

Abstract
The KIX domain of the transcriptional coactivator CREB binding protein (CBP) co-operatively mediates interactions between transcription factors. Binding of the transcription factor mixed-lineage leukemia (MLL) induces the formation of a low-populated conformer of KIX that resembles the conformation of the KIX domain in the presence of a second transcription factor molecule. NMR spin relaxation studies have previously shown that allosteric coupling proceeds through a network of hydrophobic core residues that bridge the two binding sites. Here we describe high-resolution NMR solution structures of the binary complex of KIX with MLL and the ternary complex of KIX formed with MLL and phosphorylated kinase inducible domain of CREB (pKID) as a second ligand. We show that binding of pKID to the binary complex of KIX with MLL is accompanied by a defined repacking of the allosteric network in the hydrophobic core of the protein. Rotamer populations derived from methyl group 13C chemical shifts reveal a dynamic contribution to the repacking process that is not captured by the structural coordinates and exemplify the dynamic nature of allosteric communication in the KIX domain.
Co-operativity plays a central role for the regulation of gene transcription.1−3 Transcription factors along with transcriptional coactivators assemble co-operatively on DNA promoter sequences, and transcription is stimulated by recruitment of RNA polymerase II.4 These interactions are mediated by transcriptional coactivators, such as CREB binding protein (CBP), its paralog p300, or Mediator coactivator, which act as a scaffold for the recruitment of the transcriptional machinery.5,6 It has been observed in various studies that a combination of several DNA-bound transcription factors results in synergistic transcriptional response,1 and more specifically, several lines of evidence indicate that co-operative interactions between transcription factors and CBP are pivotal to promote synergism in transcriptional activation.3,7 CBP participates in the regulation of gene transcription by linking transcription factors with components of the basal transcriptional machinery.5 CBP is present at only low (and possibly limiting) concentrations in vivo, suggesting that competition of different transcription factors for CBP may be crucial for the regulation of gene transcription.8 The characterization of the biophysical mechanism by which co-operativity modulates the affinities of transcription factors for binding to CBP is a prerequisite for understanding how gene transcription is regulated.
CBP is a modular protein that contains a number of structured domains, as well as long stretches that are intrinsically disordered and represent linker regions or interaction motifs that fold only upon binding their biological targets.9 From a regulatory perspective, the KIX domain is of particular interest, since it is capable of binding two transcription factors simultaneously through two different binding sites, thereby directly mediating interactions between bound transcription factors. The three-dimensional structure of the KIX domain is composed of a bundle of three α-helices and two short 310-helices, with the two binding sites for transcription factors, which are isolated from each other, being located at remote surfaces of the protein.10 Homologous KIX domains have been identified and characterized in p300 as well as in human and yeast Mediator coactivator subunits, and their structures were determined recently, revealing a high degree of functional and structural similarity.11,12
The KIX domain of CBP thus physically interlinks transcription factors by simultaneous binding through two interaction sites, and it does so in a co-operative and highly dynamic manner.13,14In vitro, binding of the mixed-lineage leukemia (MLL) activation domain to KIX co-operatively enhances the interaction with the activation domain of the transcription factor c-Myb: KIX in complex with MLL displays ∼2-fold higher affinity for c-Myb than the KIX domain alone.15 Likewise, positive co-operativity has been demonstrated for the interaction of the activation domain of pKID (the phosphorylated kinase inducible domain of CREB, which binds to KIX through the c-Myb interaction site) with KIX in complex with MLL.15 These co-operative effects provide a potential mechanism through which transcriptional activity might be modulated in the cell. Several observations relating to communication between transcription factors mediated by CBP have indeed been described in the literature, suggesting transcriptional synergy between various transcription factors.7,16
Using backbone and side chain nuclear magnetic resonance (NMR) relaxation dispersion techniques, we were able to monitor directly the conformational rearrangement process by which the KIX domain communicates information about the presence of a biological target at the MLL binding site to the allosterically regulated c-Myb/pKID binding site.17 NMR relaxation dispersion data revealed that binding the activation domain of MLL to KIX causes a redistribution of the relative populations of KIX conformers toward a state that adopts a conformation that is similar to that of the ternary complex. Titration experiments showed that this higher energy (excited) conformational substate of KIX·MLL, which is populated to 7% in the KIX·MLL binary complex, displays a higher affinity for c-Myb/pKID ligands than the 93% populated lower energy (ground) state of the protein complex. These results suggest that binding of c-Myb/pKID involves the selection of the higher energy conformer, whose structure is complementary to the ligand, from a pre-existing ensemble of conformations, reminiscent of the conformational selection mechanism of molecular recognition.18−20 In solution, KIX in complex with MLL is in an equilibrium between these two conformational substates within ∼3 ms. As the higher affinity conformational substate is depleted from the equilibrium upon ligand binding, the equilibrium is recovered through the allosteric transition.
The exact structural basis of the allosteric transition is, however, unknown. The relaxation dispersion data indicate that a network of hydrophobic amino acids, which bridge the two binding sites of the KIX domain, constitutes the pathway through which allosteric information is communicated. The data suggest subtle conformational differences between lower and higher energy conformers, with chemical shift differences δω between 0.4 and 1.0 ppm found for the small subset of 15N and 13C nuclei that form part of the allosteric network. This is contrasted by the remainder of the protein backbone, for which δω values <0.4 ppm were detected, indicating that the structure of the three-helix scaffold is not affected by the transition between lower and higher energy conformers. Intriguingly, side chain methyl relaxation dispersion experiments performed on isoleucine δ1 methyl 13C nuclei suggested a small but measurable conformational adaption of the part of the hydrophobic core that bridges the two binding sites. In addition, backbone amide 1H/2H exchange data showed that both (lower and higher energy) substates represent fully folded and solvent exchange protected conformers.17 Recent molecular dynamics (MD) simulations confirmed that the conformational ensemble of the KIX domain is shifted in response to the binding of MLL, leading to a preorganized c-Myb/pKID binding site.21
Here we report the NMR-derived high-resolution three-dimensional solution structures of the binary complex of KIX formed with the activation domain of MLL and the ternary complex of KIX bound to MLL and pKID, based on NOEs and backbone chemical shifts and residual dipolar couplings. Validation of the side chain conformation of valine, leucine, and isoleucine residues using methyl 13C chemical shift data establishes that these solution structures represent the highest populated rotamers of these residues in solution. However, the 13C chemical shift data also reveal a defined redistribution of the rotameric states of residues in the hydrophobic core of the KIX domain that occurs upon transition between the two complexes, which is not captured by the structural bundles. Our results thus facilitate a quantitative description of the dynamic structural adaption of the KIX domain in response to binding ligand molecules in terms of a redistribution of rotameric states of residues that form the hydrophobic core of the protein. In combination with pico- to nanosecond dynamic NMR experimental data, these results enable us to draw a comprehensive structural and dynamic picture of the molecular mechanism through which the KIX domain of CBP mediates co-operativity between transcription factors.
Materials and Methods
Sample Preparation
Unlabeled, 10% 13C-labeled, uniformly 13C-, 15N-, or 13C,15N-labeled samples of the KIX domain (residues 586–672) of human CBP were prepared by bacterial growth in standard LB and M9 minimal media and purified as described.22
Uniformly 13C,15N-labeled MLL (residues 2840–2858; with C2841 substituted with alanine) was overexpressed in Escherichia coli as an N-terminal fusion to a hexa-histidine tagged MBP. The cells were grown at 37 °C in M9 minimal media containing 15NH4Cl and 13C-glucose as sole nitrogen and carbon sources in presence of kanamycin until OD600 ≈ 0.6, then the temperature was lowered to 28 °C, and after 30 min, protein synthesis was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.8 mM. The cells were harvested 5 h after induction and resuspended in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 10 mM imidazole, and 5 mM β-mercaptoethanol. The cells were lysed using a French pressure cell and clarified by centrifugation. Subsequently, the supernatant was loaded onto a HisTrap FF crude (GE Healthcare) column. In the next step, the MBP–MLL fusion protein was applied to a Superdex 75 (GE Healthcare) equilibrated with cleavage buffer that contained 50 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, and 2 mM DTT. The His-tagged MBP was cleaved by incubation with TEV, and it was removed using nickel affinity chromatography. In the final step, MLL was purified to homogeneity by size exclusion chromatography using a Superdex 30 (GE Healthcare). Mass spectrometry was used to confirm the identity of MLL.
