

Abstract
Hydroxynitrile lyase from Arabidopsis thaliana (AtHNL) was fused to different fluorescent reporter proteins. Whereas all fusion constructs retained enzymatic activity and fluorescence in vivo and in vitro, significant differences in activity and pH stability were observed. In particular, flavin-based fluorescent reporter (FbFP) fusions showed almost 2 orders of magnitude-increased half-lives in the weakly acidic pH range compared to findings for the wild-type enzyme. Analysis of the quaternary structure of the respective FbFP-AtHNL fusion proteins suggested that this increased stability is apparently caused by oligomerization mediated via the FbFP tag. Moreover, the increased stability of the fusion proteins enabled the efficient synthesis of (R)-mandelonitrile in an aqueous-organic two-phase system at a pH of <5. Remarkably, (R)-mandelonitrile synthesis is not possible using wild-type AtHNL under the same conditions due to the inherent instability of this enzyme below pH 5. The fusion strategy presented here reveals a surprising means for the stabilization of enzymes and stresses the importance of a thorough in vitro characterization of in vivo-employed fluorescent fusion proteins.
INTRODUCTION
For scientific reasons and biotechnological purposes, it is of prime importance to label certain proteins within a cell in order to track their fate without impairing their function. To this end, fluorescent reporters can be translationally fused to the target protein (1). Ideally, the labeled target protein retains its function and at the same time becomes spectroscopically and microscopically traceable within the cell (1, 2).
The green fluorescent protein (GFP) and its derivatives have in the past been widely used as translationally fused reporter proteins for the analysis of expression, localization, folding, movement, and interaction of proteins in a wide variety of cells and tissues (for a recent review, see reference 2).
Flavin-based fluorescent proteins (FbFPs), derived from plant and bacterial light, oxygen, voltage (LOV) photoreceptors (3), represent a promising new class of fluorescent proteins with great potential for biotechnological and biomedical applications (4). In contrast to the case for the GFP family of proteins, FbFP fluorescence does not depend on the presence of molecular oxygen (5). Therefore, bacterial FbFPs have been used as reporter proteins under hypoxic and anoxic conditions in prokaryotic (5–7) and eukaryotic (8) unicellular organisms and in mammalian cells (9). Plant-derived FbFPs, such as the iLOV (LOV domain with improved fluorescent properties) protein and its derivatives (10, 11), have previously been used as reporters of plant virus infection (10). Furthermore, the monomeric iLOV protein was recently applied as a C-terminal tag fused translationally to several target proteins (12). The majority of the tested iLOV fusion proteins could be expressed in fluorescent form, and target protein functionality was proven in one case (12). To the best of our knowledge, bacterial FbFPs have so far not been used as fluorescent reporter modules in translational fusions targeting enzymatically active proteins. This issue is of particular interest, since bacterial FbFPs, in contrast to the plant iLOV system, form stable dimers in solution (13, 14) which could exert unforeseen adverse effects on folding, activity, and stability of a fused target protein.
We recently described an Escherichia coli-based whole-cell biotransformation system for the production of enantiopure cyanohydrins, employing monophasic micro-aqueous methyl tert-butyl ether (MTBE) as the reaction medium (15). This enzymatic system comprised either E. coli BL21(DE3) cells overproducing wild-type Arabidopsis thaliana hydroxynitrile lyase (AtHNL) or a translational fusion of AtHNL with a bacterial flavin-based fluorescent protein (FbFP) (15). Using this fluorescent biocatalyst, we have analyzed cellular- and fusion protein integrity in the monophasic organic solvent by monitoring whole-cell FbFP fluorescence during incubation in MTBE (15). Those analyses revealed sustained cellular- and fusion protein integrity over a reaction time of 4.5 h. Furthermore, both wild-type and FbFP-AtHNL-overproducing cells displayed excellent conversions and very good enantioselectivities over several reaction cycles (15). However, the activity, stability, biocatalytic performance, and biochemical properties of FbFP-AtHNL were not studied thoroughly in vitro.
Thus, in the present work, comparative in vitro biochemical studies regarding the influence of the FbFP tag on the enzymatic properties of the respective fusion proteins were performed. To this end, we generated a set of additional fluorescent AtHNL fusion proteins, adding the FbFP tag either to the N or C terminus of AtHNL. As a reference system, the enhanced yellow fluorescent protein (eYFP) (16) (here designated YFP) was used as an N-terminal fusion tag (nYFP-AtHNL). Whereas all generated AtHNL fusions retained fluorescence and activity, both purified FbFP-AtHNL fusions were found to be remarkably more stable at weakly acidic pH than the wild-type enzyme and the respective nYFP-AtHNL fusion. This surprising side effect, which might ultimately complicate in vivo studies using FbFP reporters, can be advantageous for a variety of biotechnological applications. In order to exploit this stabilizing effect, we applied one of the FbFP-AtHNL fusions in an aqueous-organic two-phase reaction system for the synthesis of (R)-mandelonitrile. Under those reaction conditions, wild-type AtHNL is unstable and hence cyanohydrin synthesis cannot be carried out (17, 18). Furthermore, using size exclusion chromatography, we have demonstrated that the stabilization observed for both FbFP-AtHNL fusion proteins is most probably caused by higher-order oligomerization mediated via the FbFP reporter module.
MATERIALS AND METHODS
Construction of translational fusions.
