a double-edged sword (Melcher et al., 2011): on the one hand, the immune system might rapidly eliminate infected tumor cells and inhibit replication of the virus necessary for spread within the tumor mass. On the other hand, destruction of tumor cells by OV may result in cross-priming of the immune system and lead to additional immune-mediated antitumor effects (Naik et al., 2011). Activation of the immune system might be further enhanced by arming OV with immunomodulatory proteins (Tong et al., 2012). One of the most commonly used transgenes is granulocyte-macrophage colony-stimulating factor (GM-CSF), which is known to regulate the differentiation of granulocytes and monocytes, to stimulate the proliferation and function of antigen-presenting cells (APCs) (van de Laar et al., 2012), and to enhance natural killer (NK) cell function (Richard et al., 1995). GM-CSF is a component of sipuleucel-T (Provenge; Dendreon, Seattle, WA), the first U.S. Food and Drug Administration (FDA)-approved immunotherapeutic to treat established cancer (Hammerstrom et al., 2011). It has also been introduced into several oncolytic viral vectors including adenovirus (Ad) (Choi et al., 2012), Newcastle disease virus (Janke et al., 2007), vaccinia virus (VV) (Heo et al., 2011), herpes simplex virus (HSV) (Harrington et al., 2010), and MV (Grote et al., 2003). Previous studies have shown that GM-CSF-armed adenovirus effectively induces a long-term antitumor response (Choi et al., 2012). Both GM-CSF-armed VV and HSV demonstrated promising therapeutic efficacy and are now being tested in clinical phase 3 trials (Breitbach et al., 2010). A detailed analysis of the interactions between MV and the immune system in the context of oncolytic therapy has been hampered by the fact that murine cells lack an MV receptor, allowing only the study of human tumor xenografts in immunodeficient mice. Even in severe combined immunodeficiency (SCID) mice, the expression of GM-CSF by MV resulted in improved tumor control that was explained by an increased neutrophil antitumor response (Grote et al., 2003). To study MV oncolysis in a fully immunocompetent host with a functional adaptive immune system, we have previously described a mouse model based on the murine colon carcinoma cell line MC38cea. These cells express human carcinoembryonic antigen and can therefore be infected with a recombinant MV retargeted to human carcinoembryonic antigen (CEA) (Ungerechts et al., 2007).
In this study, we use the MC38cea model to evaluate the therapeutic potential of recombinant MV expressing GM-CSF and targeted to CEA in a fully immunocompetent host. We demonstrate that treatment with MV-GMCSF delays tumor growth and mediates a relevant survival benefit in vivo, including complete tumor remission in a significant proportion of animals. Elevated levels of tumor-infiltrating T cells are associated with MV treatment. Mice in remission after MV-GMCSF treatment showed cell-mediated tumor-specific immunity and rejected reengraftment of MC38cea tumors. These findings highlight the potential to induce enduring tumor vaccination effects by combining oncolytic viruses and immunomodulation.
Materials and Methods
Cell culture
Vero African green monkey kidney cells were purchased from the American Type Culture Collection (Manassas, VA). Vero-αHis cells (kind gift of S.J. Russell, Mayo Clinic, Rochester, MN) were generated by stable transduction of the parental cell line for expression of a single-chain antibody against a hexahistidine (His6) tag (Nakamura et al., 2005). MC38cea cells (murine colon adenocarcinoma cells transgenic for human carcinoembryonic antigen) and parental MC38 cells were kindly provided by R. Cattaneo (Mayo Clinic, Rochester, MN). All cell lines were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Darmstadt, Germany) with 10% (w/v) fetal bovine serum (FBS; Invitrogen) in a humidified atmosphere of 5% CO2. All cell lines were routinely tested for mycoplasma contamination, using a multiplex cell contamination test (DKFZ Genomics and Proteomics Core Facility, Heidelberg, Germany) (Schmitt and Pawlita, 2009).
Generation and propagation of recombinant MV
MV genomic cDNA plasmids were constructed on the basis of pMV-EGFP carrying the measles virus Edmonston-B vaccine lineage strain (Bossow et al., 2011). The sequence containing the blinded H protein with CD46 and SLAM (signaling lymphocytic activation molecule)-ablating mutations and fused to the single-chain variable fragment against CEA (HαCEA) was isolated from pCG-HantiCEA by PacI–SpeI digestion and exchanged into pMV-EGFP, resulting in the full-length genomic plasmid pMV-EGFP-antiCEA. The murine GM-CSF open reading frame (ORF) was amplified from pUMVC1-GMCSF (kindly provided by V. Teichgräber, Heidelberg, Germany) by polymerase chain reaction (PCR), using primers providing the appropriate 5′-MluI and 3′-AsiSI restriction sites. The MluI–AsiSI-digested PCR fragment was exchanged into pMV-EGFP-antiCEA via the corresponding restriction enzymes, resulting in pMV-GMCSF-antiCEA. Recombinant MV particles were generated from cDNA constructs according to Radecke and colleagues (1995), and fully retargeted viruses were subsequently propagated on Vero-αHis cells (Nakamura et al., 2005). To prepare viral stocks, Vero-αHis cells were infected at a multiplicity of infection (MOI) of 0.03 and incubated at 32°C for 56 hr. Viral particles were harvested by one freeze–thaw cycle and centrifugation of cellular debris and resuspended in Opti-MEM (Invitrogen). All experiments were performed with viral stocks from the third passage. For titration of viruses in 96-well plates, serial 10-fold dilutions were performed in octuplicate and 104 Vero-αHis cells were added to each well. Seventy-two hours postinfection individual syncytia in all wells were counted, and titers were calculated as cell infectious units per milliliter (CIU/ml).