The human KID domain of CREB (residues 116–149) was prepared as a N-terminal fusion to a hexa-histidine tagged thioredoxin. Uniformly 13C,15N-labeled samples for NMR studies were prepared by growing E. coli cells in M9 minimal media containing 15NH4Cl and 13C-glucose as sole nitrogen and carbon sources at 37 °C. Protein overexpression was induced at OD600 ≈ 0.8 by adding IPTG to a final concentration of 0.7 mM. Cells were harvested after 5 h and trx–KID purification followed the same three-step procedure as established for MBP–MLL. Ser133 of KID was phosphorylated in vitro by incubation of 60 μM purified KID with 0.05 μM PKA catalytic subunit and 1 mM ATP in a 25 mM Tris-HCl (pH 7) buffer containing 10 mM MgCl2 and 2 mM DTT for 24 h at 30 °C. pKID was purified by size exclusion chromatography using a Superdex 30 (GE Healthcare). Mass spectrometry was used to confirm that pKID was fully phosphorylated.
NMR Spectroscopy
NMR spectra were recorded at 27 °C on Varian Inova 500 and 800 MHz and Varian Direct Drive 600 MHz spectrometers. Data were processed using NMRPipe23 and analyzed using CcpNmr.24 NMR samples contained 1 mM of labeled protein or peptide, unlabeled ligands in at least 2-fold excess, 50 mM potassium phosphate buffer, pH 5.8, 25 mM NaCl, and 1 mM NaN3 in 10% D2O/90% H2O. Backbone and side chain 1H, 13C, and 15N chemical shift assignments of KIX, pKID, and MLL were obtained using standard triple-resonance experiments: HNCA/HN(CO)CA, HNCO/HN(CA)CO, CBCA(CO)NH/HNCACB, 15N-edited TOCSY-HSQC, (H)CCONH-TOCSY, and HCCH-TOCSY. For the KIX domain in the complexes KIX·MLL and KIX·MLL·pKID, stereospecific assignment of the prochiral methyl groups of valine and leucine residues were obtained using the 10% fractional labeling method of Neri et al.25 Intramolecular distance restraints were obtained from 3D 1H–1H-NOESY–15N/13C-HSQC experiments. Additional intramolecular distance restraints were obtained from 3D 13C,13C-methyl NOESY experiments.26 Intermolecular distance restraints were obtained from a ω1-13C-filtered simultaneous inter- and intramolecular three-dimensional 1H–1H NOESY–13C-HSQC experiment.27 Residual dipolar couplings (RDCs) of KIX in both KIX·MLL and KIX·MLL·pKID and of pKID were measured on samples partially aligned using strain-induced alignment in a 4% polyacrylamide gel.28 HN–N RDCs (1D1H,15N) were measured using an in-phase/anti-phase (IPAP) 1H,15N-HSQC experiment29 or a 3D best-type HNCO experiment.30
Heteronuclear 1H,15N NOE, rotating-frame longitudinal relaxation time T1ρ and the longitudinal relaxation time T1 were measured for KIX in the states KIX, KIX·MLL, KIX·pKID and KIX·MLL·pKID at 27 °C and 800 MHz 1H Larmor frequency. The heteronuclear 1H,15N NOE was obtained by recording, in an interleaved manner, one spectrum with a delay of 2 s followed by proton saturation for 3 s and another spectrum with a delay of 5 s without proton saturation. Relaxation delays of 10.9, 54.4, 108.9, 217.6, 326.4, 435.2, 598.4 and 707.2 ms were used for T1 experiments, and delays of 10.0, 20.0, 30.0, 40.0, 60.0, 80.0, and 100.0 ms were used for the T1ρ measurements. The spherical diffusion tensor was determined by the method of Brüschweiler et al.31 using the program quadric_diffusion.32 The internal dynamics and overall tumbling were fit with the program FAST-ModelFree.33
Structure Calculation and Refinement
Backbone dihedral angle restraints were set to ϕ = −60(±10)° and ψ = −45(±10)° for residues that were predicted to be α-helical based on 13Cα, 13Cβ, 13C′, 1Hα, 15N, and 1HN, using the software Talos plus.34 Distance restraints derived from the 3D 13C,13C-methyl NOESY experiments were all set to an upper bound of 5.5 Å. All other intra- and intermolecular distance restraints were calibrated using the Aria2.3 program.35 An initial structural ensemble was used to determine the alignment tensor of KIX and pKID using the PALES software.36 The experimentally determined distance, dihedral angle, and dipolar coupling restraints were used in a torsion angle simulated annealing protocol using CNS1.2/Aria2.335,37 to solve the solution structure of the binary KIX·MLL complex and the ternary KIX·MLL·pKID complex. The final NMR ensembles were refined in an explicit water shell.38 The 20 lowest-energy solution structures (out of 100 calculated) were selected as a final representative ensemble of KIX·MLL and KIX·MLL·pKID.
Results
Solution Structures of Binary and Ternary Complexes of KIX
NMR solution structures of the KIX domain of CBP (residues 586–672) were determined in the presence of the activation domain of the transcription factor MLL, as well as in the presence of the activation domains of both MLL and CREB (pKID), using triple-resonance-resolved NMR spectroscopic techniques, using backbone chemical shifts, backbone amide NH dipolar couplings, and NOEs as input restraints for the structure determination. Site-specific and stereospecific assignments of KIX isoleucine, valine, and leucine side chain methyl groups were obtained to enable a structural anlysis of the hydrophobic cores of the KIX domain in the binary and the ternary complexes. For KIX·MLL and KIX·MLL·pKID, all isoleucine side chain γ2 and δ1 methyl resonances are resolved in two-dimensional [1H,13C] correlation experiments and could be assigned individually. In addition, we obtained stereospecific assignments of valine and leucine side chain methyl resonances for all 15 valine and leucine residues. Three-dimensional 13C,13C-methyl NOESY experiments26 were used to evaluate internuclear distances in the hydrophobic core of KIX in binary and ternary complexes KIX·MLL and KIX·MLL·pKID. Structural statistics are shown in Table 1. The KIX backbone structure of these complexes (Figure 1) is very similar to the NMR structures of binary KIX complexes with pKID,39 c-Myb,40 and the PCET motif from HEB,41 as well as the ternary KIX complex formed with the MLL and c-Myb activation domains.10 The central scaffold of the KIX domain is formed by a bundle of three α-helices (residues 597–611, α1; residues 623–642, α2; residues 646–669, α3) together with two short 310-helices (residues 591–593, G1; residues 617–620, G2). The three helices α1−α3 pack together in an antiparallel fashion to form an extended hydrophobic core, which is capped by the first 310-helix G1 on one side, while the loop L12, which encompasses the 310-helix G2 and connects helices α1 and α2, partly caps the other side of the hydrophobic core.10
Table 1. NMR and Refinement Statistics for the KIX·MLL and KIX·MLL·pKID Complexes.