The gene fragments cFbFP and nFbFP, encoding the respective YtvA-LOV domain region (cFbFP, residues 1 to 126; nFbFP, residues 25 to 147) were PCR amplified from a pET28a vector containing the full-length ytvA gene bearing a single codon substitution, which results in the amino acid exchange C62A (5). All PCRs were carried out using Pfu polymerase (Fermentas GmbH, St. Leon-Rot, Germany). Sequences of all oligonucleotides used in this study (Thermo Fisher Scientific GmbH, Ulm, Germany) are listed in Table S1 in the supplemental material. For amplification of cFbFP and nFbFP, the oligonucleotides cFbFP_fw and cFbFP_rev, as well as nFbFP_fw and nFbFP_rev, were used, respectively. To enable directional cloning of the nFbFP gene fragment, restriction endonuclease recognition sites for NdeI and SacI were included in the respective oligonucleotide primers. For cFbFP, restriction sites for SacI and XhoI were added during PCR amplification. The respective PCR products were digested using the appropriate restriction endonucleases and cloned into a similarly digested pET28a vector (Novagen, Darmstadt, Germany) using standard techniques. This yielded the plasmids pETnFbFP and pETcFbFP. Insertion of the gene fragment coding for AtHNL was achieved by digesting the FbFP-carrying pET28a plasmid using the restriction endonuclease Ecl136II, which generates “blunt-ended” DNA fragments. Ecl136II is an isoschizomer of SacI. Since SacI was used to clone the respective FbFP fragment into the pET28a vector, the AtHNL-encoding gene fragment could be inserted into the FbFP-carrying pETnFbFP and pETcFbFP plasmid constructs. The HNL-encoding gene fragment was PCR amplified from a previously described expression plasmid (19). To generate the cFbFP-AtHNL-encoding gene fusion, the oligonucleotides cHNL_NheI_fw and cHNL_blunt_rev were used. For nFbFP-AtHNL, nHNL_blunt_fw and nHNL_blunt_rev were employed. The respective PCR-amplified gene fragments were either cloned “blunt ended” (nFbFP-AtHNL) or in a directional manner (cFbFP-AtHNL; NheI/blunt) into the similarly hydrolyzed pETnFbFP and pETcFbFP vectors. The nYFP-AtHNL construct was assembled in two steps by overlap extension PCR (20). In the first PCR round, YFP was amplified from the commercially available pEYFP-C1 plasmid (Clontech, Mountain View, CA, USA) containing the eYFP open reading-frame (16) using the oligonucleotides nYFP_NdeI_for and nYFP_GS_rev. The AtHNL gene fragment was amplified using the oligonucleotides nYHNL_GS_for and nYHNL_NotI _rev. The overlapping sequence which is used to assemble the two fragments in the second round of the PCR encodes a flexible 3-fold Gly-Gly-Gly-Ser repeat linker. Amplification of the full-length gene fusion encoding nYPF-AtHNL was carried out with the oligonucleotides nYFP_NdeI_for and nYHNL_NotI_rev. The resulting PCR product was digested using the restriction endonucleases NdeI and NotI and cloned into a similarly hydrolyzed pET28a vector. For both FbFP and YFP fusion constructs, cloning into pET28a resulted in the in-frame addition of 20 codons encoding an N-terminal hexahistidine tag (His6 tag) and a thrombin protease cleavage site. All sequences were verified (Seqlab GmbH, Göttingen, Germany) prior to transformation of E. coli BL21(DE3) as an expression host.
Heterologous protein expression.
E. coli BL21(DE3) seed cultures containing the appropriate plasmids for expression of nFbFP-AtHNL, cFbFP-AtHNL, or nYFP-AtHNL were grown overnight at 37°C in 5 ml LB medium. For maintenance of plasmids, kanamycin (50 μg/ml) was added to all cultures. Expression on a larger scale was carried out in 1 liter autoinduction medium (21) inoculated to an optical density at 600 nm (OD600) of 0.025 to 0.05 with an overnight seed culture. After 3 h of incubation at 37°C under constant agitation (120 rpm), the culture was further incubated at 15°C for an additional 72 h. Subsequently, cells were harvested at 6,750 × g (30 min, 4°C). Wild-type AtHNL was expressed and purified as described previously (19).
Protein purification.
Cell pellets containing the respective AtHNL fusion were resuspended in 50 mM potassium phosphate buffer (pH 7.5) to a final concentration of ca. 15% (wt/vol). Cells were disrupted by passing the suspension three times through a chilled French press cell (40K cell; Thermo Electron Corporation, Bonn, Germany) employing a constant pressure of 300 × 105 Pa. Cell debris were removed by centrifugation at 18,000 × g for 30 min at 4°C. The cleared crude extract was subsequently purified using immobilized metal ion affinity chromatography (nickel-nitrilotriacetic acid [Ni-NTA] resin; Qiagen, Hilden, Germany). All purification steps were performed using an Äkta-basic fast-protein liquid chromatography (FPLC) system (GE Healthcare, Munich, Germany). The column was equilibrated with potassium phosphate buffer (50 mM, pH 7.5). After application of the crude extract onto the column and elution of nonspecifically bound proteins, a linear gradient (5 column bed volumes; 250 ml) of imidazole (0 to 250 mM) in potassium phosphate buffer, pH 7.5, was used for elution of the His6-tagged fusion proteins. Target protein-containing fractions were identified using the mandelonitrile cleavage assay, pooled, and desalted (Sephadex G 25; GE Healthcare, Munich, Germany). As storage buffer, 10 mM potassium phosphate buffer, pH 7.5, was used. All samples were lyophilized and stored at 4°C until further use. Protein concentrations were determined using the Bradford assay (22) employing bovine serum albumin (BSA) as a standard.
In vivo fluorescence measurements.
For qualitative in vivo fluorescence measurements, 500 mg of E. coli BL21(DE3) cells expressing the different fluorescent AtHNL fusion proteins were suspended in 500 μl of phosphate-buffered saline, pH 7. Aliquots of 10 μl were illuminated with blue light (365 nm), and in vivo fluorescence was documented photographically.