Virus infection
Cell culture supernatant was substituted by Opti-MEM (Invitrogen) containing viral particles at the indicated MOI. Cells were incubated at 37°C for 2 hr with gentle rocking every 15 min. Viral inoculum was replaced with fresh medium and cells were incubated until the indicated time points. Syncytium formation was monitored daily.
Viral growth characteristics
MC38cea and Vero-αHis cells (5×104) seeded in 12-well plates were infected in triplicate at an MOI of 3 with MV-EGFP, MV-EGFP-antiCEA, and MV-GMCSF-antiCEA, respectively. After a 2-hr adsorption, the inoculum was removed, cells were washed, and medium was renewed. At the indicated time points cells were scraped into their supernatants and progeny viral particles were titrated after one freeze–thaw cycle as described previously.
Determination of GM-CSF expression
For quantification of GM-CSF expression a conventional ELISA (R&D Systems, Wiesbaden, Germany) was performed. Vero-αHis and MC38cea cells (2×104 per well) were cultivated in 12-well plates and infected with MV-GMCSF-antiCEA or MV-EGFP-antiCEA at an MOI of 1. At the depicted time points postinfection, supernatants were harvested and equal volumes were analyzed. For in vivo quantification, mice were treated as described below and blood was collected via the vena saphena from mice 1 day before as well as 5, 10, and 15 days after the first MV application. Serum was prepared after clotting at 37°C for 1 hr and centrifugation at 3000×g for 10 min; equal volumes (20 μl) were analyzed.
Assessment of therapeutic efficacy in vivo
All animal experimental procedures were approved by the responsible animal protection officer at the German Cancer Research Center (Heidelberg, Germany) and by the regional council according to the German Animal Protection Law. Six- to 8-week-old C57BL/6 mice (Harlan Laboratories, Rossdorf, Germany) received 106 syngeneic colon adenocarcinoma MC38cea cells subcutaneously into the right flank. Mice were treated intratumorally on four consecutive days with MV (106 CIU per dose in 100 μl of Opti-MEM) as soon as tumors reached an average volume of 30–50 μl. To evaluate the therapeutic efficacy of engineered oncolytic MV, four groups were established receiving the carrier fluid Opti-MEM (n=10), ultraviolet (UV)-inactivated MV-GMCSF-antiCEA (UV-MV, n=10), MV-EGFP-antiCEA (n=10), or MV-GMCSF-antiCEA (n=9). Because of a treatment-unrelated incident, one mouse in the MV-GMCSF-antiCEA group died 31 days after tumor implantation. Thus, this mouse was not included in the Kaplan–Meier survival curve. For tumor rechallenge of mice with complete tumor remission, 105 MC38cea cells were injected subcutaneously into the opposite flank 10 weeks after the initial tumor implantation. Because mock- and control virus-treated animals died of tumor burden at earlier time points, 16- to 18-week-old naive C57BL/6 mice that received MC38cea for the first time were used as controls and tumor volumes were measured every third day. For the experiment with subsequent primary and secondary engraftment of tumors mock-treated mice (n=6), mice treated with MV-EGFP-antiCEA (n=5), and mice treated with MV-GMCSF-antiCEA (n=6) were rechallenged with 106 MC38cea cells 9 days after the first implantation. Tumor diameters were measured every third day with a caliper, and the volume of each tumor was calculated according to the following formula: (largest diameter)×(smallest diameter)2×0.5. The animals were sacrificed by cervical dislocation when tumors reached a volume of 1500 μl or ulceration occurred.
Lactate dehydrogenase assay
The cytolytic activity of splenocytes was determined by lactate dehydrogenase (LDH) assay (Promega, Mannheim, Germany). Spleens from mice treated with carrier fluid (n=7), UV-MV (n=4), MV-EGFP-antiCEA (n=8), or MV-GMCSF-antiCEA (n=9) were isolated aseptically 15 days postimplantation and passed through a 100-μm cell strainer (Becton Dickinson, Heidelberg, Germany) three times. Red blood cells were lysed with ACK lysing solution (Lonza, Basel, Switzerland). Splenocytes (3×107) were cultivated in 10 ml of RPMI 1640 supplemented with 10% FBS, 1% penicillin–streptomycin, and 50 μM 2-mercaptoethanol. For prestimulation, MC38cea cells were cultivated in the presence of mitomycin C (20 μg/ml; Sigma-Aldrich, Taufkirchen, Germany) for 2 hr, washed three times, transferred onto splenocytes, and cocultivated for 5 days in the presence of recombinant murine interleukin (IL)-2 (20 U/ml; Becton Dickinson). Subsequently, splenocytes (effector cells) were recovered and cultivated in quadruplicate at various ratios with MC38cea (target) cells. Effector-to-target (E:T) ratios at 50:1, 20:1, 10:1, 5:1, 1:1, and 0.5:1 were analyzed. LDH-based cytotoxic T lymphocyte (CTL) assays were performed according to the manufacturer's instructions. Briefly, after 4 hr of incubation supernatants were harvested and LDH activity was measured. Spontaneous target cell LDH release was evaluated by cultivation without effector cells, maximal target cell release was determined by adding lysis solution, and effector cell spontaneous release was analyzed after cultivation of splenocytes alone. Medium alone served as background control. The percentage of cytolytic activity was calculated as 100×(experimental−effectorspontaneous−targetspontaneous)/(targetmaximum−targetspontaneous).