| KIX·MLL | KIX·MLL·pKID | |
|---|---|---|
| NMR Restraints | ||
| distance restraints | ||
| total NOE | 1016 | 934 |
| intraresidue | 445/80 | 309/74/92 |
| inter-residue | ||
| sequential (|i – j| = 1) | 242/32 | 180/32/55 |
| medium-range (|i – j| ≤ 4) | 124/3 | 77/3/12 |
| long-range (|i – j| ≥ 5) | 70/0 | 50/0/0 |
| intermolecular restraints | 20 | 23/27 |
| total dihedral restraints | 184 | 225 |
| ϕ | 79/13 | 79/13/20 |
| ψ | 79/13 | 79/13/21 |
| total 1D1H–15N RDCs | 80/0 | 79/0/21 |
| Structure Statistics | ||
| violations (RMSD and SD) | ||
| distance restraints | 0.016 ± 0.005 | 0.073 ± 0.02 |
| dihedral restraints | 1.2 ± 0.1 | 1.0 ± 0.2 |
| 1D1H–15N RDCs | 0.74 ± 0.04 | 0.90 ± 0.10 |
| deviation from idealized geometry | ||
| bond lengths (Å) | 0.0041 ± 0.0001 | 0.0042 ± 0.0002 |
| bond angles (deg) | 0.56 ± 0.02 | 0.59 ± 0.02 |
| Ramachandran statistics | ||
| residues in most favored regions | 92.5% | 89.4% |
| residues in additional allowed regions | 6.9% | 8.9% |
| residues in generously allowed regions | 0.5% | 1.5% |
| residues in disallowed regions | 0.1% | 0.3% |
| Average Pairwise rms Deviation (Å) | ||
| heavy atoms (2° struct) | 1.62 ± 0.23 | 1.82 ± 0.25 |
| backbone atoms (2° struct) | 0.78 ± 0.17 | 0.92 ± 0.22 |
| heavy atoms (all residues) | 2.71 ± 0.59 | 2.94 ± 0.86 |
| backbone (all residues) | 2.18 ± 0.64 | 2.47 ± 0.95 |
Figure 1.

NMR solution structures of binary and ternary complexes of KIX. (A) Lowest energy structure of KIX in complex with MLL (residues 2840–2858); helices are displayed as ribbons. Secondary structure elements are labeled as according to Radhakrishnan et al.39 and color-coded (KIX, gray; helices, blue; MLL, green). (B) NMR ensemble of 20 representative structures of KIX·MLL (PDB ID 2LXS). (C) Lowest energy structure of KIX in complex with MLL (residues 2840–2858) and pKID (residues 116–149). Secondary structure elements are color-coded as in panel a (with pKID shown in magenta). (D) NMR ensemble of 20 representative structures of KIX·MLL·pKID (PDB ID 2LXT). All figures were generated using PyMOL.64
In the binary complex KIX·MLL, as well as in ternary KIX·MLL·pKID, residues 2847–2855 of MLL form an amphipathic helix that binds to the hydrophobic groove on the surface of the KIX domain at the C-terminus of helix α1, similar to the ternary complex formed by KIX with the activation domains of MLL and c-Myb.10 Through the insertion of MLL into the hydrophobic groove, several hydrophobic residues (I2849, M2850, F2852, V2853, and L2854) of MLL are in the position to form hydrophobic contacts with a number of KIX residues (I611, F612, A619, R624 (aliphatic region), L628, Y631, I660, and L664), which are part of the hydrophobic groove. In addition, the NMR structures indicate electrostatic interactions between the side chain of N2856 of MLL and two KIX side chains, T614 and D616. The structures of KIX·MLL and KIX·MLL·pKID complexes show no significant difference with respect to the binding mode of the MLL ligand peptide. Comparison of the KIX·MLL and KIX·MLL·pKID complexes with the binary complex between KIX and PCET reveals a slight difference in the orientation of the bound ligand peptide PCET versus MLL.41 This is reflected in a lower number of intermolecular contacts between KIX and PCET compared with MLL and has been related to the relatively low affinities of various PCET peptides for KIX.41 Of note, our NOE data do not indicate binding of the MLL peptide to additional binding surfaces on the KIX domain.42
In the ternary complex KIX·MLL·pKID, the pKID peptide binds into a shallow hydrophobic pocket on the surface of the KIX domain that is formed by side chains of amino acid residues in helices α1 (N-terminal part) and α3 (central part). Bound pKID forms two almost perpendicular helices αA and αB, as observed previously for the binary complex of KIX formed with pKID.39 The side chains of helix αB residues I137, L138, and L141 make close hydrophobic contacts with the pKID binding pocket formed by KIX helix α3 residues Y650, A654, I657, and Y658, while the side chain of pKID residue A145 loosely packs against the N-terminus of KIX helix α1 (L599 and L603). For the αA helix of pKID intermolecular NOEs were only found for residues I127 and L128, which make van der Waals contacts with Y658. In addition, the hydroxyl group of Y658 forms a hydrogen bond with the phosphate group of pS133 of pKID.39 Importantly, there are no direct contacts between the bound activation domains of MLL and pKID through which co-operativity between transcription factors could be mediated.
Structural Comparison
The pairwise rmsd between backbone atoms of the well-structured parts of the KIX domain (residues 589–669) in KIX·MLL and KIX·MLL·pKID is 1.07, which is comparable to the rms of the structural bundles of the two complexes (0.61 and 0.87, respectively). This is consistent with the observation of only small chemical shift differences of backbone (15N, 1HN, 13Cα) resonances between the binary and ternary complexes.17 It is therefore evident from the NMR structural analysis of the two complexes that the KIX protein backbone is not significantly affected by binding pKID. In addition, the methyl group NOE patterns are generally fairly similar for the binary and ternary complexes, consistent with well-defined and tightly packed hydrophobic cores. However, a number of significant differences between the NOE patterns of KIX·MLL and KIX·MLL·pKID indicate that the binding of pKID to binary KIX·MLL is accompanied by a small but measurable rearrangement of the hydrophobic core: for example, while relatively strong NOEs are observed, in the binary complex KIX·MLL, between the δ1 methyl group of I660 and both methyl groups of L607, these NOEs are absent in the ternary complex (along with their symmetry-equivalent NOEs in the three-dimensional experiments). Likewise, the NOE signal intensity between the δ1 methyl group of I657 and the backbone amide proton indicates a change in orientation of the I657 side chain upon binding pKID.
In Figure 2, the hydrophobic cores of KIX·MLL and KIX·MLL·pKID complexes are compared. In the binary complex KIX·MLL, the δ1 methyl group of I660 is packed to the γ1 methylene group of I611 and the methyl group of A610. Binding of pKID, however, is accompanied by a change of the I660 side chain χ1 torsion angle, leading to an increase of the distance between the δ1 methyl group of I660 and the side chain methyls of residues I611, A610, and L607. Concomitantly, upon transition from binary KIX·MLL to ternary KIX·MLL·pKID, the side chain χ1 torsion angle of isoleucine I657 is rotated from gauche– to gauche+, thereby linking the conformational adaption of the hydrophobic core to the c-Myb/pKID binding surface of the KIX domain. In KIX·MLL·pKID, the δ1 methyl group of I657 forms close hydrophobic contacts with the side chains of I137 and L141 of the ligand peptide pKID.
Figure 2.
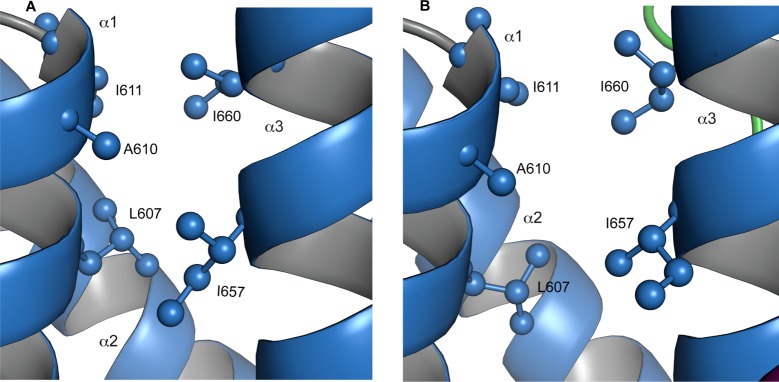
Close-up view of the hydrophobic core residues L607, A610, I611, I657, and I660 in (A) KIX·MLL and (B) KIX·MLL·pKID.
