

Abstract
Replicating in vitro the complex in vivo tissue microenvironment has the potential to transform our approach to medicine and also our understanding of biology. In order to accurately model the 3D arrangement and interaction of cells and extracellular matrix, new microphysiological systems must include a vascular supply. The vasculature not only provides the necessary convective transport of oxygen, nutrients, and waste in 3D culture, but also couples and integrates the responses of organ systems. Here we combine tissue engineering and microfluidic technology to create an in vitro 3D metabolically active stroma (∼1 mm3) that, for the first time, contains a perfused, living, dynamic, interconnected human capillary network. The range of flow rate (μm/s) and shear rate (s−1) within the network was 0–4000 and 0–1000, respectively, and thus included the normal physiological range. Infusion of FITC dextran demonstrated microvessels (15–50 μm) to be largely impermeable to 70 kDa. Our high-throughput biology-directed platform has the potential to impact a broad range of fields that intersect with the microcirculation, including tumor metastasis, drug discovery, vascular disease, and environmental chemical toxicity.
Introduction
The concept of recapitulating 3D tissue function in vitro is not new. In fact, 3D models of tumor biology (tumor spheroids) were presented nearly four decades ago.1–3 However, 2D monolayer culture (“flat biology”) is technically simple, reproducible, and inexpensive; thus, the adoption of 3D systems has been relatively slow. Recently, interest in mimicking more complex physiological functions has been spurred by a growing appreciation of 3D cell–cell and cell–matrix interactions that mediate proper differentiation and promote survival (e.g., notch signaling).4 This is, perhaps, not surprising given that human organs and their associated functions are, with few exceptions, 3D. More recently, techniques have been developed to create 3D structures of cell-dense tissue such as cell-sheet tissue engineering5 and cells in gels in paper (CiGiP).6 While these techniques produce tissues that can recapitulate some 3D structure and function, if a normoxic (i.e., normal function) environment is required, the tissues are limited to <200 μm in depth, the diffusion limit of oxygen in most tissues. In order to create realistic 3D tissues of significant and physiologically relevant depth, a network of human vessels is required. Such models can be used to enhance our understanding of normal and pathological angiogenesis in physiologically relevant 3D environments, but can also be used to advance fields such as drug discovery or chemical toxicity screening.
Integrating microfluidics into in vitro cell culture has the potential to improve the physiological relevance by introducing convective flow, and thus more in vivo-like conditions for 3D systems.7,8 For example, endothelial cells (ECs) have been seeded in fabricated microchannels on both nonbiological9,10 substrates or within biological substrates11,12 to mimic vessels; however, while this approach offers some insight into the effects of convective forces on cells, the channels are not living vessels and thus are unresponsive to the dynamic metabolic demands of the surrounding environment.
An alternative approach to provide convective flow in an in vitro microenvironment is bulk perfusion (interstitial flow) of cell-seeded scaffolds with adjacent microfluidic channels.13–15 This approach permits control over both diffusive and convective mass transport, and thus manipulates interstitial biomechanical forces (e.g., shear stress) and biochemical cues (e.g., gradients of solutes). Interstitial flow significantly influences the development of capillaries,16 cell migration,17 stromal cell remodeling of the extracellular matrix,18 and sprout formation.11 Recently, a few notable studies have demonstrated that sprouting ECs from a cell-lined fabricated channel can invade the matrix and form nominal vessel structures.19–22 In two of these studies,20,21 large tubular structures (d> 50 μm) were perfused. However, these models have not demonstrated an interconnected, branching microvascular network. The ability to form such networks in vitro will be a critical step in advancing our understanding of angiogenesis and vascular remodeling.
Drawing from 3D cell culture techniques and microfluidic technology, our approach creates an environment with minimal artificial architecture. We provide matrix, cells, and angiogenic stimuli that allow capillaries to self-assemble into a continuous capillary network. The capillaries then anastomose with adjacent fluidic channels, allowing subsequent perfusion of the vessel network. In this work we report for the first time the perfusion of an in vitro capillary network of living, dynamic human microvessels at physiological flow and shear rates.