Interferon-γ enzyme-linked immunospot assay
For determination of activated T cells, interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) assays (U-CyTech, Utrecht, The Netherlands) were performed. Splenocytes were isolated as described previously and cultivated for 24 hr in the presence of MC38cea cells and viral particles as stimuli, respectively. Subsequently, serial 2-fold dilutions of splenocytes (2×105, 105, 5×104, 2.5×104, and 1.25×104) were cultivated in anti-IFN-γ-precoated 96-well plates including stimuli in triplicate for an additional 24 hr at 37°C. The IFN-γ ELISPOT assay was performed according to the manufacturer's instructions. Briefly, cells were aspirated and washed three times with phosphate-buffered saline (PBS). Subsequently, biotinylated detection antibodies were added to each well and incubated at 4°C for 12 hr. Plates were washed three times with PBS containing 0.05% Tween 20. Addition of substrate solution allowing spot formation reveals the site of IFN-γ secretion. Colored IFN-γ spots were counted with an automated colony-counting device (Cellular Technology Limited, Bonn, Germany). Wells were normalized by comparing experimental spot development with negative control spots.
Immunohistochemical staining
Tissues were sectioned with a Vacutome (HM 550; Microm, Walldorf, Germany) and hemalaun staining of separate sections was done. Tissue specimens were immunohistochemically analyzed for their infiltration with CD3- and CD8-positive immune cells. The sections were dried and then fixed with paraformaldehyde (PFA) for 20 min (the latter followed by epitope retrieval with citrate for CD3). A rabbit monoclonal antibody recognizing mouse CD3 (clone SP7; Abcam, Cambridge, UK) and a rat monoclonal antibody recognizing CD8 (clone 53-6.7; BioLegend, London, UK) were used as primary antibodies applied at room temperature for 30 min. For CD3, a Bond polymer refine detection kit with anti-rabbit secondary antibody (kit from Novocastra, Berlin, Germany) and for CD8 a “rat on mouse” horseradish peroxidase (HRP)–polymer kit (Zytomed/Biocare Medical, Berlin, Germany) were used. Antigen detection was performed on the basis of a color reaction with 3,3′-diaminobenzidine (DAB chromogen; Leica, Berlin, Germany). The staining procedure was carried out with a BOND-MAX system (Leica). The sections were counterstained with hematoxylin (Bond refine detection kit; Leica) and mounted with Aquatex (Merck, Darmstadt, Germany).
Evaluation of immunohistochemical variables by quantitative microscopy
The number of stained immune cells was counted by a computerized image analysis system consisting of an NDP NanoZoomer (Hamamatsu Photonics, Hamamatsu City, Japan) attached to a personal computer as described previously (Halama et al., 2009). Complete microscopic images of full tissue sections were automatically obtained for later automatic and visual control (virtual microscopy). The invasive margin was defined as the border between tumor cells and adjacent skin tissue, and a band of 100 μm in each direction from the border was taken for analysis of the invasive margin region. High-precision quantification of cell densities in various cell compartments was performed as described previously (Halama et al., 2010), using the VIS software package (Visiopharm, Hørsholm, Denmark) to avoid observer bias. All evaluations were visually checked for consistency.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (version 5.04; GraphPad Software, San Diego, CA). Results represent means, with positive and negative error bars indicating the SD. Data were analyzed by two-sample t tests and analysis of variance (ANOVA) including Dunnett tests for pair-wise comparisons between groups. Survival data were analyzed by the Kaplan–Meier method and the log-rank test was used to analyze significance between treated groups. P values less than 0.05 were considered statistically significant.
Results
Generation and characterization of GM-CSF-armed and CEA-targeted MV
For targeted infection of murine adenocarcinoma MC38cea cells (Robbins et al., 1991), we have engineered MV vectors blinded for the natural receptors CD46 and SLAM and redirected against CEA via a single-chain antibody (Hammond et al., 2001) fused to the attachment protein H containing a C-terminal histidine hexapeptide (Fig. 1A). To implement immunotherapy, we generated recombinant MV encoding murine GM-CSF in an additional transcription unit upstream of the nucleocapsid gene, resulting in MV-GMCSF-antiCEA. In addition, MV encoding enhanced green fluorescent protein (EGFP) in combination with CEA-targeted H (MV-EGFP-antiCEA) and unmodified H (MV-EGFP) were generated as controls. When transgenic MC38cea and parental MC38 cells were inoculated with the engineered MV variants, CEA-positive MC38cea cells but not the parental CEA-negative MC38 cells were infected with the retargeted viral variants (data not shown), demonstrating CEA-dependent entry. Because MV naturally infects primate cells, murine cells support only limited replication of MV; syncytium formation on MC38cea cells was less pronounced compared with the MV producer cell line Vero-αHis (Nakamura et al., 2005). Levels of MV-mediated GM-CSF expression from infected cells were quantified by ELISA (Fig. 1B). Levels of GM-CSF secretion increased over time both in Vero-αHis and MC38cea cells infected with MV-GMCSF-antiCEA. In contrast, GM-CSF expression in cells infected with control virus was equal to GM-CSF basic levels in uninfected cells.
FIG. 1.

Recombinant measles virus, transgene expression, and virus kinetics. (A) Schematic representation of vectors harboring either unmodified hemagglutinin (H) or fully retargeted H against carcinoembryonic antigen (CEA) (listed at bottom, middle) and encoding transgene X, either granulocyte-macrophage colony-stimulating factor (GM-CSF) or enhanced green fluorescent protein (EGFP), in an additional transcription unit (bottom left) upstream of the nucleocapsid protein (N) gene. Corresponding names of viruses are listed at bottom right. (B) MV producer cell lines Vero-αHis (left) and murine MC38cea (right) were infected in triplicate with MV-EGFP-antiCEA (circles) or MV-GMCSF-antiCEA (squares) at a multiplicity of infection (MOI) of 1. GM-CSF concentrations in supernatants were determined at the indicated time points. (C) One-step growth curves of either the parental MV-EGFP (solid circles) or the targeted MV-EGFP-antiCEA (open symbols) and MV-GMCSF-antiCEA (solid symbols). Vero-αHis cells (left) and MC38cea cells (right) were infected at an MOI of 3. At the designated time points progeny viral particles were determined in octuplicate by serial dilution titration assay on Vero-αHis cells. The detection limit of progeny particles lies below 12.5 cell infectious units (CIU)/ml.