Structural Validation Using 13C Chemical Shifts
We used methyl carbon 13C chemical shifts to validate the NOE-based hydrophobic packing of the KIX domain in the two complexes. Isoleucine (χ2), leucine (χ2), and valine (χ1) rotameric distributions were determined from 13C chemical shifts using quantitative expressions that are available for these residues.43−45 A comparison of the chemical shift based populations of χ2 and χ1 rotamers with the dihedral angles of the NMR solution structures of binary KIX·MLL and ternary KIX·MLL·pKID is shown in Table 2, indicating a clear correspondence between the rotameric states observed in the three-dimensional structures and the 13C chemical shift data in both complexes. For example, only for two (out of nine) leucine residues, L599 and L628, the χ2 gauche+ conformer is present in both complexes, while all other leucine residues populate the χ2 trans conformer. This is in very good agreement with the 13C chemical shift data, from which χ2 gauche+ populations exceeding 50% are only predicted for L599 and L628 (with the exception of the highly dynamic C-terminal L672, for which a distribution of χ2 rotamers is found in ternary KIX·MLL·pKID, and L620, which populates the unusual gauche– conformer in binary KIX·MLL). Likewise, all three isoleucine residues are found to populate the χ2 trans conformer in our structures in both complexes, and all five well structured valine residues are found in χ1 trans conformation (only for the highly dynamic N-terminal V587 a distribution of χ1 rotamers is found in both complexes), consistent with their methyl group 13C chemical shifts. Taken together, these data clearly show that the conformation in the solution structures of both complexes (binary KIX·MLL and ternary KIX·MLL·pKID) represents the most populated side chain rotameric states of isoleucine, leucine, and valine residues in all cases.
Table 2. Comparison of Side Chain Rotameric Distributions (χ2 in Leucine and Isoleucine, χ1 in Valine) as Derived from Methyl 13C Chemical Shiftsa with the NMR Solution Structures of KIX·MLL (2LXS) and KIX·MLL·pKID (2LXT).
| KIX·MLL | KIX·MLL·pKID | ||||
|---|---|---|---|---|---|
| χ2 t/g+ (%) from 13C shifts | χ2 rotamer in 2LXS | χ2 t/g+ (%) from 13C shifts | χ2 rotamer in 2LXT | change of χ2 t/g+ (%) | |
| L599 | 33:67 | g+ | 32:68 | g+ | 1 |
| L603 | 81:19 | t | 73:27 | t | 8 |
| L607 | 57:43 | t | 62:38 | t | 5 |
| L620 | 71:29 | g– | 71:29 | t | 0 |
| L628 | 43:57 | g+ | 29:71 | g+ | 14 |
| L652 | 88:12 | t | 87:13 | t | 1 |
| L653 | 59:41 | t | 67:33 | t | 8 |
| L664 | 80:20 | t | 80:20 | t | 0 |
| L672 | 69:31 | t | 69:31 | g+/t/g– | 0 |
| χ2 t/g– (%) from 13C shifts | χ2 rotamer in 2LXS | χ2 t/g– (%) from 13C shifts | χ2 rotamer in 2LXT | change of χ2 t/g– (%) | |
| I611 | 87:13 | t | 83:17 | t | 4 |
| I657 | 74:26 | t | 67:33 | t | 7 |
| I660 | 89:11 | t | 93:7 | t | 4 |
| χ1 t/g+/g– (%) from 13C shifts | χ1 rotamer in 2LXS | χ1 t/g+/g– (%) from 13C shifts | χ1 rotamer in 2LXT | change of χ1 t/g+/g– (%) | |
| V587 | 70:9:21 | t/g– | 70:9:21 | g+/t/g– | 0 |
| V595 | 90:0:10 | t | 89:0:11 | t | 1 |
| V604 | 87:13:0 | t | 87:13:0 | t | 0 |
| V608 | 73:27:0 | t | 75:25:0 | t | 2 |
| V629 | 90:10:0 | t | 90:10:0 | t | 0 |
| V635 | 98:0:2 | t | 99:0:1 | t | 1 |
t, trans, g–, gauche–; g+, gauche+; populations are given in percent (%), and the most populated rotameric state is printed in bold face.
Pico- to Nanosecond Time Scale Dynamics of KIX and KIX Complexes
To characterize the dependence of the conformational flexibility of the KIX domain on the presence of bound ligands, we determined backbone amide order parameters, which are sensitive reporters of pico- to nanosecond time scale dynamics,46 for four different (complexed) states of the KIX domain: unliganded KIX, binary KIX·MLL, binary KIX·pKID, and ternary KIX·MLL·pKID (Figure 3). In binary and ternary complexes of KIX, the three-helix scaffold of the protein is well-ordered in solution, with backbone order parameters of ca. 0.9 for helices α1−α3. Order parameters of the unliganded KIX domain are measurably lower (the average of all order parameters in helices α1−α3, Savg2, is 0.85), suggesting a higher level of flexibility on the pico- to nanosecond time scale. Significantly lower than average order parameters are also found for the N-termini (including the 310 helix G1) and the C-termini of the protein in all four states, consistent with the dynamic fraying of these segments of the protein that is observed in the structural bundles. In addition, the order parameters of the backbone amides in helix α3 suggest that the C-terminal half of this helix is more rigid in complexes containing MLL compared with unliganded KIX and KIX·pKID. This observation agrees with the low degree of dynamic fraying that is observed for the C-terminal half of helix α3 in the structural bundles of KIX·MLL and KIX·MLL·pKID.10
Figure 3.
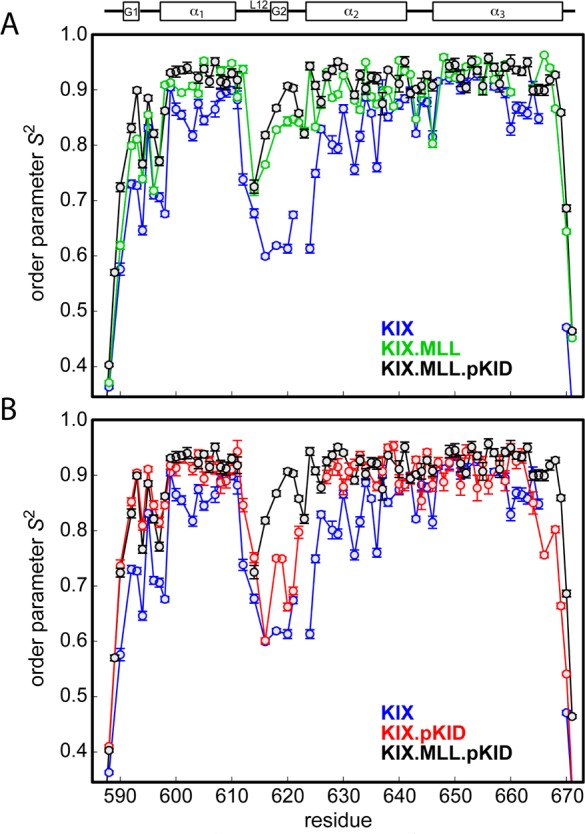
Comparison of backbone amide 15N order parameters S2 in (A) KIX (blue), binary KIX·MLL (green), and ternary KIX·MLL·pKID (black) and (B) KIX (blue), binary KIX·pKID (red), and ternary KIX·MLL·pKID (black). Error bars are shown and lines between data points are drawn except in cases where S2 could not be determined due to resonance overlap for two or more consecutive backbone amides.