Determination of native molecular weights.
Native molecular weights were determined using high-performance liquid chromatography (HPLC)-based size exclusion chromatography (SEC). For separation, a BioSep-SEC-S2000 column (Phenomenex, Aschaffenburg, Germany) was connected to an LC-10Ai series (Shimadzu, Kyoto, Japan) liquid chromatography system equipped with an SPD-M10Avp photodiode array detector (PDA) and an RF-10AXL fluorescence detector. As a mobile phase, 200 mM sodium phosphate buffer, pH 7.5, containing 10 mM NaCl was employed. Elution of protein was detected at 280 nm. FbFP (excitation, 450 nm; emission, 495 nm) and YFP (excitation, 450 nm; emission, 527 nm) fluorescence emission was monitored using the RF-10AXL fluorescence detector connected to the HPLC system. All analyzed protein samples had a concentration of about 2 mg/ml and were dissolved in the buffer used as a mobile phase. For determination of the native molecular weights (MW), a mixture of standard proteins (each 1 mg/ml) (aqueous SEC1 [Phenomenex], thyroglobulin [MW, 660,000], IgG [MW, 150,000], Ovalbumin [MW, 44,000], myoglobin [MW, 17,000], and uridine [MW, 244]) dissolved in the same buffer was used. Subunit molecular weights were calculated based on the protein sequence using the ProtParam Web server (web.expasy.org/protparam/).
Photometric mandelonitrile cleavage assay for quantification of HNL activity.
Mandelonitrile cleavage reactions were carried out as described previously (19). In brief, the accumulation of benzaldehyde, which is formed as one reaction product of the HNL-catalyzed cleavage of rac-mandelonitrile, was photometrically monitored at 280 nm. All measurements were performed in 10-mm quartz cuvettes at 25°C in 50 mM acetate buffer (pH 5.5) using about 0.06 mg/ml of the respective enzyme. Total assay volume was 1 ml. Protein determination was performed according to the method of Bradford (22). Reactions were started by addition of rac-mandelonitrile (67 mM in citrate phosphate buffer, pH 3.5) to a final concentration of 13.4 mM in the assay. To account for nonenzymatic mandelonitrile cleavage, the respective control in buffer (without enzyme) was subtracted from all measurements. All measurements were performed at least in triplicate. Enzymatic activity was calculated using the molar extinction coefficient of benzaldehyde (ε280 = 1,376 liters mmol−1 cm−1). One unit of HNL activity is defined as the amount of enzyme which converts 1 μmol (R)-mandelonitrile per min in the given buffer at 25°C.
Determination of pH optima.
pH-dependent initial rate activities were determined using the mandelonitrile cleavage assay in the range from pH 3.5 to pH 6. All measurements were performed in triplicate. Controls without enzyme for each variation of the pH value were measured in duplicate. The rate of the nonenzymatic reaction increased continuously with increasing pH values. Above pH 6, the nonenzymatic mandelonitrile cleavage is too fast, so that enzymatic activity cannot reliably be measured.
Stability investigations.
Enzyme solutions with a concentration of 0.6 mg/ml were prepared in 10 mM potassium phosphate buffer, pH 7.5. For pH-dependent stability measurements, the respective protein sample was diluted 1:10 using 50 mM acetate buffer adjusted to pH 4.5 or pH 4.75, respectively. Samples were incubated up to 6 h at the respective pHs, aliquots were withdrawn at defined intervals, and residual activity was measured using the mandelonitrile cleavage assay. The final protein concentration in the assay was ca. 0.06 mg/ml. Half-lives (τ1/2) were derived from a single exponential fit of the experimental data, and the mean lifetime τ value obtained as a fit parameter was converted into a half-life by the following relation: τ1/2 = (ln2) × τ.
HNL-catalyzed cyanohydrin synthesis in an aqueous-organic two-phase system.
The general reaction setup for enantiopure cyanohydrin synthesis has been described previously (18). In brief, 20 mg of lyophilized enzyme was dissolved in 1 ml 50 mM acetate buffer (pH 4.75, 4.5, or 5.0). The protein solution (8 mg/ml enzyme) was placed in the reaction vessel, and the reaction was started immediately by adding 1 ml of a 1.5 to 2 M hydrogen cyanide (HCN) solution dissolved in MTBE, 0.5 mmol of the respective aldehyde (benzaldehyde or 2-chlorobenzaldehyde), and 0.1 mmol dodecane as an internal standard. All reactions were carried out at 25°C in an airtight vessel under argon atmosphere. The reaction mixture was constantly mixed using a magnetic stirrer. At defined time intervals, samples were withdrawn and (R)-mandelonitrile or (R)-chloromandelonitrile formation was monitored by chiral GC as described previously (18, 19).
RESULTS
Construction and heterologous overexpression of fluorescent AtHNL fusion proteins.
The photoreceptor protein YtvA from Bacillus subtilis consists of a canonical light, oxygen, voltage (LOV) sensor domain, flanked by an N-terminal helical cap and a C-terminal α-helical linker element. In full-length YtvA, the C-terminal linker connects the sensor LOV domain with the effector sulfate transporter anti-sigma factor antagonist (STAS) domain (Fig. 1) (23). In this study, we exploited this modularity, which is an intricate feature of most LOV photoreceptor proteins (24), for the construction of translational fusion proteins. In particular, the N- and C-terminal helical elements outside the canonical LOV core domain were used here as a naturally occurring linker to facilitate separation of the FbFP module from the fused target enzyme (Fig. 1). N- and C-terminal FbFP fusion constructs of AtHNL were generated (Fig. 1). As a control, we fused YFP N-terminally to AtHNL via a flexible 3×GGGS linker (Fig. 1) (25).