GM-CSF arming and CEA targeting do not alter viral growth characteristics
One-step growth curves with MV-EGFP, MV-EGFP-antiCEA, and MV-GMCSF-antiCEA were performed on Vero-αHis and MC38cea cells (Fig. 1C). All viruses displayed similar growth kinetics resulting in similar titers at the indicated time points after infection of Vero-αHis cells. As expected, infection of murine MC38cea cells with the untargeted MV-EGFP variant resulted in progeny particle titers below the detection limit. Targeted MV-EGFP-antiCEA and MV-GMCSF-antiCEA replicated to similar titers at designated time points in MC38cea cells. Thus, viral growth kinetics were not altered by CEA targeting and insertion of the GM-CSF transgene.
GM-CSF-armed MV prolongs survival in syngeneic colon carcinoma model
To analyze the therapeutic efficacy of GM-CSF-armed MV in vivo, immunocompetent C57BL/6 mice with subcutaneous MC38cea tumors were treated with MV-GMCSF-antiCEA or with the respective controls via four intratumoral injections on days 5–8 postimplantation. Control groups received carrier fluid or UV-inactivated MV-GMCSF-antiCEA (UV-MV). Serum levels of GM-CSF were assessed by ELISA. Elevated levels of GM-CSF were detected in the serum of MV-GMCSF-antiCEA-treated mice on day 1 after the last virus application, indicating efficient short-term expression of GM-CSF in vivo (Fig. 2A). MV-GMCSF-antiCEA application led to a significant delay in tumor growth compared with all other treatment groups (Fig. 2B). On day 16, mice treated with GM-CSF-armed MV showed a mean tumor volume of 94 μl including the majority of individuals with tumor volumes less than 50 μl (tumor stasis), whereas MV-EGFP-antiCEA-treated mice displayed a mean tumor volume of 260 μl (p=0.037 by two-sample t test) (Fig. 2C). MV-GMCSF-antiCEA treatment led to a significant increase in median overall survival compared with mock treatment or treatment with UV-inactivated MV or control virus (35.5 days vs. 19, 28, or 26.5 days postimplantation), whereas MV-EGFP-antiCEA-treated groups showed no significant survival benefits compared with controls (Fig. 2D). Moreover, in each of two independent experiments, three of eight mice treated with GM-CSF-armed MV demonstrated complete tumor remission. In addition, no tumor recurrence was observed until sacrifice 6 months postimplantation.
FIG. 2.

In vivo antitumor activity of MV-GMCSF-antiCEA in an immunocompetent murine colon carcinoma model. MC38cea cells (106) were implanted subcutaneously into the right flank of 6- to 8-week-old C57BL/6 mice. When tumor volumes reached 30–50 μl, mice were treated intratumorally with carrier fluid (mock; n=10), 106 particles of UV-inactivated MV-GMCSF-antiCEA (UV-MV; n=10), 106 particles of MV-EGFP-antiCEA (n=10), or 106 particles of MV-GMCSF-antiCEA (n=9), each on four consecutive days (days 5, 6, 7, and 8 postimplantation). Tumor volumes were measured every third day. (A) Blood was collected at the indicated time points after tumor implantation and an ELISA was performed to evaluate GM-CSF serum levels. (B) Tumor volume was measured every third day. (C) Distribution of tumor volumes on day 16 postimplantation. Each dot represents one mouse. (D) Kaplan–Meier plot documenting survival effects of MV-GMCSF-antiCEA and MV-EGFP-antiCEA compared with mock- or UV-MV-treated mice. Mice were sacrificed when tumors reached a volume of 1500 μl or when severe ulceration occurred. Circles, mock treatment; asterisks, UV-MV; triangles, MV-EGFP-antiCEA; squares, MV-GMCSF-antiCEA.
Antitumor immunity induced by GM-CSF-armed MV
To assess possible cellular mechanisms of the enhanced antitumor effect mediated by GM-CSF-armed MV, effector cells isolated from spleens were cocultivated at various ratios with target MC38cea cells and tumor-specific CTL activity was determined by LDH assay (Decker and Lohmann-Matthes, 1988). Splenocytes obtained from MV-GMCSF-antiCEA-treated mice induced significantly enhanced MC38cea-specific lysis compared with MV-EGFP-antiCEA-treated mice, whereas tumor-specific cell lysis was only at background level in mock and UV-MV groups (Fig. 3A). Tumor cell killing mediated by splenocytes from mice treated with carrier fluid, UV-inactivated MV, MV-EGFP-antiCEA, and MV-GMCSF-antiCEA amounted to 8, 12, 23, and 60%, respectively, at an E:T ratio of 20:1. At a higher E:T ratio of 50:1, tumor cell killing at nearly the same extent was observed. Unspecific stimulation was excluded by cocultivation of splenocytes with syngeneic B16 melanoma cells, which led to LDH release at background levels (data not shown). Similar tumor-specific cell killing as observed for MC38cea cells could be confirmed for parental MC38 cells (Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/hum), indicating MC38 antigen-specific cytotoxicity independent of CEA expression after treatment with MV-GMCSF-antiCEA.