Materials and Methods
Cell culture
Human endothelial colony forming cell-derived ECs (ECFC-ECs) were isolated from cord blood as previously described1 and expanded on gelatin-coated flasks in endothelial growth medium-2 (EGM-2; Lonza, Wakersfield, MD). Normal lung human fibroblast (NHLFs) purchased from Lonza were cultured in fibroblast growth media (Lonza). NHLFs (used at passage 5–8) and ECFC-ECs (used at passages 4–6) were both grown in a 37°C/5% CO2/20% O2 incubator in 100% humidified air.
Microfabricating the device
The microfluidic device was created and bonded to a 1-mm-thick polydimethylsiloxane (PDMS) sheet using standard PDMS micromolding.23 First, a layer of (100 μm thick) SU-8 was spin-coated on to a Si-wafer (RCA-1 cleaned and 2% HF treated). Then, a single mask photolithography step patterned the tissue chamber, communication pores, and fluidic channels. The microfabricated SU-8 mold was then silanized with trichlorosilane (C8H4Cl3F13Si) in a vacuum chamber. A 8-mm-thick layer of PDMS was molded on the SU-8 mold to create the tissue chamber. The PDMS microfluidic device was de-molded and holes punched followed by plasma bonding to a 1-mm-thick PDMS sheet. Another 1-mm glass slide was then bonded to the other side of the 1-mm-thick PDMS sheet for mechanical support.
The device consisted of two fluidic channels separated by a central channel of 12 daisy-chained mm-sized diamond tissue chambers (1×2 mm) (Fig. 1a). The fluidic channels connected on either side of the tissue chamber via a single connecting pore 30 μm wide and 100 μm on each side (Fig. 1b). The pores were designed with curved boundaries to prevent fibrin gel leakage into the fluidic channel during fibrin gel loading. The curved boundary mimicked a capillary burst valve in which the surface tension at the fibrin–air interface increases as the fibrin flows into the pore until it equals the hydrostatic pressure difference across the boundary.24 At this point the liquid stops and does not enter any further into the fluidic channel. A hydrostatic pressure difference (10 mm H2O) using large reservoirs was used to control the direction and magnitude of convective flow through the channels and interstitial flow (0.8–10 μm/s) in the microtissues (Fig. 1c).
FIG. 1.

(a) The high-throughput microfluidic platform is constructed of optically clear polydimethylsiloxane (PDMS) and consists of outer microfluidic channels that connect to a series of central microtissue chambers through a communication pore on each side. A portion of the fluidic system includes a removable “jumper” that is in place during vessel network formation, but can be removed at a later time point to facilitate the characterization of the vessel network. (b) The central chamber is loaded with endothelial cells and stromal cells embedded in a fibrin matrix (cross section of the tissue chamber at the pore inlets as indicated by the vertical dotted black line in panel a). (c) The microfluidic channels connect to large media reservoirs that provide the hydrostatic pressure head needed for flow of media, as well as vessel network formation and later during perfusion of the vessel network. The platform supports the development of a robust interconnected microvessel network within 14–21 days (panel d is the bright-field image of the fluorescent image in panel e, scale bar=200 μm). Vessel structures enter the communication pores and form points of anastomosis with the microfluidic channel (panels f, g and h, scale bar=50 μm). (h) Fluorescent red microspheres (1 μm in diameter) introduced into the high-pressure fluid stream were visualized with confocal microscopy (white arrows) confirming patent and perfused vessels. Color images available online at www.liebertpub.com/tec
Device design
Two variations of the microfluidic device were used to investigate the development of perfused vessels under a broad range of pressures within the microtissue. Finite element stimulations were performed with COMSOL Multiphysics® 3.5a to predict initial pressure gradients and subsequent interstitial flow in the absence of a formed microvascular network as described previously.25 In both designs, the high-pressure arterial microfluidic channel connected consecutively to the microtissue compartments through the communication pores. In one adaptation (VP), the arterial microfluidic line from the last microtissue chamber (#12) connected immediately to the venular side of the same compartment and then consecutively to the remaining compartments in reverse numerical order (Fig. 2a). This design created a similar mean pressure within the microtissue chambers, and thus a negligible longitudinal (x-direction) interstitial velocity, but a wide variation in the transverse (y-direction) pressure drop and interstitial velocity across the microtissue chambers (Fig. 2c). During vessel network formation, the interstitial velocity and pressure drop, respectively, ranged from ∼8.5 μm/s (supraphysiological) and 3.5 mm H2O in compartment #1 to ∼0.8 μm/s (low physiological) and 0.3 mm H2O in compartment #12.
