

Abstract
The Escherichia coli regulator MarR represses the multiple-antibiotic resistance operon marRAB and responds to phenolic compounds, including sodium salicylate, which inhibit its activity. Crystals obtained in the presence of a high concentration of salicylate indicated two possible salicylate sites, SAL-A and SAL-B. However, it was unclear whether these sites were physiologically significant or were simply a result of the crystallization conditions. A study carried out on MarR homologue MTH313 suggested the presence of a salicylate binding site buried at the interface between the dimerization and the DNA-binding domains. Interestingly, the authors of the study indicated a similar pocket conserved in the MarR structure. Since no mutagenesis analysis had been performed to test which amino acids were essential in salicylate binding, we examined the role of residues that could potentially interact with salicylate. We demonstrated that mutations in residues shown as interacting with salicylate at SAL-A and SAL-B in the MarR-salicylate structure had no effect on salicylate binding, indicating that these sites were not the physiological regulatory sites. However, some of these residues (P57, R86, M74, and R77) were important for DNA binding. Furthermore, mutations in residues R16, D26, and K44 significantly reduced binding to both salicylate and 2,4-dinitrophenol, while a mutation in residue H19 impaired the binding to 2,4-dinitrophenol only. These findings indicate, as for MTH313, the presence of a ligand binding pocket located between the dimerization and DNA binding domains.
INTRODUCTION
Multiple-antibiotic resistance in Escherichia coli can arise from reduction of the intracellular concentration of antibiotics by upregulation of the expression of a drug efflux pump and by a decrease in the outer membrane permeability (1–4). The E. coli transcriptional regulator MarR represses the multiple-antibiotic resistance marRAB operon (Fig. 1) by interacting as a homodimer with two palindromic DNA regions, called site 1 (S1) and site 2 (S2), located in the promoter region (5) (Fig. 1). Both MarR binding sites are required for full transcriptional repression, but either site alone permits partial repression (5). MarR binding to its target DNA is reversed when a ligand such as sodium salicylate, menadione, plumbagin, 2,4-dinitrophenol (DNP), or the metabolite 2,3-dihydroxybenzoate interacts with the regulator (6–10). Consequent inactivation of MarR allows transcription of the marRAB operon. The resulting increased expression of MarA mediates multidrug resistance by activating the expression of a tripartite resistance-nodulation-division (RND) efflux pump encoded by acrAB and tolC (1, 11, 12). MarA also increases expression of small noncoding RNA micF, a translational inhibitor of ompF porin mRNA, and consequently decreases drug entry (2, 13, 14) (Fig. 1). marB, located just downstream of marA in the operon, is of unknown function, although data have suggested that it somehow enhances the transcription of marA (15).
Fig 1.
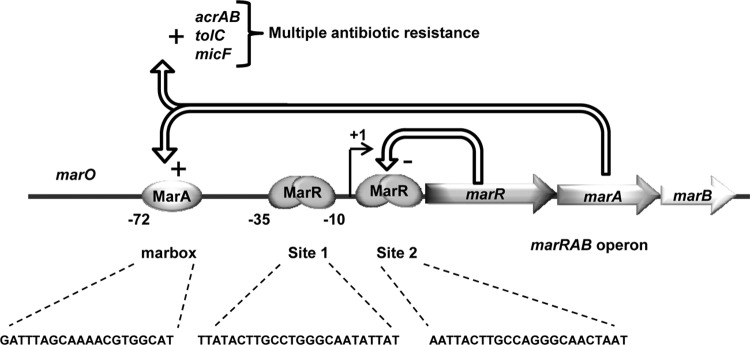
The marRAB operon. The MarR dimer binds marO at two different sites, site 1 and site 2, while MarA activates marRAB transcription by interacting with the marbox. MarA also activates the transcription of micF, acrAB, and tolC, leading to multiple-antibiotic resistance. The role of MarB is unknown.