FIG. 3.

Induction of an antitumor immune response. MC38cea cells (106) were engrafted subcutaneously. When tumor volumes reached 30–50 μl, mice were treated intratumorally on four consecutive days with carrier fluid (mock), 106 particles of UV-inactivated MV-GMCSF-antiCEA (UV-MV), 106 particles of MV-EGFP-antiCEA, or 106 particles of MV-GMCSF-antiCEA per day. Spleens were harvested aseptically 15 days after tumor implantation. (A) Lactate dehydrogenase (LDH) assay. MC38cea cells were cocultivated with prestimulated splenocytes obtained from mock-treated mice and mice treated with UV-MV, MV-EGFP-antiCEA, or MV-GMCSF-antiCEA. Mean values of n=7 (mock), n=8 (MV-EGFP-antiCEA), n=9 (MV-GMCSF-antiCEA), and n=4 (UV-MV) mice are shown, and the standard deviations. (B) ELISPOT assay for tumor-specific interferon (IFN)-γ secretion. Splenocytes isolated from treated mice were cocultivated with MC38cea tumor cells for 24 hr. Spots of IFN-γ-secreting cells were counted with an automated colony-counting device. (C) Virus-specific IFN-γ secretion. Splenocytes were cocultivated with MV-GMCSF-antiCEA or MV-EGFP-antiCEA at an MOI of 0.5 for 24 hr. Circles, mock treatment; asterisks, UV-MV; triangles, MV-EGFP-antiCEA; squares, MV-GMCSF-antiCEA. **p≤0.01; n.s., not significant.
In addition, the proportion of IFN-γ-secreting splenocytes was assessed by ELISPOT (De Rose et al., 2005). In the presence of MC38cea tumor cells, IFN-γ secretion by splenocytes obtained from mice treated with MV-GMCSF-antiCEA was significantly higher than in the case of MV-EGFP-antiCEA-treated individuals (on average, 60 spot-forming units [SFU]/105 splenocytes vs. 18 SFU/105 splenocytes; p≤0.01; Fig. 3B), indicating tumor-specific enhancement of immune effector cell activation by MV-GMCSF-antiCEA.
Adaptive antiviral immunity was assessed by IFN-γ ELISPOT after prestimulation with viral particles. In contrast to mock-treated animals, mice treated with the MV variants or with UV-irradiated MV showed MV-dependent IFN-γ secretion. Splenocytes of MV-GMCSF-antiCEA-treated mice demonstrated a slight but not significant increase in MV antigen-specific IFN-γ secretion, suggesting that GM-CSF also contributed to an enhanced antiviral immune response (p>0.05; Fig. 3C).
Subcutaneous tumors treated with MV-GMCSF are infiltrated with T lymphocytes
Representative MC38cea tumor samples from mock-treated mice and mice treated with control virus and MV-GMCSF-antiCEA were harvested and frozen on day 15 after tumor implantation, that is, 7 days after the last viral treatment. Complete tissue sections were subjected to immunohistochemistry and immune cell quantification (Halama et al., 2011). Tumors treated with MV-EGFP-antiCEA and MV-GMCSF-antiCEA showed increased CD3+ tumor-infiltrating T lymphocytes compared with mock-treated animals. T cell densities at the invasive margin of MC38cea tumors showed remarkable differences between virus- and mock-treated mice (Fig. 4). When CD8+ tumor-infiltrating cells were quantified, we observed a trend to a higher density after MV-GMCSF-antiCEA treatment compared with mock- and MV-EGFP-antiCEA-treated animals (103 vs. 68 and 85 CD8+ cells/mm2, respectively; data not shown), but this difference was not statistically significant.
FIG. 4.
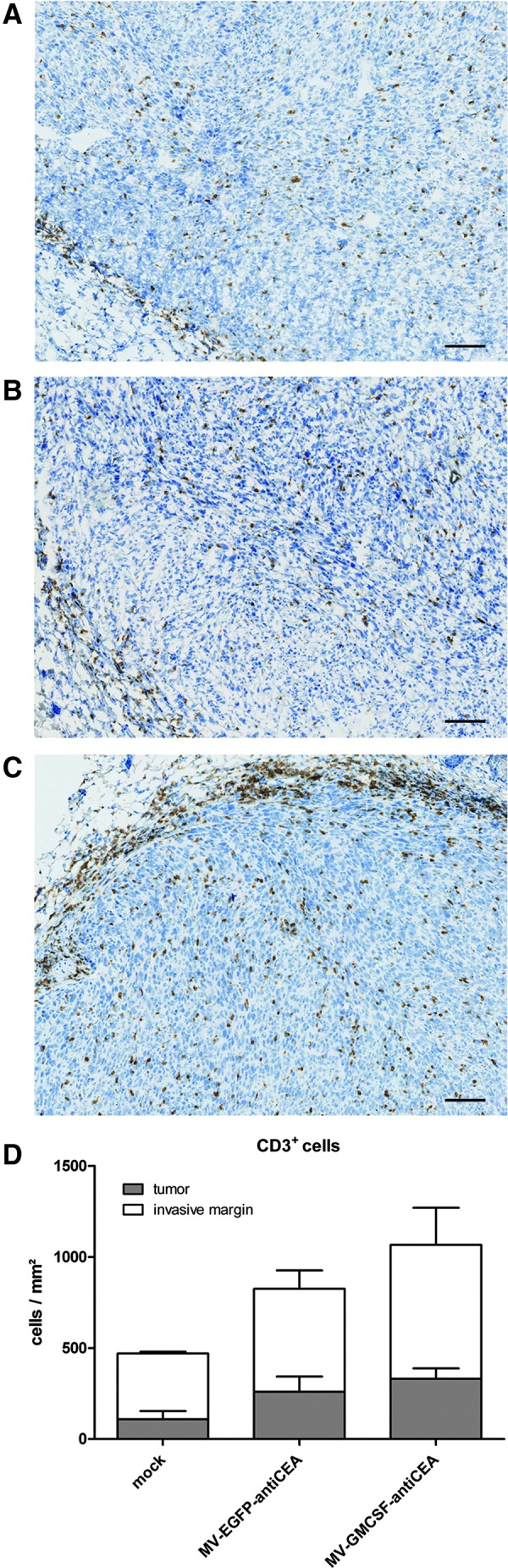
Immunohistological staining of CD3+ lymphocytes. Remaining tumors were isolated 7 days after the last virus application. The numbers of stained immune cells in tumors and at the invasive margin of (A) mock-treated, (B) MV-EGFP-antiCEA-treated, and (C) MV-GMCSF-antiCEA-treated mice were quantified automatically. Scale bars: 100 μm. (D) The average total number of CD3+ cells per tumor and invasive margin, respectively, in each group.