FIG. 2.

Finite element simulations demonstrate the ability to control the pressure distribution (a, b) and subsequent interstitial flow distribution within the device. In the first design the transverse pressure gradient across the microtissue chamber is variable (VP), or (ΔP1>ΔP12) while the mean pressure is constant (c). The alternate design creates equal (EQ) pressure gradients across the tissue microchambers (ΔP1=ΔP12), although the mean pressure of the chambers decreases as chamber number increases (d). Color images available online at www.liebertpub.com/tec
In the alternate design (EQ), the arterial microfluidic line from the last microtissue chamber connects initially to the venular side of the first compartment (#1), and then consecutively to the remaining microtissue chambers (in the same order as the arteriolar channel, Fig. 2b). This design created a larger pressure drop between (y-direction) adjacent compartments, and a large, yet near constant, transverse (x-direction) pressure drop (∼2 mm H2O) and interstitial flow (5–7 μm/s) across the microtissues Fig. 2d). For each microchamber in the EQ and VP design, the average volume flow was 26.1±2.8 nL/h and varied from 2.8 to 53.9 nL/h, respectively.
Seeding the device
The stromal chamber was prepared using a modified procedure for construction of in vitro vessel networks.1,4,25–27 To prepare cell–matrix solution, bovine fibrinogen (Sigma-Aldrich, St. Louis, MO) was dissolved in 1× Dulbecco's Phosphate Buffered Saline to a final concentration of 10 mg/mL. NHLFs and ECFC-ECs were suspended in the fibrinogen solution at a ratio of 2:1 to make a final cell density of 7.5×106 cells/mL total solution. The fibrinogen–cell solution was mixed with thrombin (50 U/mL) for a final concentration of 3 U/mL and quickly pipetted into the microchambers, where it was allowed to fully polymerize. Cells in the devices were maintained in fully supplemented EGM-2 in a 20% O2 incubator for 12 h to allow for cell attachment. After 12 h, the media in the devices was replaced with EGM-2 without vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) and for the remainder of the study the devices were cultured at 5% O2. Media in the reservoirs was leveled every other day where the maximum pressure variation was 21 μm H2O/h. This media change frequency created a maximum flow of 2.6 μL/h in the side channels. Cell viability was confirmed using Live/Dead assay (Invitrogen, Carlsbad, CA) at 40 days (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tec).
Capillary network growth and anastomosis
To promote the organization of the vessel network and anastomosis with the side channels, cells in the tissue chamber were grown under interstitial flow and in the absence of VEGF and bFGF for the first 14 days. The addition of exogenous growth factors was considered unnecessary and potentially confounding during development given that fibroblasts have been shown to secrete a variety of growth factors when cocultured with ECs.4,6,28 In order to stimulate a connection with the fluidic channel, vessels were encouraged to grow toward both pores on either side by alternating the direction of interstitial flow. After 1 week, the direction of the interstitial flow in the tissue channel was reversed by reversing the pressure head in the adjacent microfluidic channels. After the second week, the interstitial flow direction was again reversed, and fully supplemented (with VEFG and bFGF) EGM-2 was introduced into the fluidic channels and used for the remaining week of the experiment.
Immunofluorescent staining of devices and image analyses
To observe the vessel networks, microtissues were immunofluorescently stained with mouse anti-human CD31 antibody (Dako, Carpinteria, CA) and Alexa-Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) following a modified indirect staining procedure.1 Microtissues were first fixed by flowing formalin through the fluidic channel for 6 h. Blocking, washing and antibody incubation steps were also all conducted by flowing solutions through the fluidic channels for 1–3 days. Images of stained vessels were taken using a Zeiss LSM 510 Meta laser scanning confocal microscope (Zeiss, Jena, Germany). Vessels stained positive for CD31 were manually selected using ImageJ software (NIH, Bethesda, MD) and vessel network area as a percentage of the chamber area was assessed. Images of CD31 stained vessels were also used to measure vessel diameter and an index of network connectedness was determined using the following formula:
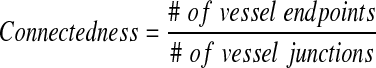 |
Values for connectedness approaching 0 indicate a well-connected network.