Biochemical studies have been performed and molecular structures have been determined for several MarR homologues. How the binding of anionic compounds interferes with DNA binding remains unclear since, of the structures solved (9, 16–21), only a few show the structure both with and without an anionic ligand. Stable crystals grown in the presence of a high concentration of sodium salicylate (250 mM) were used to solve the structure of MarR (9). Two salicylate molecules were observed bound to each MarR subunit: the one at SAL-A appears to be involved in forming crystal packing contacts, while the other, at SAL-B, is solvent exposed (Fig. 2A). The hydroxyl of the salicylate at SAL-A is hydrogen bonded to the hydroxyl chain of Thr-72 in the H4 helix, and its carboxylate is hydrogen bonded to the guanidinium group of Arg-86. It also interacts with MarR through van der Waals contacts from Pro-57. The salicylate at SAL-B binds to the protein through hydrogen bonds with the backbone carbonyl of Ala-70 and Asp-42, van der Waals interactions with Ala-70 and Met-74, and an electrostatic contact with Arg-77 (Fig. 2A). Crystals grown in the absence of salicylate were poorly ordered, and removal of salicylate by soaking after crystal growth led to a disordering of diffraction (9). The DNA binding domains of the MarR dimer in the presence of salicylate are too closely apposed to be docked to a model of the DNA palindrome. However, since no structure of MarR has been obtained in its apo and DNA-bound conformation, any change in conformation caused by the occupancy of these sites by salicylate is unclear.
Fig 2.

Crystal structure of E. coli MarR and M. thermautotrophicum MTH313. (A) Ribbon representation of MarR-salicylate complex (Protein Data Bank [PDB] identity code 1JGS). Salicylate binds at two sites in each monomer. (B) Ribbon representation of MTH313 dimer interacting with salicylate (PDB identity code 3BPX). (C) Representation of the residues lying in the putative binding pocket in MarR by analogy with MTH313 structure (PDB identity code 1JGS). In each panel, one monomer is represented with colored secondary structures and the second monomer is represented in gray. H, alpha helix; SAL, salicylate. The software used for making the drawing was Swiss-PdbViewer (47).
Methanobacterium thermautotrophicum MTH313 is a regulatory protein in the MarR family for which the structure of the salicylate complex has been resolved (17). In the structure, a salicylate molecule (at SAL-1) was buried between the dimerization and DNA binding domains at a site containing mainly hydrophobic and basic residues (Fig. 2B). In the binding site, the salicylate interacts with the guanidinium group of Arg-16 and with the side chain amino group of Lys-8. The alkyl function of Arg-44, located in subunit B, might contribute to the hydrophobicity of the binding site (Fig. 2B). The second salicylate molecule, not well resolved, was positioned asymmetrically from SAL-1 and did not seem to impart a large conformational change to its subunit (17). SAL-1 was therefore suggested to be the biologically relevant site in MTH313. Comparison with the salicylate-free MTH313 structure indicated that the binding of salicylate produced a large conformational change in which the DNA binding lobes of the dimer were both pushed apart and twisted, leading to an open conformation unable to bind DNA (17). Interestingly, the authors of the study indicated a similar pocket conserved in the MarR structure. By analogy with MTH313 structure, Arg-16, His-19, and Lys-44 appeared to be good candidates in forming the analogous pocket of MarR (Fig. 2C).
Previously, no mutagenesis analysis had been performed to test which residues were truly involved in salicylate binding and response. In this study, we investigated, by site-directed mutagenesis, the role of the residues interacting with salicylate in MarR (see structures Fig. 2A and C). We also included the superrepressor D26N and G95S mutants in our study. Asp-26, located in helix H1, lies at the interface of the dimerization and DNA binding domains, while Gly-95 is located in the wing (Fig. 2C). Both mutants were previously obtained by chemical mutagenesis, with selection for inability to respond to salicylate (22). In our studies, we first analyzed the activity of each mutant in vivo with and without salicylate using a marO-lacZ fusion. We then purified numerous mutated proteins and examined their properties of binding to DNA and to ligands salicylate and 2,4-dinitrophenol (DNP).