GM-CSF-armed MV provides long-term tumor protection
To evaluate long-term antitumor immunity, six mice in complete remission after treatment with MV-GMCSF were rechallenged with 105 MC38cea cells 10 weeks after initial tumor implantation. Tumor engraftment was not successful in any of the rechallenged mice, whereas four of six mice that received MC38cea cells for the first time developed solid tumors (Fig. 5A). To assess tumor-specific immune effectors involved in tumor rejection, splenocytes of mice in complete remission were isolated 6 months after the initial experiment (i.e., 14 weeks after rechallenge) and an LDH assay was carried out. The CTL activity of splenocytes against MC38cea cells from mice treated with MV-GMCSF-antiCEA demonstrated tumor-specific killing of 50% at an E:T ratio at 50:1 (Fig. 5B). Altogether, this indicates robust immune clearance and tumor-specific long-term protection of MV-GMCSF-antiCEA-treated mice.
FIG. 5.

Memory immune response. (A) Percentage of naive and MV-GMCSF-antiCEA-pretreated mice, rechallenged with MC38cea cells, that demonstrate tumor engraftment. (B) Mice that demonstrated complete tumor remission were sacrificed 6 months after the initial experiment. Spleens were isolated and an antitumor-specific LDH assay was performed. LDH release of MC38cea cells cocultivated with prestimulated splenocytes was measured and calculated as the percentage of dying MC38cea cells. Circles, mock; squares, MV-GMCSF-antiCEA.
Tumor treatment with MV-GMCSF-anti-CEA abolishes secondary engraftment of tumors
To further support the notion of establishing an antitumor immune response, an experiment with sequential engraftments of MC38cea tumors was performed. Here, primarily implanted tumors (primary engraftment) were treated as described above and 106 MC38cea cells were implanted into the opposite (left) flank 2 days after the last viral treatment (secondary engraftment). Mice treated with carrier fluid demonstrated 100% secondary engraftment (Fig. 6). Mice treated with the control virus MV-EGFP-antiCEA demonstrated secondary engraftment with delayed tumor growth. One of five MV-EGFP-antiCEA-treated mice demonstrated no tumor reengraftment of the secondary tumor. Importantly, mice treated with MV-GMCSF-antiCEA completely rejected secondary engraftment. Altogether, this indicates the potential of the combined immunovirotherapy for elimination of tumors and for prevention of recurrence.
FIG. 6.

Secondary engraftment of MC38cea tumors. Tumor cells (106) were implanted subcutaneously into the right flank of C57BL/6 mice (primary engraftment) and treated intratumorally with Opti-MEM (n=6), MV-EGFP-antiCEA (n=5), or MV-GMCSF-antiCEA (n=6) on four consecutive days as soon as tumors reached a volume of approximately 30 μl. Two days after the last virus treatment, 106 MC38cea cells were injected into the left flank (secondary engraftment). Tumor volume development of each individual is shown after primary engraftment (right) and secondary engraftment (left) of groups treated with carrier fluid, MV-EGFP-antiCEA, or MV-GMCSF-antiCEA. Two of six mice treated with MV-GMCSF-antiCEA demonstrated complete tumor remission of the primary engraftment. Arrows indicate the time point of secondary tumor implantation.
Discussion
Until now, few studies have evaluated immune modulation in combination with oncolytic measles virus therapy. This work demonstrates the potential of an immunovirotherapy based on MV expressing the cytokine GM-CSF.
Combining oncolytic agents and immunotherapy is not uncontroversial. In an immunocompetent host, antiviral immune responses will arise, with the risk of premature clearance of virus and infected target cells before adaptive antitumor mechanisms can develop (Prestwich et al., 2008). Nevertheless, the combination of virus-encoded danger signals and release of tumor-associated antigens (TAAs) provides potential for induction of adaptive antitumor immunity, that is, cell-mediated long-term immune control. Innate immune mechanisms such as acute inflammation, neutrophil infiltration, and NK and DC activation may also contribute to tumor regression.
Arming OV with cytokines, for example, IL-2, IL-12, or most prominently GM-CSF, can support the attraction of immune effector cells to the tumor site and improves the induction of a tumor-specific immune response (Fukuhara et al., 2005; Lee et al., 2006; Dempe et al., 2012; Lemay et al., 2012). In addition to its key role in granulocyte and monocyte differentiation, GM-CSF attracts neutrophils and induces maturation of dendritic cells. Direct effects on T cells may be limited, but as a potent stimulator for antigen-presenting cells (APCs), GM-CSF enhances T cell-mediated specific immune responses (Dranoff et al., 1993).