The NMR relaxation data also indicate significant pico- to nanosecond time scale dynamics in the loop region L12, along with a clear difference between different complexes of KIX. For this part of the KIX backbone, average order parameters vary in a stepwise manner upon binding ligands, with Savg2 values of 0.64 in the free form of the protein and 0.85 in the ternary complex KIX·MLL·pKID and intermediate levels of dynamics for binary complexes KIX·MLL and KIX·pKID (Savg2 of 0.80 and 0.71, respectively). Contrary to the stepwise changes of the order parameters in the L12 loop, which forms direct interactions with MLL in binary KIX·MLL and ternary KIX·MLL·pKID, the three-helix scaffold of the binary KIX complexes rigidifies upon binding a single ligand (either MLL or pKID) and shows no further rigidification upon binding an additional ligand molecule to form the ternary complex KIX·MLL·pKID. This behavior is most prominent for helices α1 and α2 of the KIX domain, helix α1 (KIX Savg2 = 0.84; KIX·MLL Savg2 = 0.90; KIX·pKID Savg2 = 0.90; KIX·MLL·pKID, Savg2 = 0.91) and α2 (KIX Savg2 = 0.82; KIX·MLL Savg2 = 0.90; KIX·pKID, Savg2 = 0.91; KIX·MLL·pKID Savg2 = 0.91). For helix α3, resonance overlap in unliganded KIX prevents the measurement of order parameters for four C-terminal residues of helix α3 (666–669). Comparison of the order parameters of backbone amides in the N-terminal part of helix α3 (646–665), however, suggests that this part of the three-helix scaffold is less dynamic on a pico- to nanosecond time scale in the unliganded protein and only slightly changes its dynamic properties when ligand(s) are bound to the protein, helix α3 (KIX Savg2 = 0.90; KIX·MLL Savg2 = 0.92; KIX·pKID Savg2 = 0.91; KIX·MLL·pKID Savg2 = 0.93).
The KIX backbone amide order parameter data display a relatively large variation for α2 helix in the unliganded form of the protein. An oscillatory behavior of the order parameters is observed for the segment of helix α2 between residues 625–637 with lowest order parameters found for residues M625, L628, V629, K632, K633, and E636 and higher order parameters for residues between these. Of note, the more dynamic backbone amides in helix α2 of unliganded KIX are located on the side of the helix that contacts helix α1. We have previously shown that helix α1 is conformationally less stable than helices α2 and α3, with a tendency to unfold and refold (in milliseconds) in the unliganded form of the protein,47 suggesting a dynamic coupling of microsecond to millisecond and picosecond to nanosecond motions. The local unfolding–refolding process is suppressed if MLL or pKID is bound to the KIX domain.17 Likewise, upon binding ligand (MLL, pKID, or both), the oscillatory behavior of the order parameters in helix α2 is lost, and helix α2 displays relatively uniform order parameters in ligand bound states.
Taken together, the average order parameters of residues in helical segments of the KIX domain (α1−α3) increase from 0.85 in free KIX to 0.91 and 0.90 in the binary complexes KIX·MLL and KIX·pKID, respectively, suggesting significant rigidification of the KIX backbone upon peptide binding. Unlike for the loop L12, formation of the ternary complex KIX·MLL·pKID (average order parameter in α1−α3, Savg2 = 0.91) is not accompanied by further rigidification of the three-helix bundle scaffold.
Discussion
NMR relaxation dispersion experiments suggest that allosteric coupling in the KIX domain involves conformational selection of a ca. 7% low-populated (excited-state) conformer in the binary complex formed by KIX with MLL whose structure resembles the ternary complex.17 In agreement with these experimental data, molecular dynamics studies showed that MLL binding to the KIX domain results in a redistribution of the conformational substates of this protein toward conformers that favor the binding of the second, remote ligand.21 Side chain methyl group dynamic NMR data indicate that allosteric communication between the MLL and pKID/c-Myb binding sites proceeds through a conserved hydrophobic network in KIX that is formed by the side chains of aliphatic residues. A comparison of binary KIX·MLL and ternary KIX·MLL·pKID complexes of KIX is shown in Figure 1. It is evident that conformational changes of the KIX backbone upon binding of pKID to the binary KIX·MLL complex are indeed minor, which is in agreement with the small magnitude of backbone amide 15N and backbone carbon 13Cα chemical shift changes that are observed upon ligand binding.17 Our NMR solution structures show, however, that binding of the pKID activation domain to binary KIX·MLL is accompanied by a small but significant repacking of the hydrophobic core of the KIX domain, involving residues L607, L628, and I660 (Figure 2). Concomitantly, the side chain of residue I657 flips around its χ1 dihedral angle from gauche– to gauche+, positioning its δ1 methyl group at the surface of the KIX domain in the ternary complex. In the ternary complex of KIX with MLL and pKID, the I657 side chain is part of the hydrophobic groove on the surface of KIX that represents the docking interface for pKID, where it hydrophobically packs to residues I137 and L141 of the pKID amphipathic helix αB (see below). Both I137 and L141 are part of the consensus motif for ligands that bind to the KIX domain.48
Allosteric Communication through Redistribution of Rotameric States
It was recently shown that the carbon 13C chemical shifts of methyl groups in isoleucine, leucine, and valine provide a very sensitive measure of the side chain conformations of these residues.49 Assuming that conformational sampling of side chain dihedral angles (χ2 in leucine and isoleucine, χ1 in valine) can be described in terms of jumps between a limited number of rotameric states, the methyl group 13C chemical shifts of these residues can be used to determine the populations of these rotameric states. For example, with respect to leucine side chain χ2 dihedral angles, to a very good approximation only the trans and gauche+ conformers are sampled in solution, and their populations can be determined from the difference of their δ-methyl 13C chemical shifts.43 Similar approaches are available for isoleucine χ2 and valine χ1 rotameric distributions.44,45
The NMR solution structures of the binary and ternary complexes of the KIX domain that are presented here are based on backbone chemical shifts, NOEs, and residual dipolar couplings as experimental restraints, while isoleucine, leucine, and valine methyl group chemical shifts were not used as restraints in the structure calculations. It is therefore possible to validate the structures by comparison with the rotameric states of isoleucine, leucine, and valine that can be derived from methyl 13C chemical shifts (Table 2). We find excellent agreement between the methyl 13C chemical shifts and the side chain rotamers that are present in the solution structures of both complexes (binary KIX·MLL and ternary KIX·MLL·pKID), suggesting the conformation that is present in the NMR structural bundles represents the most populated rotameric state of isoleucine, leucine, and valine residues in all cases.
It is clear from the chemical shift data, however, that the three-dimensional solution structures of KIX complexes do not fully capture the dynamic nature of this protein. As evident from Table 2, populations of alternative rotameric states of 30% and more are not uncommon, which is in agreement with other proteins.43,44 Intriguingly, however, the chemical shift data suggest that the distributions of isoleucine and leucine χ2 dihedral angles are not identical in binary KIX·MLL and ternary KIX·MLL·pKID: for a subset of residues (L603, L607, I611, L628, L653, I657, and I660) conformational sampling is different in the two complexes, indicating a redistribution of their individual χ2 rotamers by 4–14% upon binding of the pKID ligand peptide, as shown in Figure 4. These residues cluster together in the molecular structure of the KIX domain and form part of the hydrophobic core of the protein. This is contrasted by residues L599, L620, L652, L664, L672, and all six valines, for which rotamer populations are identical (≤1% change) in the two structures and which are located outside the hydrophobic core of KIX. The 13C chemical shift data thus suggest that the conformational transition between the two complexes is accomplished through a relatively subtle (<14%) redistribution of the rotameric states of residues that form the hydrophobic core of the protein.
Figure 4.