Perfusion analyses
Flow in formed capillaries was assessed in the third week of culture by adding 1 μm polystyrene fluorescent microspheres (Invitrogen) into the arteriole microfluidic channel and observed as they entered the capillaries via the communication pores. Images were captured at 10 Hz using a Nikon eclipse TE300 inverted microscope and a B/W CCD digital camera (Hamamatsu ORCA). Microspheres were manually tracked in perfused tissues from three separate microfluidic chips using ImageJ. To assess vessel permeability fluorescent dextran (70 and 150 kDa; Sigma-Aldrich) was introduced into the arteriole microfluidic channel at day 21, and visualized in the fluidic lines and central tissue chambers using fluorescent microscopy.
Statistical analyses
To determine significant differences between the data of the two designs, Student's t-test was performed where p<0.05 was considered significant. The data are expressed as mean±standard deviation.
Results
We constructed a microfluidic device of PDMS consisting of two fluid-filled outer microfluidic channels, mimicking the arteriole and venule, which connect via single communication pores on either side of 12 daisy-chained mm-sized diamond-shaped tissue microchambers. To induce vessel development and subsequent perfusion of the capillary network, anastomosis of the capillaries with the microfluidic channels was encouraged by manipulating both mechanical (pressure gradients, interstitial flow) and chemical stimuli (hypoxia, nutrient deprivation, and VEGF). Cells in the devices were cultured and remained viable through 40 days. Between days 14 and 21, the ECFC-EC, supported by cocultured NHLFs, formed an interconnected network of capillaries measuring from 15 to 50 μm in diameter, 29.1±14 for VP and 33.0±17 for EQ (Fig. 1d, e). Confocal imaging of the vessels stained for CD31 (EC marker) and DAPI (nuclei marker) demonstrated anastomosis of the vessels with the connecting pore leading to the fluidic channels (Fig. 1f, g). Networks formed in both designs, VP and EQ, were well connected, with connectedness values of 0.06±0.02 and 0.05±0.02, respectively. No significant differences in capillary network (diameter and connectedness) were observed between the two designs. Both designs had similar robust vessel networks whose area encompassed more than a third of the chamber area (VP vessel area: 35%±6%; EQ vessel area: 40%±6%).
To characterize the observed flow in the capillaries, 1-μm-diameter fluorescent microspheres were introduced in the arterial microfluidic channel and tracked as they entered the capillaries via the communication pores. In some experiments, the jumper was removed and convective flow in the arterial and venular channels were not coupled, but controlled independent of each other, and flow could be in the opposite direction as during vessel network development. Microspheres were noted to travel longitudinally as well as laterally in the vessel reflecting pressure gradients and resistance within the vessels (Fig. 3).
FIG. 3.

Flow in human microvessels formed in vitro. Flow was confirmed in both designs (EQ and VP) by tracking fluorescent red microspheres (1 μm diameter) flowing through the capillaries. The different tracked paths of a small subset of these microspheres over time are depicted as different colored lines (a, b); scale bar=50 μm. The microsphere velocity (μm/s) varies with linear distance within a given path (c, d). The wall shear rate (s−1) averages for the representative tissue of each design (“Tissue Average”) as well as the averages for groups of tissue analyzed for each design (“Group Average”), assuming laminar flow, along each path are also shown in panel (e, f). For real-time visualization of microspheres moving through the microvessels, please see Supplementary Video S1 and Supplementary Video S2. Color images available online at www.liebertpub.com/tec
In devices with varying transverse pressure gradients, VP (Fig. 3a), during vessel network formation, only the chambers in the supraphysiological interstitial range demonstrated perfusion of the formed capillaries (e.g., chambers #1–#3). Microspheres tracked as they moved through microvessels revealed fluid velocities ranging from near zero (when the microsphere appears to stop before restarting) to >1000 μm/s at any particular position. There was significant variation in flow velocity with linear distance along a single microsphere path (representative tissue of this design shown in Fig. 3c, e, and Supplementary Video S1). Assuming laminar flow, the shear rate in the fluid at the vessel wall, γw, can be estimated by 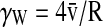 , where
, where 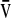 is the velocity at a single linear position in a vessel radius of R. Thus, the γw ranged from near zero to ∼200 s−1. The mean (±SD) velocity over all linear paths and γw for 19 beads in 3 perfused was 250±130 μm/s and 110±75 s−1 (Fig. 3e).