To date, measles virus encoding GM-CSF has been studied only in an SCID model, where MV-GMSCF treatment in lymphoma xenografts delayed tumor progression associated with neutrophil infiltration via activation of innate immunity (Grote et al., 2003). Therefore, we chose the C57BL/6 MC38cea model for evaluation of MV-GMCSF in a fully immunocompetent host with a functional adaptive immune system. Our results suggest that arming MV with GM-CSF had beneficial therapeutic effects in terms of tumor-free survival. Strikingly, complete tumor remission was achieved in 37.5% of mice treated with MV-GMCSF-antiCEA.
In the present study, UV-MV- and MV-EGFP-antiCEA-treated mice demonstrated no significant differences in antiviral immunity. Compared with UV-MV and control virus, MV-GMCSF-antiCEA treatment resulted in slightly, but not significantly, increased antiviral immunity, suggesting that GM-CSF-armed MV enhances not only an antitumor immune response. Immunomodulation with GM-CSF did not abrogate oncolytic effects. Instead, our results support the notion of a cell-mediated antitumor immune response induced by treatment with MV-GMCSF-antiCEA via increased intratumoral infiltration of CD3+ lymphocytes. In addition, T cell densities at the invasive margin showed remarkable differences between mock- and virus-treated mice. The higher proportion of CD8+ tumor-infiltrating cells may contribute to the notable therapeutic effect of MV-GMCSF.
Tumor-specific memory T cells are thought to provide long-term protection against cancer recurrence (Ostrand-Rosenberg, 2005). In this study, assessment of tumor-specific cytotoxicity 6 months after treatment demonstrated a durable antitumor effect of MV-GMCSF-antiCEA. Importantly, mice treated with MV-GMCSF-antiCEA were completely protected against MC38cea reengraftment in terms of a tumor vaccination effect, whereas mock-treated animals demonstrated 100% secondary engraftment of MC38cea tumors. However, one of five mice treated with MV-EGFP-antiCEA demonstrated no tumor reengraftment. This underlines the potential of oncolytic viruses to mediate immunity against TAAs that may be liberated during oncolysis, rendering immune escape phenomena less likely compared with conventional tumor vaccination approaches that might target only selected antigens.
Taking advantage of the C57BL/6 MC38cea model, we have previously used low-dose cyclophosphamide (CPA) as an immunosuppressive agent to support oncolytic efficacy (Ungerechts et al., 2007). Thereby, CPA modulates the immune system in at least two different ways: suppression of regulatory T cells might break tumor tolerance, and suppression of innate immune responses can abolish the antiviral status supporting viral replication. The fact that, on the one hand immune suppression by CPA and on the other hand immune activation by GM-CSF both result in better treatment efficacy, demonstrates the complexity of interactions between oncolysis and the immune system and shows the need for further detailed studies in the established immunocompetent model.
Our results are in line with other combined immunovirotherapy approaches, employing oncolytic viruses including HSV and VV encoding GM-CSF, which also successfully recruited the adaptive immune system. In small-animal models, HSV-GMCSF proved an effective tumor-eliminating agent and evoked antitumor immunity by a T cell-dependent mechanism (Derubertis et al., 2007). In the case of GM-CSF-armed vaccinia virus JX-594, treated tumors regressed and were infiltrated by CD4+ and CD8+ T cells (Kim et al., 2006).
An immunosuppressive tumor microenvironment might often impede immune clearance of cancer cells. Studies demonstrate the potential of blocking distinct inhibitory pathways to overcome immune tolerance (Zhou et al., 2012). In a clinical phase 3 trial for metastatic melanoma, treatment with ipilimumab (humanized antibody against CTLA-4) showed a remarkable long-term, complete response rate. Consequently, combining counter actors to inhibitory factors such as anti-CTLA-4 (Margolin et al., 2012) or anti-PD-L1 (Brahmer et al., 2012) with immunostimulatory MV vectors may be a promising strategy for virotherapy of cancer.
Although our immunocompetent MV oncolysis mouse model will probably enable us to further elucidate some of the complex interactions between immune system and viral agent, relevant differences regarding the role of the immune system in tumor biology between mice and humans are obvious. On the basis of the results of this study, MV-GMCSF will follow the lead of GM-CSF-armed VV and HSV and will be tested in a clinical immunovirotherapy trial to finally determine whether the encouraging results in mice can be translated into clinical benefit for cancer patients.
Supplementary Material
Acknowledgments
The authors thank Jessica Albert, Stefanie Sawall, Anna Spille, and Silke Hamzaoui-Nord for valuable technical assistance, and Manuela Brom and Felix Bestvater (light microscopy facility, German Cancer Research Center). The authors acknowledge Roberto Cattaneo and Stephen J. Russell (Mayo Clinic, Rochester, MN) for providing MC38cea, MC38, and Vero-αHis cells and Volker Teichgraeber for providing the GM-CSF plasmid. This work was supported by German Cancer Aid, Max Eder grant 108307 (G.U.).
Author Disclosure Statement
No competing financial interests exist.