MATERIALS AND METHODS
Compounds, bacterial strains, and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. Kanamycin, chloramphenicol, spectinomycin, isopropyl-1-thio-β-d-galactopyranoside (IPTG), sodium salicylate, 2,4-dinitrophenol, dithiothreitol (DTT), NaCl, HEPES, Na2HPO4, NaH2PO4, EDTA, imidazole, Tris, and boric acid were purchased from Sigma-Aldrich (St. Louis, MO). 4-(2-Aminoethyl)-benzylsulfonylfluoride hydrochloride (AEBSF) was purchased from Gold Biotechnology (St. Louis, MO). Restriction enzymes were purchased from New England BioLabs (Ipswich, MA). Synthetic oligonucleotides (see Table S1 in the supplemental material) were obtained from Integrated DNA Technology (Coralville, IA).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| DH5α | F− (ϕ80lacZΔM15) Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 (rK− mK+) phoA supE44 thi-1 gyrA96 relA1 | Laboratory collection |
| Rosetta (DE3) | lon and ompT protease-deficient E. coli carrying pRARE; expresses six rare tRNAs which facilitate expression of genes that contain rare E. coli codons; Camr; p15a ori | Merck KGaA, Darmstadt, Germany |
| SPC105 | MC4100 (ΔlacU169 araD rpsL relA thi flbB) containing a chromosomal PmarII::lacZ fusion at the λ attachment site; wild-type mar locus | 6 |
| SPC107 | SPC105 with a 39-kb deletion that includes the mar locus | 23 |
| AG100 | argE3 thi-1 rpsL xyl mtl supE44 λ lysogen | 24 |
| AG112 | AG100 marR mutant | 25 |
| Plasmids | ||
| pGEM-T Easy | Vector for direct cloning of PCR fragments; Ampr | Promega, Madison, WI |
| pET28a | Expression cloning vector; Kanr; the transcription of the cloned gene is driven by the T7 RNA polymerase and controlled by the LacI repressor; ori ColE1 | Merck KGaA |
| pET21b | Expression cloning vector; Ampr; the transcription of the cloned gene is driven by the T7 RNA polymerase and controlled by the LacI repressor; ori ColE1 | Merck KGaA |
| pACT7Sp | T7 RNA polymerase regulated by lacUV5; Spr; this plasmid is used in conjunction with pET28a; ori p15a | 26 |
| pET13a-G95S | pET13a carrying G95S variant of marR; Kanr | 22 |
| pET13a-D26N | pET13a carrying D26N variant of marR; Kanr | 22 |
| pVDMarR | pET28a carrying wild-type marR; Kanr | This study |
| pVD57 | pET28a carrying P57A variant of marR; Kanr | This study |
| pVD72 | pET28a carrying T72A variant of marR; Kanr | This study |
| pVD86 | pET28a carrying R86A variant of marR; Kanr | This study |
| pVD74 | pET28a carrying M74A variant of marR; Kanr | This study |
| pVD77 | pET28a carrying R77A variant of marR; Kanr | This study |
| pVD5772 | pET28a carrying P57A-T72A variant of marR; Kanr | This study |
| pVD7477 | pET28a carrying M74A-R77A variant of marR; Kanr | This study |
| pVD26 | pET28a carrying D26N variant of marR; Kanr | This study |
| pVD95 | pET28a carrying G95S variant of marR; Kanr | This study |
| pVD16 | pET28a carrying R16A variant of marR; Kanr | This study |
| pET28a-MarR-H19A | pET28a carrying H19A variant of marR; Kanr | This study |
| pET28a-MarR-K44A | pET28a carrying K44A variant of marR; Kanr | This study |
| pVDOR | pET21b carrying ompR nucleotide sequence cloned in frame with a polyhistidine tag at the C-terminal end | This study |
Cloning and site-directed mutagenesis.
Wild-type marR nucleotide sequence was amplified by PCR using primers marR-ncoI and marR-xhoI (see Table S1 in the supplemental material) and E. coli AG100 chromosomal DNA as the template. The fragment was cloned into the pGEM-T Easy vector using the manufacturer's protocol. The resulting plasmid was used as a template to introduce the desired mutation into the marR coding sequence using the overlap extension PCR method (27) and the FW and RV mutagenic primers listed in Table S1. Briefly, mutated fragments were generated using marR-ncoI plus RV primers (PCR1) and marR-XhoI plus FW primers (PCR2). A third PCR was performed mixing PCR1 and PCR2 and using marR-ncoI plus marR-xhoI as primers. Each mutated fragment was cloned into the pET28a vector using the restriction sites NcoI and XhoI. A stop codon was included in marR-xhoI so that the construction allowed the expression of native MarR with no His tag. Also, an Ala codon was added to the marR-ncoI primers to maintain frame with the ATG located in the NcoI restriction site. For the D26N and G95S substitutions, the corresponding nucleotide sequence was amplified using plasmids pET13a-D26N and pET13a-G95S as the template and primers marR-ncoI and marR-xhoI. The amplified fragment was then cloned into pET28a.