Fig 1.
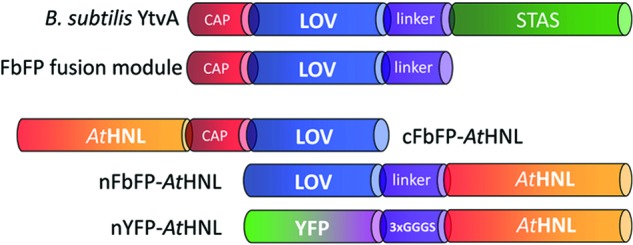
Structural organization of FbFP- and YFP-based AtHNL fusions. Abbreviations: LOV, light, oxygen, voltage domain, here derived from B. subtilis YtvA; STAS, sulfate transporter anti-sigma factor antagonist domain; YFP, enhanced yellow fluorescent protein; AtHNL, hydroxynitrile lyase from Arabidopsis thaliana; CAP, N-terminal α-helical cap; linker, α-helical linker; 3×GGGS, flexible linker polypetide, consisting of a 3-fold GGGS repeat. cFbFP-AtHNL/nFbFP-AtHNL stand for C/N-terminal fusion of FbFP with AtHNL. nYFP-AtHNL represents the N-terminal fusion of YFP and AtHNL.
All constructs contained an N-terminal His6 tag to enable protein purification. Cells expressing both FbFP and YFP fusion proteins showed FbFP- and YFP-specific fluorescence emission in vivo (Fig. 2A; see also see Fig. S2 in the supplemental material). Furthermore, all purified fusion proteins were enzymatically active (Fig. 2B).
Fig 2.
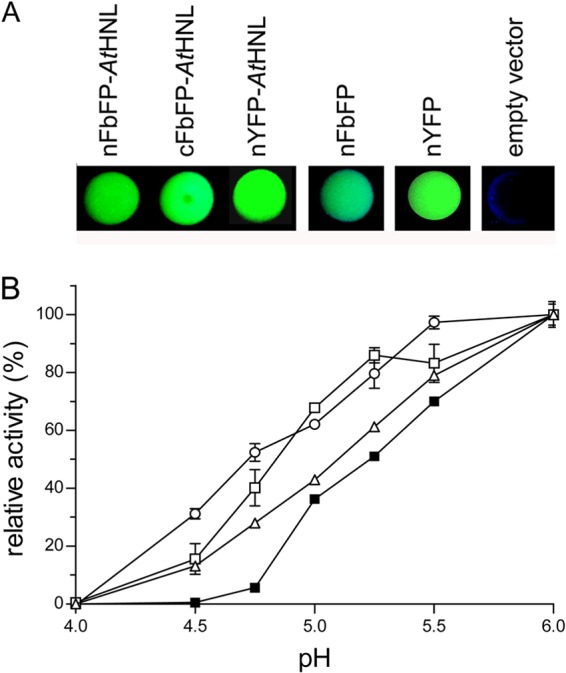
(A) In vivo fluorescence of FbFP and YFP fusions with AtHNL. Fluorescence of cells expressing the indicated FbFP or YFP (fusion) protein was visualized by illuminating drops of cell suspensions under blue light (365 nm). (B) Influence of pH on the relative initial rate activity. All enzyme preparations were diluted in 50 mM acetate buffer, adjusted to the respective pH values. All samples were preequilibrated for 5 min at 25°C. AtHNL activity was quantified photometrically by monitoring the release of benzaldehyde from rac-mandelonitrile. To account for nonenzymatic mandelonitrile cleavage, the respective control, containing only buffer and substrate, was subtracted. ■, AtHNL; ○, cFbFP-AtHNL; △, nFbFP-AtHNL; □, nYFP-AtHNL. Error bars represent the standard deviations derived from three independent measurements.
pH optima and stability.
The stereoselectivity of HNL-catalyzed hydrocyanation reactions is strongly influenced by the employed reaction conditions (see Fig. S1 in the supplemental material) (26). In aqueous media, the limiting factor, compromising the optical purity of the enzymatic cyanohydrin formation, is a nonenzymatic hydrocyanation reaction which results in the formation of racemic product (see Fig. S1) (19). This nonenzymatic reaction can be suppressed by using low pH values, by lowering the reaction temperature, and by decreasing the water content in the reaction system (18).
Wild-type AtHNL is rapidly and irreversibly inactivated at a pH of <5.4 (19). Thus, pH control of the nonenzymatic side reaction is not feasible for the isolated wild-type enzyme. Since reporter protein fusion might result in altered enzymatic properties, we have studied all fusion constructs with respect to their pH optima (Fig. 2B) and their pH-dependent stability (Table 1). Figure 2B shows that the activity of wild-type AtHNL in the weakly acidic range of pH 4.5 to pH 5.0 is significantly lower than those for the fusion proteins. For example, wild-type AtHNL is barely active at pH 4.75, whereas, the cFbFP-AtHNL, nFbFP-AtHNL, and nYFP-AtHNL fusions show 10-, 5-, and 7-fold-higher initial rate activities in the cleavage reaction in aqueous solution using rac-mandelonitrile as a substrate (Fig. 2B).
Table 1.
pH-dependent half-lives of wild-type AtHNL and the AtHNL fusion proteins used in this studya
| pH | τ1/2 (min) |
|||
|---|---|---|---|---|
| AtHNL | nFbFP-AtHNL | cFbFP-AtHNL | nYFP-AtHNL | |
| 4.75 | 2.2 ± 0.5 | 55.0 ± 13.0 | 95.5 ± 0.7 | 4.5 ± 0.8 |
| 4.5 | 0b | 5.1 ± 1.4 | 5.7 ± 0.2 | 1.2 ± 0.1 |
Residual enzymatic activity was measured using the mandelonitrile cleavage assay after incubation of the respective enzyme variant in 50 mM acetate buffer at 25°C at the indicated pH. Half-lives and standard deviations were calculated from a single exponential fit of experimental data using three independent replicates.