References
- Bossow S. Grossardt C. Temme A., et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18:598–608. doi: 10.1038/cgt.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J.R. Tykodi S.S. Chow L.Q., et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach C.J. Reid T. Burke J., et al. Navigating the clinical development landscape for oncolytic viruses and other cancer therapeutics: No shortcuts on the road to approval. Cytokine Growth Factor Rev. 2010;21:85–89. doi: 10.1016/j.cytogfr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Choi K.J. Zhang S.N. Choi I.K., et al. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012;19:711–723. doi: 10.1038/gt.2011.125. [DOI] [PubMed] [Google Scholar]
- Decker T. Lohmann-Matthes M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Dempe S. Lavie M. Struyf S., et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: Implication of leucocyte recruitment. Cancer Immunol. Immunother. 2012;61:2113–2123. doi: 10.1007/s00262-012-1279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rose R. Taylor E.L. Law M.G., et al. Granulocyte contamination dramatically inhibits spot formation in AIDS virus-specific ELISpot assays: Analysis and strategies to ameliorate. J. Immunol. Methods. 2005;297:177–186. doi: 10.1016/j.jim.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Derubertis B.G. Stiles B.M. Bhargava A., et al. Cytokine-secreting herpes viral mutants effectively treat tumor in a murine metastatic colorectal liver model by oncolytic and T-cell-dependent mechanisms. Cancer Gene Ther. 2007;14:590–597. doi: 10.1038/sj.cgt.7701053. [DOI] [PubMed] [Google Scholar]
- Dranoff G. Jaffee E. Lazenby A., et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara H. Ino Y. Kuroda T., et al. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65:10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]
- Galanis E. Hartmann L.C. Cliby W.A., et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote D. Cattaneo R. Fielding A.K. Neutrophils contribute to the measles virus-induced antitumor effect: Enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63:6463–6468. [PubMed] [Google Scholar]
- Halama N. Zoernig I. Spille A., et al. Estimation of immune cell densities in immune cell conglomerates: An approach for high-throughput quantification. PLoS One. 2009;4:e7847. doi: 10.1371/journal.pone.0007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama N. Zoernig I. Spille A., et al. Quantification of prognostic immune cell markers in colorectal cancer using whole slide imaging tumor maps. Anal. Quant. Cytol. Histol. 2010;32:333–340. [PubMed] [Google Scholar]
- Halama N. Michel S. Kloor M., et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011;71:5670–5677. doi: 10.1158/0008-5472.CAN-11-0268. [DOI] [PubMed] [Google Scholar]
- Hammerstrom A.E. Cauley D.H. Atkinson B.J., et al. Cancer immunotherapy: Sipuleucel-T and beyond. Pharmacotherapy. 2011;31:813–828. doi: 10.1592/phco.31.8.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond A.L. Plemper R.K. Zhang J., et al. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J. Virol. 2001;75:2087–2096. doi: 10.1128/JVI.75.5.2087-2096.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington K.J. Hingorani M. Tanay M.A., et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. 2010;16:4005–4015. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- Heo J. Breitbach C.J. Moon A., et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: Preclinical and clinical demonstration of combination efficacy. Mol. Ther. 2011;19:1170–1179. doi: 10.1038/mt.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke M. Peeters B. de Leeuw O., et al. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007;14:1639–1649. doi: 10.1038/sj.gt.3303026. [DOI] [PubMed] [Google Scholar]
- Kim J.H. Oh J.Y. Park B.H., et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006;14:361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Lee Y.S. Kim J.H. Choi K.J., et al. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006;12:5859–5868. doi: 10.1158/1078-0432.CCR-06-0935. [DOI] [PubMed] [Google Scholar]
- Lemay C.G. Rintoul J.L. Kus A., et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012;20:1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin K. Ernstoff M.S. Hamid O., et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012;13:459–465. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- Melcher A. Parato K. Rooney C.M., et al. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msaouel P. Iankov I.D. Allen C., et al. Oncolytic measles virus retargeting by ligand display. Methods Mol. Biol. 2012;797:141–162. doi: 10.1007/978-1-61779-340-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik J.D. Twelves C.J. Selby P.J., et al. Immune recruitment and therapeutic synergy: Keys to optimizing oncolytic viral therapy? Clin. Cancer Res. 2011;17:4214–4224. doi: 10.1158/1078-0432.CCR-10-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T. Peng K.W. Harvey M., et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005;23:209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S. CD4+ T lymphocytes: A critical component of antitumor immunity. Cancer Invest. 2005;23:413–419. [PubMed] [Google Scholar]
- Prestwich R.J. Harrington K.J. Pandha H.S., et al. Oncolytic viruses: A novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008;8:1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radecke F. Spielhofer P. Schneider H., et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard C. Baro J. Bello-Fernandez C., et al. Recombinant human granulocyte-macrophage colony stimulating factor (rhGM-CSF) administration after autologous bone marrow transplantation for acute myeloblastic leukemia enhances activated killer cell function and may diminish leukemic relapse. Bone Marrow Transplant. 1995;15:721–726. [PubMed] [Google Scholar]
- Robbins P.F. Kantor J.A. Salgaller M., et al. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3657–3662. [PubMed] [Google Scholar]
- Russell S.J. Peng K.W. Bell J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M. Pawlita M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res. 2009;37:e119. doi: 10.1093/nar/gkp581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A.W. Senzer N. Cerullo V., et al. Oncolytic viruses for induction of anti-tumor immunity. Curr. Pharm. Biotechnol. 2012;13:1750–1760. doi: 10.2174/138920112800958913. [DOI] [PubMed] [Google Scholar]
- Ungerechts G. Springfeld C. Frenzke M.E., et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol. Ther. 2007;15:1991–1997. doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- van de Laar L. Coffer P.J. Woltman A.M. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood. 2012;119:3383–3393. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- Zhou J. Yuen N.K. Zhan Q., et al. Immunity to the melanoma inhibitor of apoptosis protein (ML-IAP; livin) in patients with malignant melanoma. Cancer Immunol. Immunother. 2012;61:655–665. doi: 10.1007/s00262-011-1124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.