ompR nucleotide sequence was amplified by PCR using chromosomal DNA from E. coli strain AG100 as the template and primers OmpR-FW and OmpR-RV (see Table S1 in the supplemental material). The amplified DNA fragment was cloned into the pGEM-T Easy vector by following the manufacturer's instructions. The resulting plasmid was digested with NdeI and XhoI (New England BioLabs, Ipswich, MA), and the DNA fragment containing ompR was ligated to the similarly cut vector pET21b, yielding plasmid pVDOR, in which ompR was cloned in frame with a C-terminal His tag.
All nucleotide sequences were verified at the Tufts University Core Facility (Tufts University School of Medicine, Boston, MA).
Determination of MIC.
E. coli AG112 transformants carrying pACT7Sp and pET28a derivative plasmids were isolated on LB agar plates supplemented with 50 μg/ml of kanamycin and 50 μg/ml of spectinomycin. After suspension of several isolated colonies in LB medium to reach an optical density at 600 nm of 0.1, the bacteria were spread with a swab on LB agar plates supplemented with the appropriate antibiotics. MICs of ampicillin, tetracycline, and chloramphenicol were determined using Etest strips (bioMérieux, Durham, NC) at 37°C.
β-Galactosidase assays.
To characterize the activity of each mutant, we expressed MarR in SPC107 using the pET28a plasmid and the helper vector pACT7Sp. In pET28a, marR was regulated by a T7/lacO promoter, while pACT7Sp carried the T7 polymerase gene regulated by the lacUV5 promoter (26); both promoters were inducible by IPTG. An overnight culture of a strain containing a marO-lacZ fusion grown in LB medium with appropriate antibiotics was diluted 1/100 in identical medium for growth. When the optical density at 600 nm reached 0.4, the culture was divided into two subcultures and salicylate was added to one of them. Since a high concentration of salicylate affects the growth of the bacteria, 1 mM salicylate was used, as it allows a good induction of LacZ expression with minimal effect on growth. Both cultures were continued, and β-galactosidase (LacZ) activity was assayed by permeabilizing the cells with 0.005% sodium dodecyl sulfate and 0.05% chloroform and expressed in Miller units as previously described (28). All assays were carried out at least in three independent experiments.
Detection of MarR by Western blotting.
To measure MarR protein expression level, 10 ml of cells grown to an optical density at 600 nm of 0.4 was recovered by centrifugation for 10 min at 4,000 × g. After suspension in 500 μl of buffer (20 mM sodium phosphate, 2 mM DTT, 0.5 mM EDTA, 0.5 mM AEBSF, 200 mM NaCl [pH 7.4]), the cells were lysed by sonication and the supernatant was recovered after centrifugation for 10 min at 4,000 × g. Protein content was quantified using the Pierce 660-nm protein assay reagent (Thermo Fisher Scientific, Waltham, MA). Ten micrograms of protein was then separated by 15% SDS-PAGE and electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). The membrane was incubated overnight at 4°C in Tris-borate-saline buffer (TBS) supplemented with 3% milk powder. The membrane was then incubated for 2 h at room temperature with serum from rabbit polyclonal anti-MarR antibodies (22) diluted in TBS (1/10,000). After three 15-min washes with TTBS (TBS supplemented with 0.5% Tween 20), the membrane was incubated for 2 h with goat alkaline phosphatase-coupled secondary anti-rabbit antibodies (Bio-Rad Laboratories, Hercules, CA) diluted 1/10,000 in TBS, followed by three 15-min washes with TTBS. MarR was visualized by adding 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium by following the manufacturer's instructions (Promega, Madison, WI).
Expression and purification of proteins.