is the velocity at a single linear position in a vessel radius of R. Thus, the γw ranged from near zero to ∼200 s−1. The mean (±SD) velocity over all linear paths and γw for 19 beads in 3 perfused was 250±130 μm/s and 110±75 s−1 (Fig. 3e).
In devices designed for identical pressure gradients, EQ, across the tissues during vessel network formation, microspheres traversed exclusively from the arterial microfluidic channel side of the tissue chamber to the venular side, despite a modest pressure gradient in the longitudinal direction (Fig. 3b, d). Microspheres tracked in these devices again revealed a wide range for velocity from near zero to >4000 μm/s at any particular position. The shear rate demonstrated a similar large range from near zero to ∼1000 s−1. (representative tissue of this design shown in Fig. 3d, f, and Supplementary Video S2). The mean (±SD) velocity over all linear paths and γw for 21 beads in 4 perfused tissues was 690±660 μm/s and 320±267 s−1 (Fig. 3f).
While the microspheres demonstrate anastomosis of the vessels to the microfluidic channels and facilitate assessment of fluid velocity, they cannot evaluate vessel permeability. To assess permeability of the vessels, fluorescent dextran (70 and 150 kDa) was introduced into the arterial microfluidic channel at day 21 (Fig. 4). The dextran entered through anastomosis of the vessel network to the pore. Perfusion of the vessel network was confirmed by observing that the fluorescent dextran traveled through the vessels connecting all the tissue chambers and reached capillaries in distal tissue chambers before the dextran appeared in the corresponding microfluidic channel adjacent to the distal chambers on the arterial side (Fig. 4a). It is also evident that there is variability in the degree of permeability to 70-kDa dextran along the length of the vessels (Supplementary Video S3), although all vessels appear to be impermeable to 150 kDa (Fig. 4a, right side). Fluorescent dextran (70 kDa) was observed to travel from the arterial side through the network and empty into the venular microfluidic channel demonstrating anastomosis on both sides of the tissue chamber (Fig. 4b).
FIG. 4.

Flow in the developed vessel network was verified using FITC-dextran. After 21 days of development, FITC-dextran was introduced into the high-pressure arterial fluidic channel of a series of perfused microtissues. (a) Fluorescent image 30 min after introducing dextran (150 kDa, VP design) shows that fluorescence is observed in the capillaries, not in the fluidic channel on the arterial (top) side (right panel). This observation demonstrates that that FITC-dextran was transported down the hydrostatic pressure gradient (left to right) from the neighboring upstream compartment and the vessels are impermeable to 150-kDa dextran. (b) Experiments using 70-kDA FITC-dextran in the EQ design yield similar results, demonstrating that the vast majority of vessels are impermeable to lower molecular weight proteins. FITC-dextran introduced on the arterial side traveled through the vessel network and emptied out on the venular channel side (hydrostatic gradient right to left) revealing anastomotic connections on both the arterial and venular side of the tissue chamber.
Discussion
Respiratory gas exchange and filtration are the primary functions of the microcirculation (capillary network). Adequate respiratory gas exchange requires transit times (and thus velocities) that are low enough for the red blood cell to equilibrate oxygen and carbon dioxide concentrations but high enough to maintain shear rates that inhibit clot formation. Normal blood velocities and shear rates in human capillaries range from 500 to 1500 μm/s and 100 to 500 s−1 and depend on numerous factors, including the specific organ physiology, hematocrit, and metabolic demand.29–32 Our system demonstrates fine control over pressure to not only dictate interstitial flow during vessel formation, but also provides the driving force for fluid flow within the developed network of vessels. By controlling the pressure, we demonstrate a wide range of lumen velocities (from near 0 to >1000 μm/s) and shear rate (near 0 to 200 s−1) that encompass the normal physiologic range. Furthermore, filtration requires a semi-permeable vessel wall that can selectively retain large proteins. Capillaries normally retain protein >70 kDa33 consistent with our experimental observations.