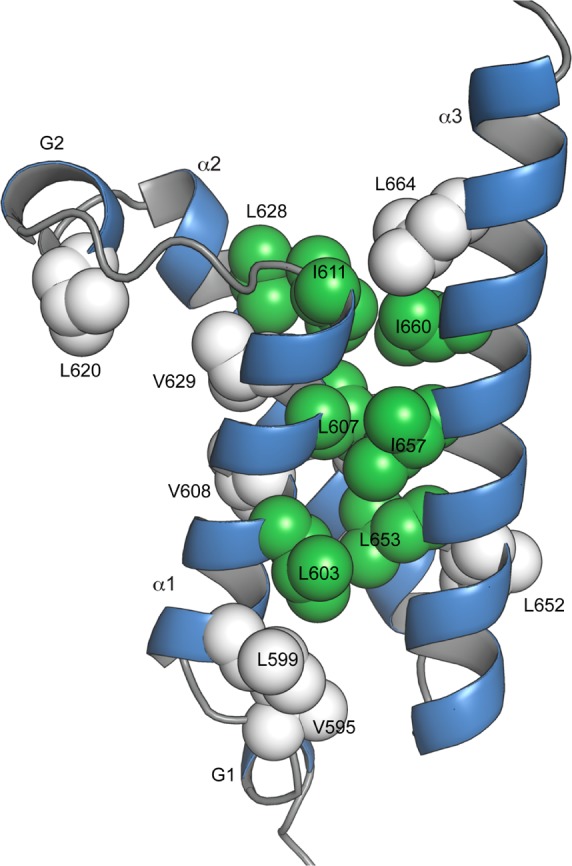
Conformational sampling of side chain dihedral angles (χ2 in leucine and isoleucine, χ1 in valine) in binary KIX·MLL and ternary KIX·MLL·pKID. The backbone of KIX·MLL is shown as ribbon. Leucine, isoleucine, and valine side chains are displayed as spheres and color-coded in green (change of rotamer distribution ≥4% between KIX·MLL and KIX·MLL·pKID) and white (change of rotamer distribution <2%).
It is intriguing that the hydrophobic core residues for which we observe the repacking upon ligand binding include the ones that were previously identified to be part of the hydrophobic cluster that constitutes the allosteric network in KIX.15,17 Using side chain methyl relaxation dispersion experiments, we found a clear correspondence between the hydrophobic core chemical shifts of the ternary KIX complex and the shifts of the excited state in the binary complex of KIX with MLL.17 This shows that the dynamic repacking process of the hydrophobic core that we observe already occurs in binary KIX·MLL (i.e., before pKID binds) during the transition between the 93% populated ground and the 7% populated excited states. Taken together, the chemical shift and the structural data provide a quantitative dynamic picture of allosteric communication between the two remote ligand binding sites in the KIX domain.
Figure 5 shows a close-up view of the KIX/pKID binding surface in the ternary complex KIX·MLL·pKID. The δ1 methyl group of KIX residue I657 is exposed to the surface of the domain and forms part of the hydrophobic pocket that accommodates the amphipathic helix αB of the ligand peptide pKID.39 The hydrophobic side chains of two residues of pKID, I137 and L141, pack to the I657 of the KIX domain. I137 and L141 of pKID are known to be critical for the molecular recognition of ligands binding to the KIX domain: both I137 and L141 are part of the conserved ΦXXΦΦ sequence motif, where Φ is a hydrophobic residue and X is an arbitrary residue, which is characteristic for ligands that bind to KIX.48 Any conformational change at the pKID/c-Myb hydrophobic binding groove will thus very likely modulate the efficiency with which hydrophobic contacts with ligand peptides are formed and affect their affinities. With respect to allosteric coupling, the presence of a higher affinity (excited state) conformer, even if populated only to a low extent, can act as a driving force for the ligand binding process. Recognition and binding of peptides will predominantly occur through the higher affinity conformer, whose structure is complementary to the ligand, while the thermodynamic equilibrium between ground and excited states will be re-established through the allosteric conformational transition, resulting in a redistribution of the conformational substates toward the structure of the higher affinity state. The relaxation dispersion data suggest that in the 7% populated excited state in the binary complex KIX·MLL residue I657 is indeed present in a conformation that is similar to the ternary complex.17
Figure 5.
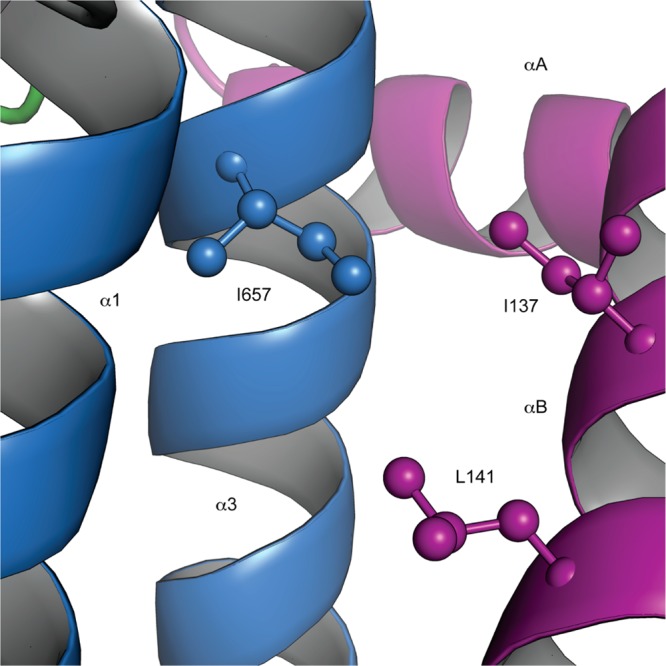
Close-up view of the solution structure of the ternary complex KIX·MLL·pKID showing the interaction surface of the KIX domain with the pKID peptide. The backbone of the KIX domain is displayed as blue ribbon, along with the side chain heavy atoms of I657. The pKID backbone (magenta ribbon) and the side chains of pKID residues I137 and L141, which interact with the hydrophobic side chain of I657, are displayed.
Previous reports have indeed shown that the complementarity of the hydrophobic interactions between the pKID binding pocket and peptide ligands plays a critical role for the affinity with which these ligands are bound to the KIX domain.40 NMR solution structures of KIX bound to the activation domain of the transcription factor c-Myb indicate that the positions that are equivalent to I137 and L141 in pKID are taken by two leucine residues in c-Myb (L298 and L302, both part of the c-Myb ΦXXΦΦ motif). It is evident from the KIX·c-Myb structures that the side chain of L302 of c-Myb penetrates more deeply into the hydrophobic pocket, which is formed by the KIX domain, than the equivalent L141 of pKID,39 enabling the formation of hydrophobic contacts of its methyl groups with the side chain of KIX residue L607 at the bottom of the pocket. Based on these observations, it was suggested that the hydrophobic interactions between pKID and KIX are less optimal than those between pKID and c-Myb, which explains the relatively low affinity of unphosphorylated KID to KIX.40
A crystallographic study of KIX bound to a covalently linked small-molecule ligand underlines the remarkable ability of this protein to adapt its hydrophobic core in response to a binding partner.50 In this structure, an aromatic ring that is part of the ligand molecule penetrates into the hydrophobic core of the KIX domain, forming contacts with residues that are part of the allosteric network (I611, L607, L628). Binding assays showed that in this small-molecule complex of KIX allosteric communication is disrupted, most likely by engaging the residues that constitute the allosteric network of the protein. Indeed, in the crystal structure of this complex, the side chain χ1 torsion angle of isoleucine 657 occupies the gauche– conformation, similar to the lower affinity complex KIX·MLL. It thus appears that constraining the hydrophobic core by insertion of a ligand molecule attenuates the malleability of the allosteric network that is required for communication between the binding sites, in agreement with our proposed dynamic allosteric mechanism.
Allosteric Communication in the Opposite Direction
While our relaxation dispersion experiments in combination with the high-resolution structures presented here provide insights into the mechanism by which KIX propagates information about the presence of MLL to the c-Myb/pKID binding site, the molecular mechanism of communication in the opposite direction (from the c-Myb/pKID binding site to the MLL binding site) is not obvious.17 The lack of significant contributions of micro- to millisecond time scale dynamic processes to NMR spin relaxation rates indicates that either the population of any higher energy state(s) that might be present in KIX·pKID or KIX·c-Myb binary complexes is too low or the time scale of the process is outside the micro- to millisecond window that can be studied by relaxation dispersion NMR.