To express each protein variant, a fresh transformant of E. coli Rosetta (DE3) carrying the pET28a derivative plasmid was grown at 37°C in 500 ml of LB supplemented with 50 μg/ml of kanamycin and 30 μg/ml of chloramphenicol. The high expression of the protein was obtained by adding 0.5 mM IPTG to the culture when the optical density at 600 nm reached 0.6. Growth of the culture was then continued for 3 h. Cells were recovered by centrifugation for 15 min at 6,000 × g and suspended in 10 ml of buffer A (20 mM Na2HPO4-NaH2PO4 [pH 7.4] and 2 mM DTT). After sonication of the cells, the soluble lysate was recovered by centrifugation for 30 min at 18,000 × g and loaded onto 10 ml of SP-Sepharose Fast Flow matrix (GE Healthcare, Pittsburgh, PA) previously equilibrated in buffer A. After the matrix was washed, MarR was eluted with 20 ml of 300 mM NaCl in buffer A. Eluted MarR was diluted 1:1 (vol/vol) in buffer A to decrease the NaCl concentration to 150 mM and loaded onto a 1-ml Hi-Trap Heparin HP column (GE Healthcare) previously equilibrated with 150 mM NaCl in buffer A. MarR was eluted with 4 ml of 300 mM NaCl in buffer A. MarR was subsequently dialyzed twice against 20 volumes of storage buffer (20 mM HEPES [pH 7.4], 250 mM NaCl, 5% glycerol, 5 mM DTT, 0.5 mM EDTA, 0.5 mM AEBSF) and then concentrated to ∼1 mg/ml using an Amicon Ultra-4 centrifugal unit (Merck KGaA, Darmstadt, Germany). Aliquots were stored at −20°C, used once, and discarded. Wild-type MarR and mutants were purified to greater than 95% homogeneity as determined by SDS-PAGE (see Fig. S2A in the supplemental material).
OmpR-His protein was expressed as described above for MarR proteins using E. coli Rosetta (DE3). Cells harvested from a culture of 250 ml by centrifugation were suspended in buffer B (20 mM sodium phosphate, 500 mM NaCl, 2 mM DTT [pH 7.0]). After sonication and elimination of the insoluble extract by centrifugation, the soluble extract was loaded onto a 5-ml nickel-nitrilotriacetic acid (Ni-NTA) chelating column (Qiagen, Germantown, MD) precharged with 50 mM NiSO4 and equilibrated with buffer B. The column was washed with 50 mM imidazole (pH 8.0), and OmpR-His protein was eluted with 25 ml of 250 mM imidazole (pH 8.0). The protein was dialyzed twice against 20 volumes of storage buffer (20 mM NaH2PO4-Na2HPO4 buffer, 200 mM NaCl, 2 mM DTT, 0.5 mM EDTA, 0.5 mM AEBSF [pH 7.0]), concentrated by ultrafiltration to 1 mg/ml, aliquoted, and stored at 4°C. OmpR-His was purified to greater than 95% homogeneity, as determined by SDS-PAGE (see Fig. S2A in the supplemental material).
Protein concentration determination.
Protein concentration was determined using the Pierce 660-nm protein assay reagent and bovine serum albumin (BSA) as a standard (Thermo Fisher Scientific, Waltham, MA).
Electrophoretic mobility shift assay (EMSA).
The synthetic oligonucleotides used in this study were obtained from Integrated DNA Technologies (Coralville, IA). The melting temperatures (Tms) of S2 and M2A oligonucleotides were 62 and 59°C, respectively (see Fig. 5). To generate the double-stranded DNA, 25-μl quantities of the forward and reverse complementary single-stranded oligonucleotides at 100 μM (see Fig. 5A for sequences) were mixed, denatured, and annealed using a 2720 thermal cycler (Applied Biosystems, Carlsbad, CA) with the following steps: 10 min at 95°C, 30 s at 85°C, 30 s at 75°C, 30 s at 65°C, 30 s at 55°C, 30 s at 45°C, 30 s at 35°C, and 10 min at 25°C. The annealed DNA was always used within 4 h after annealing. Binding reaction mixtures contained reaction buffer (20 mM HEPES [pH 7.4], 50 mM NaCl, 10% [vol/vol] glycerol, 0.5 mg/ml of AEBSF, 100 μg/ml of BSA, 5 μg/ml of herring sperm DNA, 1 mM DTT), annealed DNA, and purified MarR. After incubation at 4°C for 20 min, 10×-concentrated loading buffer (60% glycerol, 0.01% xylene cyanol, and 0.01% bromophenol blue) was added and electrophoresis was performed using an 8% polyacrylamide nondenaturing gel. After electrophoresis, the gel was stained for 30 min with SYBR green (Life Technologies, Carlsbad, CA) diluted 1/10,000 in TBE buffer and visualized using a GBox Chemi XT4 (Syngene, Frederick, MD).