The ability to study a variety of interstitial flows on a single chip makes our system a potentially useful tool to examine microvessel network development in physiological environments characterized by altered interstitial flow, such as wound healing, exercise, or the tumor microenvironment. Previous work has demonstrated that cells suspended in a 3D matrix respond to interstitial shear stress through membrane-bound receptors.34 However, despite differences in microchamber interstitial flow created by the design, an interconnected network of vessels developed under all conditions. By 3 weeks, no significant differences in terms of vessel density or diameter were observed between VP and EQ design. Although a relatively large range in interstitial flows was investigated (∼0.8–8.5 μm/s), more extreme values may be needed to observe mass transport effects on network development.25
Interestingly, while we observed the formation of vessel networks in all chambers, only chambers exposed to a supraphysiological flow (∼8.5 μm/s of perfused vessels) produced a perfused network (i.e., resulted in anastomosis between the vessel network and the microfluidic channel). While interstitial flow is important for normal filtration, pathophysiological changes in interstitial flow may serve to elicit a vascular remodeling response. For example, increased interstitial flow of 4–10 μm/s in a collagen matrix can upregulate the production of TGF-β1, leading to the differentiation of fibroblasts into myofibroblasts and increased collagen production.18,35 In our study, cell migration toward the pores may have been stimulated by elevated interstitial flow. It is also possible that cells exposed to a higher interstitial flow develop a vascular network faster due to enhanced mass transport via convection of solutes or nutrients.25 Finally, interstitial flow has previously been reported to induce weak matrix anisotropy, suggesting that interstitial forces may induce changes in the matrix.36 Furthermore, alignment of matrices has been demonstrated to influence vessel sprouts.37 Thus, a higher interstitial flow may preferentially align the matrix in a manner that favors vessel growth toward the connecting pore, thereby encouraging anastomosis with the microfluidic channel.
Our experimental protocol focused on manipulating the pressure gradients and interstitial flow along the series of microchambers. However, the same approach could also be used to control additional variables relevant to the tissue microenvironment, including oxygen tension, pH, nutrient and growth factor concentration, and waste products. As such, the system may useful to determine optimal conditions for vascular network growth or cell proliferation in the tissue microenvironment, or the screening of toxins or drugs under a wide range of concentrations.
The use of microbeads to detect flow allowed us to estimate flow velocities. One major limitation in this method is the observation that the microbeads can adhere temporarily to the vessel wall. The adherence and surface interaction of the microbead to the vessel wall could thus lead us to underestimate the actual flow velocities. An alternate estimate of the flow velocity using laminar pressure–flow relationships is not possible due to the complex geometry of the vascular networks.
In summary, we have demonstrated perfusion of a living network of dynamic human capillaries in a microphysiological (volume<1 mm3) system that has potentially broad diagnostic and therapeutic applications. For example, humans are exposed to therapeutic drugs or chemical toxins primarily through skin absorption, inhalation into the lungs, or ingestion. While an acute therapeutic or toxic response to the initial organ of exposure can occur, drugs have the potential to be absorbed by the microcirculation and either impact the cardiovascular system directly or be delivered by convection to a distant site (e.g., liver, brain). Hence, the ability to screen new drugs or chemical toxins to determine the permeability across human capillaries or a direct effect on the microcirculation is of enormous importance in understanding the integrated effect of an individual compound. Finally, the development of new microphysiological systems that mimic complex and metabolically demanding organ functions (e.g., cardiac muscle, pancreas, liver) will require a functional capillary network to provide nutrients and remove waste products.
Supplementary Material
Acknowledgments
The authors acknowledge the continuous support of Dr. Tatiana Krasieva for confocal microscopy and also acknowledge Mr. Zachary Campagna for his assistance in quantifying the capillary network. This research was supported by National Institutes of Health RC1 ES018361-01 and the National Heart, Lung and Blood Institute (MM, F32HL105055). C.C.W.H. is supported by Award Number P30CA062203 from the National Cancer Institute.