Indeed, the NMR spin relaxation data reported here argue for a contribution of pico- to nanosecond time scale dynamic processes to the mechanism of allosteric coupling from the c-Myb/pKID binding site to the MLL binding site. Upon binding of pKID to the KIX domain, we observed a change in pico- to nanosecond dynamics for the KIX backbone for segments of the protein that are remote from the binding site of the peptide. In particular, two of the three helices (α1 and α2) of the KIX domain that form the three-helix scaffold of the protein rigidify, along with the connecting linker loop L12 and the N-terminus of the domain. These findings are in agreement with the ITC data published by Goto et al.,15,17 who showed that binding of pKID to KIX decreases the entropic penalty for MLL binding. Furthermore, molecular dynamics studies suggest that c-Myb binding to the c-Myb/pKID binding site of KIX indeed rigidifies the loop L12 and G2 region.21 This is contrasted by the pKID binding surface itself, which is mostly formed by residues in helix α3 and which does not display a significant change in pico- to nanosecond time scale dynamics upon binding. The rigidification of the linker L12, which has no direct contacts with pKID, is particularly interesting, because it is believed to play a critical role for binding MLL.10 Based on the observation of chemical shift averaging and narrow backbone amide resonances, it has been suggested that the linker L12 is conformationally flexible in order to enable the interaction of this binding surface of the KIX domain with a variety of disparate ligand sequences, while in complex with MLL, the conformational flexibility is reduced.10,39 In addition, this part of the protein is considerably displaced upon MLL binding, allowing the side chain of KIX residue F612 to form hydrophobic contacts with the MLL ligand. In light of the significance of this loop domain for binding MLL,21 this long-range dynamic coupling between the two binding sites is likely of functional significance.
A markedly different behavior is found for MLL binding to KIX. While the presence of bound MLL significantly increases the order parameters of amino acid residues that are part of the hydrophobic MLL binding pocket (loop L12) and the helices α1 and α2 of the three-helix scaffold of the protein as well as the C-terminal end of helix α3, we find no significant changes of order parameters at the pKID binding site. Thus, binding of the activation domain of MLL appears to cause local rigidification but has no long-range effect on dynamic processes on the pico- to nanosecond time scale. Interestingly, however, the loss of flexibility on the pico- to nanosecond time scale upon MLL binding of the L12 loop is accompanied by a significant increase of dynamics on a millisecond time scale (the allosteric conformational transition) that involves a contiguous network of residues connecting the L12 loop to the remote pKID binding site of the KIX domain.17
Dynamics and Allostery
Dynamic allosteric coupling has been reported for a variety of proteins.51−55 For example, various variants of the catabolite activator protein (CAP) differentially populate active and inactive DNA-binding states.56 Allosteric regulation of CAP is entirely of an entropic nature and involves the redistribution of the conformational substates of the protein in response to the presence of a ligand molecule.57 Likewise, phosphorylation of the bacterial nitrogen regulatory protein C shifts the balance of populations of the different forms of this protein from the inactive to the active form,58 and the PBX1 homeodomain transiently folds into a conformation in which the binding sites for DNA and the transcription factor Hox are preorganized even in the absence of ligands.59 In PDZ3, binding function is regulated through an entirely dynamic allosteric mechanism that lacks measurable conformational changes.60 Together with our results for the KIX domain of CBP, these data underscore the mechanistic significance of dynamic equilibria for allosteric regulation and ligand recognition. Of note, such population shift mechanisms enable the modulation of binding affinities in a versatile manner by the extent to which the higher affinity conformer is populated, as well as by the difference in binding affinities of the different conformers.
It is noteworthy that the KIX domain displays a nonuniform distribution of aromatic and aliphatic residues in its hydrophobic core. The interior of the hydrophobic core (within the three-helix scaffold) is formed by nonaromatic residues and contains mostly isoleucines, leucines, valines, and alanines, while at the surface of KIX, aromatic residues are enriched, making van der Waals contacts with the bound ligand peptides.10,39,41 A possible involvement of these aromatic residues at the protein–ligand interface in allosteric coupling has been inferred from relaxation dispersion and 19F NMR measurements,17,61 but their exact mechanistic role is not evident. Considering the dynamic nature of the repacking process of isoleucine, leucine, and valine side chains in the KIX hydrophobic core, it is tempting to speculate that aliphatic side chains could be more amenable for such processes than aromatic side chains. Sequence comparison showed that the aliphatic residues in the KIX hydrophobic core are conserved to a higher level than the aromatic residues,17 arguing for an evolutionary conservation of such functional dynamics.62,63 However, further experimental studies of allosteric proteins, including techniques that probe the dynamic behavior of aromatic side chains, will be necessary to comprehensively understand the structural prerequisites for dynamic allosteric coupling.
Acknowledgments
We are grateful to D. Flemming Hansen (University College London) for insightful discussions. We thank Paul Schanda (IBS Grenoble, France) for preliminary alignment experiments. This work was supported by the Austrian Science Fund FWF (P22735).
Accession Codes
Structural coordinates and NMR restraints have been deposited in the Protein Data Bank and BioMagResBank with the following accession numbers: KIX·MLL (PDB ID 2LXS; BMRB 18694) and KIX·MLL·pKID (PDB ID 2LXT; BMRB 18695).
Author Present Address
§ S. Brüschweiler: Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA.
The authors declare no competing financial interest.
References
- Carey M. (1998) The enhanceosome and transcriptional synergy. Cell 92, 5–8. [DOI] [PubMed] [Google Scholar]
- Panne D. (2008) The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242. [DOI] [PubMed] [Google Scholar]
- Ptashne M.; Gann A. (1997) Transcriptional activation by recruitment. Nature 386, 569–577. [DOI] [PubMed] [Google Scholar]
- Lemon B.; Tjian R. (2000) Orchestrated response: A symphony of transcription factors for gene control. Genes Dev. 14, 2551–2569. [DOI] [PubMed] [Google Scholar]
- Goodman R. H.; Smolik S. (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev. 14, 1553–1577. [PubMed] [Google Scholar]
- Kornberg R. D. (2005) Mediator and the mechanism of transcriptional activation. Trends. Biochem. Sci. 30, 235–239. [DOI] [PubMed] [Google Scholar]
- Merika M.; Thanos D. (2001) Enhanceosomes. Curr. Opin. Genet. Dev. 11, 205–208. [DOI] [PubMed] [Google Scholar]
- Vo N.; Goodman R. H. (2001) CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 6, 197–208. [DOI] [PubMed] [Google Scholar]
- De Guzman R. N.; Goto N. K.; Dyson H. J.; Wright P. E. (2006) Structural basis for cooperative transcription factor binding to the CBP coactivator. J. Mol. Biol. 355, 1005–1013. [DOI] [PubMed] [Google Scholar]
- Yang F.; Vought B. W.; Satterlee J. S.; Walker A. K.; Jim Sun Z. Y.; Watts J. L.; DeBeaumont R.; Saito R. M.; Hyberts S. G.; Yang S.; Macol C.; Iyer L.; Tjian R.; van den Heuvel S.; Hart A. C.; Wagner G.; Naar A. M. (2006) An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442, 700–704. [DOI] [PubMed] [Google Scholar]
- Thakur J. K.; Arthanari H.; Yang F.; Pan S. J.; Fan X.; Breger J.; Frueh D. P.; Gulshan K.; Li D. K.; Mylonakis E.; Struhl K.; Moye-Rowley W. S.; Cormack B. P.; Wagner G.; Naar A. M. (2008) A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452, 604–609. [DOI] [PubMed] [Google Scholar]