Fig 5.

Thermal stabilization of MarR upon binding to DNA. (A) Sequence of the DNA probes used to study MarR-DNA interaction. Arrows indicate inverted repeats. Bold type indicates the palindromic sequence. Boxes highlight a substitution of nucleic acid bases. (B) Thermal stabilization of wild-type (WT) MarR upon binding to DNA. Increasing concentrations of DNA S2 were incubated with 1.5 μM dimeric protein. Melting curves were generated by plotting SYPRO orange fluorescence as a function of temperature. Tm0 is the Tm measured in the absence of DNA. Tm5 is the Tm measured in the presence of 5 μM DNA. DNA did not display any change in fluorescence above background at any temperature tested. RFU, relative fluorescence units. (C) Change in protein Tm (ΔTm) in the presence of increasing concentrations of DNA duplex S2 and M2A. OmpR was used a negative control. (D) EMSA of DNA and wild-type (WT) MarR. Lanes: 1, 50 nM S2 DNA; 2, 50 nM S2 DNA and 100 nM MarR dimer; 3, 50 nM M2A DNA; 4, 50 nM M2A DNA and 100 nM MarR dimer. (E) Change in protein Tm in the presence of 3 μM DNA S2 and 1.5 μM dimeric protein. The data represent the means ± the standard deviations for at least 3 independent experiments. Statistically significant differences for a mutant compared to the WT are shown as double asterisks (P < 0.01).
Thermal stabilization of MarR by DNA and salicylate.
Protein thermal stabilization has been previously used to validate the quality of protein preparations and to screen for buffers, ligands, and additives that increased the stability of a protein (29–31). This assay uses SYPRO orange (Sigma-Aldrich, St. Louis, MO), a nonspecific protein-binding dye for which the fluorescence increases when the environment becomes more hydrophobic (31, 32). Thermal denaturation of protein causes exposure of normally interior hydrophobic regions and can be followed by SYPRO orange fluorescence (excitation and emission wavelengths of 465 and 610 nm, respectively) monitored using a LightCycler 480 instrument (Roche, Indianapolis, IN) (30). The protocol used in this study was optimized from reference 33. After an initial incubation for 10 s at 25°C, the temperature was increased from 25°C to 95°C with continuous increments and the acquisition rate was set at 3 readings per °C. The reaction buffer consisted of 20 mM HEPES, 50 mM NaCl, and 1 mM DTT (pH 7.4). In a clear LightCycler 480 multiwell plate (Roche), the reaction mixtures contained 10 μl of SYPRO orange diluted 1/1,000 in reaction buffer, 2 μl of MarR (0.5 mg/ml) or 3.4 μl of OmpR (0.5 mg/ml), and 2 μl of ligands or 4 μl of DNA. Reaction buffer was then added to reach a total volume of 20 μl. For every experiment, the intrinsic stability of the protein was measured in the absence of DNA or ligands. The fluorescence intensity plotted as a function of temperature gave a two-state transition sigmoidal curve following the equation
where F is the fluorescence measured, Fmax is the maximal fluorescence, Fmin is the minimal fluorescence, T is the temperature, Tm (apparent melting temperature) is the temperature halfway between Fmax and Fmin, and a is the slope of the curve (30). The data were exported as a text file (.txt) and copied into a GraphPad Prism file for further analysis (GraphPad Software Inc., La Jolla, CA). The apparent Tm, which corresponds to the inflection point of the transition curve, was calculated as the maximum of the first derivative of the equation above using ThermoQ software (http://jshare.johnshopkins.edu/aherna19/thermoq/).
Statistical analysis.
At least three determinations were made for each experiment; we report the average (mean) and the standard deviation (SD). The statistical significance of differences between two averages was determined by Student's t test (two independent samples, with two-tailed distribution) using GraphPad Prism software.