Disclosure Statement
No competing financial interests exist.
References
- 1.Chen X. Aledia A.S. Popson S.A. Him L. Hughes C.C. George S.C. Rapid anastomosis of endothelial progenitor cell-derived vessels with host vasculature is promoted by a high density of cotransplanted fibroblasts. Tissue Eng Part A. 2010;16:585. doi: 10.1089/ten.tea.2009.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Folkman J. Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138:745. doi: 10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song J.W. Munn L.L. Fluid forces control endothelial sprouting. Proc Natl Acad Sci U S A. 2011;108:15342. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman A.C. Nakatsu M.N. Chou W. Gershon P.D. Hughes C.C. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu T. Yamato M. Kikuchi A. Okano T. Cell sheet engineering for myocardial tissue reconstruction. Biomaterials. 2003;24:2309. doi: 10.1016/s0142-9612(03)00110-8. [DOI] [PubMed] [Google Scholar]
- 6.Hughes C.C. Endothelial-stromal interactions in angiogenesis. Curr Opin Hematol. 2008;15:204. doi: 10.1097/MOH.0b013e3282f97dbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung S. Sudo R. Vickerman V. Zervantonakis I.K. Kamm R.D. Microfluidic platforms for studies of angiogenesis, cell migration, and cell-cell interactions. Sixth International Bio-Fluid Mechanics Symposium and Workshop March 28–30, 2008 Pasadena, California. Ann Biomed Eng. 2010;38:1164. doi: 10.1007/s10439-010-9899-3. [DOI] [PubMed] [Google Scholar]
- 8.Powers M.J. Domansky K. Kaazempur-Mofrad M.R. Kalezi A. Capitano A. Upadhyaya A., et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol Bioeng. 2002;78:257. doi: 10.1002/bit.10143. [DOI] [PubMed] [Google Scholar]
- 9.Borenstein J.T. Weinberg E.J. Orrick B.K. Sundback C. Kaazempur-Mofrad M.R. Vacanti J.P. Microfabrication of three-dimensional engineered scaffolds. Tissue Eng. 2007;13:1837. doi: 10.1089/ten.2006.0156. [DOI] [PubMed] [Google Scholar]
- 10.Shin M. Matsuda K. Ishii O. Terai H. Kaazempur-Mofrad M. Borenstein J., et al. Endothelialized networks with a vascular geometry in microfabricated poly(dimethyl siloxane) Biomed Microdevices. 2004;6:269. doi: 10.1023/B:BMMD.0000048559.29932.27. [DOI] [PubMed] [Google Scholar]
- 11.Wong A.P. Perez-Castillejos R. Christopher Love J. Whitesides G.M. Partitioning microfluidic channels with hydrogel to construct tunable 3-D cellular microenvironments. Biomaterials. 2008;29:1853. doi: 10.1016/j.biomaterials.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chrobak K.M. Potter D.R. Tien J. Formation of perfused, functional microvascular tubes in vitro. Microvasc Res. 2006;71:185. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Cuchiara M.P. Allen A.C. Chen T.M. Miller J.S. West J.L. Multilayer microfluidic PEGDA hydrogels. Biomaterials. 2010;31:5491. doi: 10.1016/j.biomaterials.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 14.Toh Y.C. Zhang C. Zhang J. Khong Y.M. Chang S. Samper V.D., et al. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 2007;7:302. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- 15.Vickerman V. Blundo J. Chung S. Kamm R. Design, fabrication and implementation of a novel multi-parameter control microfluidic platform for three-dimensional cell culture and real-time imaging. Lab Chip. 2008;8:1468. doi: 10.1039/b802395f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helm C.L. Zisch A. Swartz M.A. Engineered blood and lymphatic capillaries in 3-D VEGF-fibrin-collagen matrices with interstitial flow. Biotechnol Bioeng. 2007;96:167. doi: 10.1002/bit.21185. [DOI] [PubMed] [Google Scholar]