Half-life of wild-type AtHNL according to the work of Guterl et al. (19).
Hereby, an increased stability of the fusion constructs in the weakly acidic range compared to that of wild-type AtHNL could explain the observed activity differences. Therefore, we analyzed all constructs with respect to their stability at pHs 4.5 and 4.75 by determining residual enzymatic activities during incubation at the respective pHs (Table 1).
nYFP-AtHNL possesses half-lives of 4.5 ± 0.8 min at pH 4.75 and 1.2 ± 0.1 min at pH 4.5, respectively. Notably, at pH 4.75, the half-life of cFbFP-AtHNL was increased 44-fold (τ1/2 = 95.5 ± 0.7 min) over that of the wild-type enzyme (τ1/2 = 2.2 ± 0.5 min). The same holds for nFbFP-AtHNL, which was 25 times more stable (τ1/2 = 55 ± 13 min) than the wild-type enzyme under the same conditions.
SEC analyses reveal an altered quaternary structure of FbFP-AtHNL fusions.
The fusion of a reporter protein module might impact on the native oligomerization state of the target protein, which might influence its function, i.e., by inhibiting subunit association or by facilitating additional nonnative protein-protein contacts. Those in turn might result in an altered quaternary structure of the fusion protein compared to that of the wild-type target protein. In order to account for such effects, we analyzed all fusion proteins by analytical size exclusion chromatography (SEC) (Fig. 3A; see also Table S2 in the supplemental material).
Fig 3.

(A) HPLC SEC analyses of all fusion constructs used in this study. In all cases, the initial protein concentration was 2 mg/ml in 10 mM potassium phosphate buffer containing 10 mM NaCl (pH 7.5). Above the main elution peak, the calculated molecular weight is depicted. Elution times for standard proteins: 1, thyroglobulin, 660,000; 2, IgG, 150,000; 3, ovalbumin, 44,000 (marked by gray lines). (B) SDS-PAGE analysis of purified fusion constructs. The theoretical molecular weights of the SEC analyzed (fusion) proteins predicted based on sequence were as follows: AtHNL, 29,216; cFbFP-AtHNL, 46,460; nFbFP-AtHNL, 46,198; nYFP-AtHNL, 59,128. The purity of all preparations was estimated based on SDS-PAGE and HPLC-SEC analyses (see also Fig. S3 in the supplemental material) to be ≥80%.
As reported previously, AtHNL predominately forms homodimers (19). Likewise, nYFP-AtHNL, which does not show an altered pH stability profile compared to that of wild-type AtHNL, appears to be predominately dimeric in aqueous solution. The somewhat more stable nFbFP-AtHNL fusion exists mainly as a dimer but contains a certain fraction of an apparent tetrameric species. In contrast, the most stable fusion construct, cFbFP-AtHNL (Table 1), appears predominately as a tetramer in SEC experiments, with a shoulder corresponding to a trimer. Thus, it appears that the fusion of an FbFP tag alters the native quaternary structure of the target enzyme compared to that of the nonfused wild-type enzyme. This is in contrast to the fusion with YFP, where the native dimeric organization of the target AtHNL is retained in the respective nYFP-AtHNL fusion protein.
The native quaternary structure of the FbFP and YFP fusions is retained under assay conditions.
SDS-PAGE analyses (Fig. 3B) revealed that all preparations exhibited comparable degrees of purity. Multiple peaks observed in the SEC chromatograms arose from the fusion protein existing in different oligomerization states (see above). As a proof, we ran HPLC-SEC analyses on a system equipped with a photodiode array detector, which allowed recording UV/visible (UV/Vis) absorbance spectra for each elution peak. Since all analyzed fluorescent fusion proteins contain a chromophore which absorbs light in the visible range of the spectrum, it is possible to distinguish the target fusion protein from possible impurities in the preparation (see Fig. S3 in the supplemental material). This allowed the unequivocal identification of all major peaks containing the respective fusion protein. Similar results were obtained when the reporter proteins' specific fluorescence emission of the HPLC elution peaks was monitored using a fluorescence detector attached to the HPLC system (see Fig. S4A). Moreover, due to the sensitivity of fluorescence detection, very low protein concentrations could be detected. To verify that the observed quaternary structures of the different fusion constructs are retained under assay conditions (at low protein concentrations), we diluted the samples to a final concentration of 0.03 mg/ml, which could still be reliably monitored using fluorescence detection (see Fig. S4B). This concentration is 2 times lower than the one employed in the rac-mandelonitrile cleavage assay which was used for pH optimum and stability determinations.
Our results thus indicate that the different oligomeric species observed in the SEC experiment (Fig. 3) represent stable oligomeric subpopulations rather than a concentration-dependent dynamic equilibrium as might be expected for weakly interacting protein assemblies.
The increased stability of FbFP-AtHNL fusions allows efficient cyanohydrin synthesis in an aqueous-organic two-phase system.
nFbFP-AtHNL and cFbFP-AtHNL showed increased stability at weakly acidic pH compared to both wild-type AtHNL and the nYFP-AtHNL fusion (Table 1). The stabilization was particularly pronounced for the cFbFP-AtHNL construct at pHs 4.5 and 4.75. To capitalize on this unexpected side effect, we applied cFbFP-AtHNL for the synthesis of (R)-mandelonitrile in an aqueous-organic two-phase system using methyl tert-butyl ether (MTBE) as the organic phase (Fig. 4). The pH of the aqueous buffer phase was adjusted to 4.5 or 4.75. At both pH values, the nonenzymatic side reaction is essentially suppressed (27).