RESULTS
Repression activity of the MarR variants.
The activity of each MarR variant was first tested using E. coli SPC105 ΔmarR (SPC107), a strain which harbors a marO-lacZ transcriptional fusion. MarR was expressed using the pET28a vector, for which the expression of the target gene is under the control of a T7 lacO promoter (see Materials and Methods for details). Even in the absence of IPTG to induce this promoter, the complemented strain SPC107/marRwt expressed low β-galactosidase activity compared to that of the control (no MarR), indicating that the basal expression of wild-type MarR was sufficient to repress LacZ expression (Fig. 3A); wild-type MarR decreased LacZ expression 19.5-fold compared to that of the control strain lacking MarR. The addition of 1 mM salicylate to SPC107/marRwt induced LacZ expression 11.5-fold (Fig. 3A). We also observed that salicylate was able to induce LacZ expression 1.5-fold in the absence of MarR, suggesting a MarR-independent induction. This effect likely depends on EmrR, since previous studies have shown that overexpression of EmrR led to repression of the marRAB operon and that EmrR was also inactivated by salicylate (34, 35). Moreover, we have found that chromosomally expressed EmrR repressed marO-lacZ about 2-fold (unpublished data).
Fig 3.

Activity and expression of each MarR variant in complemented SPC107. (A) Effect of each marR mutation on β-galactosidase activity from a marO-lacZ fusion in SPC107. SPC107 was transformed with plasmids pACT7Sp and pET28a carrying marR variants. Cells were grown in the absence of IPTG. The data represent the means ± the standard deviations for at least three independent experiments. Percentages at A = 100 × [LacZ (MarR)/LacZ (no MarR)]no salicylate. Numbers above the bars represent the LacZ (salicylate)/LacZ (no salicylate) ratio for each variant. (B) Western blot analysis using lysates of complemented SPC107 grown in the absence of IPTG. Lysate from cells carrying pACT7Sp and pET28a without insert was used as a negative control (empty vector). Each lane contained 10 μg of cell lysate. MarR was detected using polyclonal anti-MarR antibodies. WT, wild type.
We engineered and studied mutants of several residues lying in the SAL-A and SAL-B sites by alanine replacement. G95S and D26N mutants from an earlier work were also included, since they have been shown to not respond to salicylate (22). We characterized the mutants in vivo using SPC107 in the manner described above. Our analysis showed that mutations P57A, R86A, M74A, and R77A in sites SAL-A and SAL-B were not able to repress LacZ (Fig. 3A). With salicylate, the P57A, R86A, M74A, and R77A mutants displayed LacZ activity similar to that of the control SPC107 lacking marR (Fig. 3A). These results indicated either the presence of a dysfunctional MarR protein or the absence of the protein in the cell. Although MarR expression was slightly different among the strains, we were able to detect P57A, R86A, M74A, and R77A protein in SPC107 lysates (Fig. 3B). Moreover, overexpression of these mutants using 5 μM IPTG (see Fig. S1C in the supplemental material) did not cause wild-type levels of repression of LacZ expression, suggesting that these mutants were in fact inactive (see Fig. S1A and B). Our findings therefore suggested that the mutations introduced in the SAL-A and SAL-B binding sites led to a decreased DNA binding activity rather than a decrease in ligand binding. It should be noted that the addition of only 5 μM IPTG to SPC107/marRwt led to a large amount of MarR in the cell and consequently extremely low LacZ activity for SPC107/marRwt that responded poorly to salicylate (see Fig. S1), indicating a tight interaction with the DNA in the absence and in the presence of 1 mM salicylate. Thus, overexpression of MarR through addition of even 5 μM IPTG should not be used for in vivo analysis.
T72A (in SAL-1 of MarR) and R16A, H19A, and K44A (corresponding to SAL-1 of MTH313) gave an intermediate repression of LacZ expression (LacZ activity was ∼50% of that of the control strain lacking MarR). Western blot analysis showed that T72A and H19A mutant expression was less than that of wild-type MarR (Fig. 3B), which could partly explain the intermediate activity of those mutants. The addition of salicylate to SPC107/T72A induced LacZ expression to the level of the control strain lacking MarR, showing a normal response to salicylate. However, the R16A, H19A, and K44A mutants, while also showing only partial repression, did not induce well with salicylate (Fig. 3A): for these mutants, LacZ activity values in the presence of salicylate were 1,000 Miller units below that of the control lacking MarR. These results suggested that a mutation in R16, H19, and K44 decreased response to salicylate.