- 17.Shields J.D. Fleury M.E. Yong C. Tomei A.A. Randolph G.J. Swartz M.A. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11:526. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Ng C.P. Swartz M.A. Fibroblast alignment under interstitial fluid flow using a novel 3-D tissue culture model. Am J Physiol Heart Circ Physiol. 2003;284:H1771. doi: 10.1152/ajpheart.01008.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y. Chen J. Craven M. Choi N.W. Totorica S. Diaz-Santana A., et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci U S A. 2012;109:9342. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song J.W. Bazou D. Munn L.L. Anastomosis of endothelial sprouts forms new vessels in a tissue analogue of angiogenesis. Integr Biol. 2012;4:857. doi: 10.1039/c2ib20061a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon N.L. In vitro formation and characterization of a perfusable three-dimensional tubular capillary network in microfluidic devices. Lab Chip. 2012;12:2815. doi: 10.1039/c2lc40131b. [DOI] [PubMed] [Google Scholar]
- 22.Chung S. Sudo R. Mack P.J. Wan C.-R. Vickerman V. Kamm R.D. Cell migration into scaffolds under co-culture conditions in a microfluidic platform. Lab Chip. 2009;9:269. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 23.Xia Y. Whitesides G.M. Soft lithography. Annu Rev Mater Sci. 1998;28:153. [Google Scholar]
- 24.Cho H. Kim H.-Y. Kang J.Y. Kim T.S. How the capillary burst microvalve works. J Colloid Interface Sci. 2007;306:379. doi: 10.1016/j.jcis.2006.10.077. [DOI] [PubMed] [Google Scholar]
- 25.Hsu Y.H. Moya M.L. Abiri P. Hughes C.C. George S.C. Lee A.P. Full range physiological mass transport control in 3D tissue cultures. Lab Chip. 2013;13:81. doi: 10.1039/c2lc40787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X. Aledia A.S. Ghajar C.M. Griffith C.K. Putnam A.J. Hughes C.C., et al. Prevascularization of a fibrin-based tissue construct accelerates the formation of functional anastomosis with host vasculature. Tissue Eng Part A. 2009;15:1363. doi: 10.1089/ten.tea.2008.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakatsu M.N. Sainson R.C. Aoto J.N. Taylor K.L. Aitkenhead M. Perez-del-Pulgar S., et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res. 2003;66:102. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 28.Griffith C.K. Miller C. Sainson R.C. Calvert J.W. Jeon N.L. Hughes C.C., et al. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Eng. 2005;11:257. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 29.Hathcock J.J. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2006;26:1729. doi: 10.1161/01.ATV.0000229658.76797.30. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood L. Human Physiology: from Cells to Systems. Brooks/Cole, Cengage Learning. 2008 [Google Scholar]
- 31.Bollinger A. Butti P. Barras J.P. Trachsler H. Siegenthaler W. Red blood cell velocity in nailfold capillaries of man measured by a television microscopy technique. Microvasc Res. 1974;7:61. doi: 10.1016/0026-2862(74)90037-5. [DOI] [PubMed] [Google Scholar]
- 32.Dintenfass L. Blood Viscosity, Hyperviscosity & Hyperviscosaemia. Lancaster, United Kingdom; MTP Press: 1985. [Google Scholar]
- 33.Enis D.R. Shepherd B.R. Wang Y. Qasim A. Shanahan C.M. Weissberg P.L., et al. Induction, differentiation, and remodeling of blood vessels after transplantation of Bcl-2-transduced endothelial cells. Proc Natl Acad Sci U S A. 2005;102:425. doi: 10.1073/pnas.0408357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Z.D. Wang H. Tarbell J.M. Heparan sulfate proteoglycans mediate interstitial flow mechanotransduction regulating MMP-13 expression and cell motility via FAK-ERK in 3D collagen. PLoS One. 2011;6:e15956. doi: 10.1371/journal.pone.0015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng C.P. Hinz B. Swartz M.A. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci. 2005;118:4731. doi: 10.1242/jcs.02605. [DOI] [PubMed] [Google Scholar]
- 36.Ng C.P. Helm C.L. Swartz M.A. Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvasc Res. 2004;68:258. doi: 10.1016/j.mvr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Morin K.T. Tranquillo R.T. Guided sprouting from endothelial spheroids in fibrin gels aligned by magnetic fields and cell-induced gel compaction. Biomaterials. 2011;32:6111. doi: 10.1016/j.biomaterials.2011.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.