Fig 4.

Conversion of benzaldehyde and HCN to (R)-mandelonitrile by cFbFP-AtHNL in an aqueous-organic two-phase system containing methyl tert-butyl ether and 50 mM acetate buffer (pH 4.5 and pH 4.75). The organic phase contained the substrates (1 ml 1.5 to 2 M HCN and 0.5 mmol benzaldehyde). Enzymatic conversion, using 8 mg/ml enzyme, was followed using chiral gas chromatography. All reactions were carried out at 25°C. In both cases, the enantiomeric excess (ee) for (R)-mandelonitrile exceeded 96%. Error bars represent the standard deviations derived from three independent measurements.
At pH 4.75, cFbFP-AtHNL (8 mg/ml enzyme) converted 95% ± 1% benzaldehyde to (R)-mandelonitrile within 60 min. The conversion at pH 4.5 started equally fast but stopped at about 87% ± 5% conversion after 60 min, which is probably due to the lower stability of the cFbFP-AtHNL at this pH (τ1/2 = 5.7 ± 0.2 min). Both reactions yielded (R)-mandelonitrile with >96% enantiomeric excess (ee). In order to evaluate the applicability of our stabilized cFbFP-AtHNL construct for the production of other cyanohydrins, synthesis reactions were carried out using 2-chlorobenzaldehyde and HCN as substrates. Maximal conversion was reached after 5 min (78% ± 2%), but this decreased over time to 61% ± 11% after 60 min of reaction time. Likewise, the ee for (R)-2-chloromandelonitrile of >99% after 5 min decreased during the course of the reaction to about 97% after 60 min (see Fig. S5 in the supplemental material).
DISCUSSION
Bacterial FbFPs can be used as N- and C-terminal translational fusion tags.
In order to investigate the influence of N- and C-terminally fused bacterial FbFPs on AtHNL activity, we determined the pH-dependent activities for the respective fusion proteins (Fig. 2B). Apparently, the here-employed FbFP can be fused to either the N or the C terminus of AtHNL without impairing folding and activity of the enzyme. Moreover, the presence of naturally occurring N- and C-terminal α-helical linker-polypeptides outside the fluorescent FbFP protein domain might enable a straightforward attachment without the need for linker optimization.
FbFP fusion influences the enzymatic properties of AtHNL by induced oligomerization via the FbFP tag.
Comparative analyses of the different fluorescent AtHNL fusions revealed increased initial rate activities at weakly acidic pHs for all fusion proteins (Fig. 2B). Hereby, the observed differences in the initial reaction rates may reflect an overlay of rapid deactivation processes (within the time required for enzyme dilution and temperature equilibration) with the actual enzymatic reaction. Such an effect would be particularly deleterious at low pH values, where wild-type AtHNL is known to be rapidly inactivated (19). Therefore, the pH stability of all constructs was analyzed at pHs 4.5 and 4.75. Much to our surprise, both the nFbFP-AtHNL and cFbFP-AtHNL constructs showed half-lives that were increased by almost 2 orders of magnitude compared to that of the wild-type enzyme (Table 1). In contrast, the fusion of YFP to AtHNL did not influence the pH stability of the respective constructs (Table 1).
When comparing the native quaternary structures of all analyzed constructs, some notable similarities became apparent: it seems that the nYFP-AtHNL construct, which does not show an altered pH stability profile, retains the native dimeric AtHNL oligomerization state. This is in good agreement with findings of previous studies demonstrating that isolated YFP is mainly monomeric with a weak tendency to dimerize at higher concentrations (dissociation constant [Kd] = 0.1 mM) and consequently does not interfere with the formation of the native quaternary structure of the YFP-fused target protein (28). For all FbFP-AtHNL fusions, the dimeric organization of the FbFP module (13, 14) apparently results in induced higher-order oligomerization, i.e., yielding fusion proteins with variable amounts of an apparently tetrameric species. In comparison, other HNLs, such as those from Prunus species, Linum usitatissimum, and Manihot esculenta (MeHNL), are both active and stable at low pH (29). Most importantly, a comparative study of MeHNL and AtHNL previously implicated differences in their quaternary structures as a possible reason for the observed differences in stability (19). MeHNL appeared to exist in a fast-exchanging dimer-tetramer equilibrium, whereas AtHNL was found to be dimeric (Fig. 3). Moreover, for proteins of hyperthermophilic origin, strong intersubunit interactions leading to oligomerization have long since been implied to be a potential stabilization mechanism (30, 31).
Thus, the induced change in fusion protein quaternary structure compared to that of the wild-type enzyme represents the most likely explanation for the significant stabilization of the FbFP-AtHNL fusion proteins in the weakly acidic pH range. It should be noted that we cannot rule out potential stabilizing effects caused by the interdomain linker segment which was used to separate AtHNL from the fluorescent reporter protein, i.e., by influencing the quaternary structure of the corresponding construct by providing more or less structural flexibility.
Harnessing altered enzymatic properties of FbFP-AtHNL fusion proteins for the synthesis of (R)-mandelonitrile.
The observed stabilization of the FbFP-AtHNL constructs, compared to wild-type AtHNL, prompted us to test cyanohydrin synthesis using an aqueous-organic two-phase system. In such a reaction system, the pH of the aqueous buffer phase has to be adjusted to be as low as possible without compromising enzyme stability to ensure complete suppression of the nonenzymatic base-catalyzed side reaction (18). Using purified cFbFP-AtHNL at pH 4.75, nearly complete conversion of the substrates benzaldehyde and HCN to (R)-mandelonitrile was observed within 60 min (ee > 96%). Under the same reaction conditions, wild-type AtHNL cannot be used due to its inherent instability (19).