- Ernst P.; Wang J.; Huang M.; Goodman R. H.; Korsmeyer S. J. (2001) MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 21, 2249–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Marshall C. B.; Yamamoto K.; Li G. Y.; Gasmi-Seabrook G. M.; Okada H.; Mak T. W.; Ikura M. (2012) Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc. Natl. Acad. Sci. U.S.A. 109, 6078–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto N. K.; Zor T.; Martinez-Yamout M.; Dyson H. J.; Wright P. E. (2002) Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). J. Biol. Chem. 277, 43168–43174. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Reginato M. J.; Andrews N. C.; Lazar M. A. (1997) The transcriptional integrator CREB-binding protein mediates positive cross talk between nuclear hormone receptors and the hematopoietic bZip protein p45/NF-E2. Mol. Cell. Biol. 17, 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüschweiler S.; Schanda P.; Kloiber K.; Brutscher B.; Kontaxis G.; Konrat R.; Tollinger M. (2009) Direct observation of the dynamic process underlying allosteric signal transmission. J. Am. Chem. Soc. 131, 3063–3068. [DOI] [PubMed] [Google Scholar]
- Tsai C. J.; Kumar S.; Ma B.; Nussinov R. (1999) Folding funnels, binding funnels, and protein function. Protein Sci. 8, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. (1972) Ligand binding and internal equilibria in proteins. Biochemistry 11, 864–878. [DOI] [PubMed] [Google Scholar]
- Tobi D.; Bahar I. (2005) Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. U.S.A. 102, 18908–18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz E. N.; Nussinov R.; Haliloglu T. (2012) Conformational control of the binding of the transactivation domain of the MLL protein and c-Myb to the KIX domain of CREB. PLoS Comput. Biol. 8, e1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollinger M.; Kloiber K.; Agoston B.; Dorigoni C.; Lichtenecker R.; Schmid W.; Konrat R. (2006) An isolated helix persists in a sparsely populated form of KIX under native conditions. Biochemistry 45, 8885–8893. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Neri D.; Szyperski T.; Otting G.; Senn H.; Wuthrich K. (1989) Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 28, 7510–7516. [DOI] [PubMed] [Google Scholar]
- Zwahlen C.; Gardner K. H.; Sarma S. P.; Horita D. A.; Byrd R. A.; Kay L. E. (1998) An NMR experiment for measuring methyl-methyl NOEs in C-13-labeled proteins with high resolution. J. Am. Chem. Soc. 120, 7617–7625. [Google Scholar]
- Eichmüller C.; Schüler W.; Konrat R.; Kräutler B. (2001) Simultaneous measurement of intra- and intermolecular NOEs in differentially labeled protein-ligand complexes. J. Biomol. NMR 21, 107–116. [DOI] [PubMed] [Google Scholar]
- Chou J. J.; Gaemers S.; Howder B.; Louis J. M.; Bax A. (2001) A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR 21, 377–382. [DOI] [PubMed] [Google Scholar]
- Ottiger M.; Delaglio F.; Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378. [DOI] [PubMed] [Google Scholar]
- Ortega-Roldan J. L.; Jensen M. R.; Brutscher B.; Azuaga A. I.; Blackledge M.; van Nuland N. A. (2009) Accurate characterization of weak macromolecular interactions by titration of NMR residual dipolar couplings: application to the CD2AP SH3-C:ubiquitin complex. Nucleic Acids Res. 37, e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüschweiler R.; Liao X.; Wright P. E. (1995) Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268, 886–889. [DOI] [PubMed] [Google Scholar]
- Lee L. K.; Rance M.; Chazin W. J.; Palmer A. G. 3rd. (1997) Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J. Biomol. NMR 9, 287–298. [DOI] [PubMed] [Google Scholar]
- Cole R.; Loria J. P. (2003) FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. J. Biomol. NMR 26, 203–213. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. (2009) TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieping W.; Habeck M.; Bardiaux B.; Bernard A.; Malliavin T. E.; Nilges M. (2007) ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382. [DOI] [PubMed] [Google Scholar]
- Zweckstetter M.; Hummer G.; Bax A. (2004) Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys. J. 86, 3444–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- Linge J. P.; Williams M. A.; Spronk C. A.; Bonvin A. M.; Nilges M. (2003) Refinement of protein structures in explicit solvent. Proteins 50, 496–506. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I.; Perez-Alvarado G. C.; Parker D.; Dyson H. J.; Montminy M. R.; Wright P. E. (1997) Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91, 741–752. [DOI] [PubMed] [Google Scholar]
- Zor T.; De Guzman R. N.; Dyson H. J.; Wright P. E. (2004) Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J. Mol. Biol. 337, 521–534. [DOI] [PubMed] [Google Scholar]
- Denis C. M.; Chitayat S.; Plevin M. J.; Wang F.; Thompson P.; Liu S.; Spencer H. L.; Ikura M.; LeBrun D. P.; Smith S. P. (2012) Structural basis of CBP/p300 recruitment in leukemia induction by E2A-PBX1. Blood 120, 3968–3977. [DOI] [PubMed] [Google Scholar]
- Arai M.; Dyson H. J.; Wright P. E. (2010) Leu628 of the KIX domain of CBP is a key residue for the interaction with the MLL transactivation domain. FEBS Lett. 584, 4500–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder F. A. (2009) Leucine side-chain conformation and dynamics in proteins from 13C NMR chemical shifts. ChemBioChem 10, 1477–1479. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Neudecker P.; Kay L. E. (2010) Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J. Am. Chem. Soc. 132, 7589–7591. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Kay L. E. (2011) Determining valine side-chain rotamer conformations in proteins from methyl 13C chemical shifts: application to the 360 kDa half-proteasome. J. Am. Chem. Soc. 133, 8272–8281. [DOI] [PubMed] [Google Scholar]
- Mittermaier A.; Kay L. E. (2006) New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Brutscher B.; Konrat R.; Tollinger M. (2008) Folding of the KIX domain: characterization of the equilibrium analog of a folding intermediate using 15N/13C relaxation dispersion and fast 1H/2H amide exchange NMR spectroscopy. J. Mol. Biol. 380, 726–741. [DOI] [PubMed] [Google Scholar]
- Plevin M. J.; Mills M. M.; Ikura M. (2005) The LxxLL motif: A multifunctional binding sequence in transcriptional regulation. Trends Biochem. Sci. 30, 66–69. [DOI] [PubMed] [Google Scholar]
- London R. E.; Wingad B. D.; Mueller G. A. (2008) Dependence of amino acid side chain 13C shifts on dihedral angle: application to conformational analysis. J. Am. Chem. Soc. 130, 11097–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Majmudar C. Y.; Pomerantz W. C.; Gagnon J. K.; Sadowsky J. D.; Meagher J. L.; Johnson T. K.; Stuckey J. A.; Brooks C. L. 3rd; Wells J. A.; Mapp A. K. (2013) Ordering a dynamic protein via a small-molecule stabilizer. J. Am. Chem. Soc. 135, 3363–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J. F.; Gierasch L. M. (2006) The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 16, 102–108. [DOI] [PubMed] [Google Scholar]
- Kern D.; Zuiderweg E. R. (2003) The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13, 748–757. [DOI] [PubMed] [Google Scholar]
- Goodey N. M.; Benkovic S. J. (2008) Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 4, 474–482. [DOI] [PubMed] [Google Scholar]
- Csermely P.; Palotai R.; Nussinov R. (2010) Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q.; Karplus M. (2008) Allostery and cooperativity revisited. Protein Sci. 17, 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. R.; Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240. [DOI] [PubMed] [Google Scholar]
- Kalodimos C. G. (2012) Protein function and allostery: A dynamic relationship. Ann. N.Y. Acad. Sci. 1260, 81–86. [DOI] [PubMed] [Google Scholar]
- Volkman B. F.; Lipson D.; Wemmer D. E.; Kern D. (2001) Two-state allosteric behavior in a single-domain signaling protein. Science 291, 2429–2433. [DOI] [PubMed] [Google Scholar]
- Farber P. J.; Mittermaier A. (2011) Concerted dynamics link allosteric sites in the PBX homeodomain. J. Mol. Biol. 405, 819–830. [DOI] [PubMed] [Google Scholar]
- Petit C. M.; Zhang J.; Sapienza P. J.; Fuentes E. J.; Lee A. L. (2009) Hidden dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. U.S.A. 106, 18249–18254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz W. C.; Wang N.; Lipinski A. K.; Wang R.; Cierpicki T.; Mapp A. K. (2012) Profiling the dynamic interfaces of fluorinated transcription complexes for ligand discovery and characterization. ACS Chem. Biol. 7, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Ranganathan R. (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299. [DOI] [PubMed] [Google Scholar]
- Süel G. M.; Lockless S. W.; Wall M. A.; Ranganathan R. (2003) Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10, 59–69. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002), San Carlos, Ca, USA.