The AtHNL-catalyzed synthesis of chiral cyanohydrins has been carried out in the past using various purified and immobilized enzyme preparations (15, 17, 18, 26). Hereby, the enzyme has mostly been used either in micro-aqueous reaction systems as an immobilized or whole-cell biocatalyst (15, 18) or in purified form in an aqueous-organic two-phase system (26). In the latter system, the buffer pH was set to 5 and diisopropyl ether was used as the organic phase (ratio, 1:1) (26). Under those conditions, complete conversion of benzaldehyde and HCN to (R)-mandelonitrile was observed within 120 min, with the nonenzymatic side reaction amounting to about 14% (26). Hence, our stabilized FbFP-AtHNL fusions might represent an interesting alternative to wild-type AtHNL, i.e., for the conversion of more demanding substrates, such as 2-chlorobenzaldehyde and 2-bromobenzaldehyde, that show faster nonenzymatic reaction rates than benzaldehyde (26). To address this issue, we followed the conversion of 2-chlorobenzaldehyde using cFbFP-AtHNL as a catalyst. The enzyme (4 mg/ml) converted 78% of the substrates within 5 min of reaction time with an excellent ee, above 99%. With increasing reaction times, conversion and ee values dropped to about 61% and 97% after 60 min, respectively. This effect is most probably related to nonenzymatic cleavage of the produced (R)-chloromandelonitrile, which is more pronounced at the employed buffer pH than that of (R)-mandelonitrile (26). Initial reaction rates and obtained ee values are thus superior to conversion of the same substrate by our previously described whole-cell biotransformation approach, where maximally 80% 2-chlorobenzaldehyde was converted within 60 min of reaction time, with a moderate ee of 70% (wet E. coli cells expressing AtHNL wild type) in monophasic MTBE as a solvent system. The use of lyophilized cells improved ee values considerably, to about 90%, while conversion dropped to 43% under the same reaction conditions. It should be noted that initial reaction rates and final conversion values are not directly comparable between whole-cell biotransformations and the here-presented conversions using soluble enzyme in a two-phase system, because the amount of employed catalyst cannot be exactly determined for the whole-cell system. In conclusion, those proof-of-concept experiments nevertheless verify that more demanding substrates, such as 2-chlorobenzaldehyde, can successfully be converted by our stabilized cFbFP-AtHNL construct, although further process engineering would be necessary to improve product yields and enantiopurities.
Additionally, immobilization of the fluorescent FbFP-AtHNL variants could further improve their biocatalytic performances, i.e., in terms of stability and activity, as shown for sol-gel-entrapped AtHNL preparations (18).
We have here presented a detailed biochemical characterization of different fluorescent FbFP/YFP AtHNL fusions. While all constructs retained activity and fluorescence, the FbFP-AtHNL fusions displayed an altered pH stability profile compared to that of wild-type AtHNL. Both FbFP-AtHNL fusions showed markedly increased stability at weakly acidic pHs. Based on SEC data, we suggest that stabilization of the FbFP-AtHNL fusions results from higher-order oligomerization probably induced via the dimeric FbFP tag. This strategy, if transferable to other enzymes, might thus potentially be used as a tool to engineer enzymes with increased stability and hence to improve their biocatalytic performance. Experiments to rationalize this approach, as well as the transfer to other enzyme systems, are currently carried out in our laboratory.
Last but not least, our observations stress the importance of a thorough in vitro characterization of in vivo-employed fluorescent fusion proteins. This is particularly important in those cases where the used reporter proteins are not extensively characterized. Their unpredictable behavior may cause significant alterations of the biochemical properties of such fusion proteins, which may even turn out to be advantageous for biotechnological application, as demonstrated here for the synthesis of (R)-mandelonitrile at weakly acidic pH values.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Deutsche Forschungsgemeinschaft in the context of the research training group GK1166, “Biocatalysis in non-conventional media” (BioNoCo).
We thank Alexander Grünberger and Dietrich Kohlheyer (Microscale Bioengineering Group, IBG-1, Forschungszentrum Jülich) for their help with microscopic analyses.
Footnotes
Published ahead of print 31 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00795-13.
REFERENCES
- 1. Chudakov DM, Lukyanov S, Lukyanov KA. 2005. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 23:605–613 [DOI] [PubMed] [Google Scholar]
- 2. Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. 2010. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 90:1103–1163 [DOI] [PubMed] [Google Scholar]
- 3. Christie JM, Salomon M, Nozue K, Wada M, Briggs WR. 1999. LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. U. S. A. 96:8779–8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drepper T, Gensch T, Pohl M. 12 April 2013. Advanced in vivo applications of blue light photoreceptors as alternative fluorescent proteins. Photochem. Photobiol. Sci. 10.1039/C3PP50040C [DOI] [PubMed] [Google Scholar]
- 5. Drepper T, Eggert T, Circolone F, Heck A, Krauss U, Guterl JK, Wendorff M, Losi A, Gärtner W, Jaeger KE. 2007. Reporter proteins for in vivo fluorescence without oxygen. Nat. Biotechnol. 25:443–445 [DOI] [PubMed] [Google Scholar]
- 6. Choi CH, DeGuzman JV, Lamont RJ, Yilmaz O. 2011. Genetic transformation of an obligate anaerobe, P. gingivalis for FMN-green fluorescent protein expression in studying host-microbe interaction. PLoS One 6:e18499. 10.1371/journal.pone.0018499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lobo LA, Smith CJ, Rocha ER. 2011. Flavin mononucleotide (FMN)-based fluorescent protein (FbFP) as reporter for gene expression in the anaerobe Bacteroides fragilis. FEMS Microbiol. Lett. 317:67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tielker D, Eichhof I, Jaeger KE, Ernst JF. 2009. Flavin mononucleotide-based fluorescent protein as an oxygen-independent reporter in Candida albicans and Saccharomyces cerevisiae. Eukaryot. Cell 8:913–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walter J, Hausmann S, Drepper T, Puls M, Eggert T, Dihne M. 2012. Flavin mononucleotide-based fluorescent proteins function in mammalian cells without oxygen requirement. PLoS One 7:e43921. 10.1371/journal.pone.0043921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapman S, Faulkner C, Kaiserli E, Garcia-Mata C, Savenkov EI, Roberts AG, Oparka KJ, Christie JM. 2008. The photoreversible fluorescent protein iLOV outperforms GFP as a reporter of plant virus infection. Proc. Natl. Acad. Sci. U. S. A. 105:20038–20043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christie JM, Hitomi K, Arvai AS, Hartfield KA, Mettlen M, Pratt AJ, Tainer JA, Getzoff ED. 2012. Structural tuning of the fluorescent protein iLOV for improved photostability. J. Biol. Chem. 287:22295–22304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gawthorne JA, Reddick LE, Akpunarlieva SN, Beckham KS, Christie JM, Alto NM, Gabrielsen M, Roe AJ. 2012. Express your LOV: an engineered flavoprotein as a reporter for protein expression and purification. PLoS One 7:e52962. 10.1371/journal.pone.0052962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Circolone F, Granzin J, Jentzsch K, Drepper T, Jaeger KE, Willbold D, Krauss U, Batra-Safferling R. 2012. Structural basis for the slow dark recovery of a full-length LOV protein from Pseudomonas putida. J. Mol. Biol. 417:362–374 [DOI] [PubMed] [Google Scholar]
- 14. Jentzsch K, Wirtz A, Circolone F, Drepper T, Losi A, Gärtner W, Jaeger KE, Krauss U. 2009. Mutual exchange of kinetic properties by extended mutagenesis in two short LOV domain proteins from Pseudomonas putida. Biochemistry 48:10321–10333 [DOI] [PubMed] [Google Scholar]
- 15. Scholz KE, Okrob D, Kopka B, Grünberger A, Pohl M, Jaeger KE, Krauss U. 2012. Synthesis of chiral cyanohydrins by recombinant Escherichia coli cells in a micro-aqueous reaction system. Appl. Environ. Microbiol. 78:5025–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ormö M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science 273:1392–1395 [DOI] [PubMed] [Google Scholar]
- 17. Okrob D, Metzner J, Wiechert W, Gruber K, Pohl M. 2012. Tailoring a stabilized variant of hydroxynitrile lyase from Arabidopsis thaliana. Chembiochem 13:797–802 [DOI] [PubMed] [Google Scholar]
- 18. Okrob D, Paravidino M, Orru RVA, Wiechert W, Hanefeld U, Pohl M. 2011. Hydroxynitrile lyase from Arabidopsis thaliana: identification of reaction parameters for enantiopure cyanohydrin synthesis by pure and immobilized catalyst. Adv. Synth. Catal. 353:2399–2408 [Google Scholar]
- 19. Guterl JK, Andexer JN, Sehl T, von Langermann J, Frindi-Wosch I, Rosenkranz T, Fitter J, Gruber K, Kragl U, Eggert T, Pohl M. 2009. Uneven twins: comparison of two enantiocomplementary hydroxynitrile lyases with alpha/beta-hydrolase fold. J. Biotechnol. 141:166–173 [DOI] [PubMed] [Google Scholar]
- 20. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 21. Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 22. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 23. Aravind L, Koonin EV. 2000. The STAS domain—a link between anion transporters and antisigma-factor antagonists. Curr. Biol. 10:R53–R55. 10.1016/S0960-9822(00)00335-3 [DOI] [PubMed] [Google Scholar]
- 24. Herrou J, Crosson S. 2011. Function, structure and mechanism of bacterial photosensory LOV proteins. Nat. Rev. Microbiol. 9:713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson CR, Sauer RT. 1998. Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 95:5929–5934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andexer J, von Langermann J, Mell A, Bocola M, Kragl U, Eggert T, Pohl M. 2007. An R-selective hydroxynitrile lyase from Arabidopsis thaliana with an alpha/beta-hydrolase fold. Angew. Chem. Int. Ed. Engl. 46:8679–8681 [DOI] [PubMed] [Google Scholar]
- 27. von Langermann J, Guterl JK, Pohl M, Wajant H, Kragl U. 2008. Hydroxynitrile lyase catalyzed cyanohydrin synthesis at high pH-values. Bioprocess Biosyst. Eng. 31:155–161 [DOI] [PubMed] [Google Scholar]
- 28. Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
- 29. Dadashipour M, Asano Y. 2011. Hydroxynitrile lyases: insights into biochemistry, discovery, and engineering. ACS Catal. 1:1121–1149 [Google Scholar]
- 30. Schmidt PM, Attwood RM, Mohr PG, Barrett SA, McKimm-Breschkin JL. 2011. A generic system for the expression and purification of soluble and stable influenza neuraminidase. PLoS One 6:e16284. 10.1371/journal.pone.0016284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peters J, Nitsch M, Kuhlmorgen B, Golbik R, Lupas A, Kellermann J, Engelhardt H, Pfander JP, Muller S, Goldie K, Engel A, Stetter K-O, Baumeister W. 1995. Tetrabrachion: a filamentous archaebacterial surface protein assembly of unusual structure and extreme stability. J. Mol. Biol. 245:385–401 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.