The G95S mutant repressed LacZ expression 114-fold compared to that of the control strain lacking MarR and was the mutant that showed the highest repression activity (Fig. 3A). Addition of salicylate to SPC107/G95S induced LacZ expression only 1.4-fold (Fig. 3A). The D26N mutant repressed LacZ expression 24-fold. It induced only 2.1-fold with salicylate, much less than the 11.5-fold of wild-type MarR (Fig. 3A). SPC107/R16A-D26N gave a higher LacZ expression than that of the wild type, while addition of salicylate again induced LacZ expression only 2.1-fold. Therefore, the G95S and D26N mutations caused a dramatic decrease in response to salicylate.
Multiple-antibiotic susceptibility of E. coli expressing a MarR variant.
E. coli AG112, a well-characterized MAR strain, carries an inactivating mutation in MarR and consequently overexpresses the MarA regulator, leading to a decreased susceptibility to antibiotics. The antibiotic susceptibility of E. coli AG112 expressing each MarR variant was assessed by measuring the MICs for ampicillin, tetracycline, and chloramphenicol, providing an assay for the ability of the MarR variant to repress in vivo (Table 2). The susceptibility of AG112 expressing P57A, M74A, R77A, and R86A was comparable to that of the control strain carrying the pET28a vector without the insert, showing the inactivity of these MarR mutants. AG112 expressing T72A, R16A, H19A, and K44A showed intermediate antibiotic susceptibility relative to AG112. Finally, the expression of superrepressor D26N or G95S in AG112 led to antibiotic susceptibility similar to that caused by wild-type MarR in the absence of salicylate. In the presence of 1 mM salicylate, AG112/marRwt displayed a decreased susceptibility to the antibiotics tested, indicating inhibition of wild-type MarR by the ligand and the consequent expression of MarA. However, the addition of salicylate did not change the antibiotic susceptibility of AG112 carrying the G95S or D26N variant, indicating resistance of these variants to inhibition by salicylate. Only a small decrease in antibiotic susceptibility was observed, comparable to that seen for the control lacking marR. This small change in antibiotic susceptibility might be due to a MarR-independent induction of marRAB transcription by salicylate, as previously described (6).
Table 2.
Antibiotic susceptibility of E. coli carrying a MarR variant
| Straina | MIC (μg/ml)b |
||
|---|---|---|---|
| AM/+S | TC/+S | CM/+S | |
| AG112/pET28a (no marR) | 3/3.5 | 8/11 | 24/32 |
| AG112/marRwt | 1.5/3 | 2.5/5 | 6/16 |
| AG112/P57A | 3.5 | 7 | 28 |
| AG112/T72A | 2 | 4 | 14 |
| AG112/R86A | 3.5 | 7 | 28 |
| AG112/P57A-T72A | 3.5 | 5 | 24 |
| AG112/M74A | 3 | 6 | 24 |
| AG112/R77A | 3 | 6 | 24 |
| AG112/M74A-R77A | 3.5 | 6 | 24 |
| AG112/R16A | ND | 5.3 | 16 |
| AG112/H19A | ND | 4.7 | 16 |
| AG112/K44A | ND | 5.3 | 18.7 |
| AG112/D26N | 1.5/1.5 | 2.3/3 | 5.5/8 |
| AG112/G95S | 1.5/1.5 | 2/2.5 | 5/7 |
marR mutation was complemented using pACT7Sp together with pET28a carrying each marR variant. Bacteria were plated on LB agar containing kanamycin at 50 μg/ml and spectinomycin at 50 μg/ml.
Numbers represent an average of at least 4 experimental measurements. The standard errors of the means were <20% for all experiments. MICs were determined by Etest. AM, ampicillin; TC, tetracycline; CM, chloramphenicol; ND, not determined; +S, MIC determined in the presence of 1 mM sodium salicylate.
Effect of amino acid substitutions on DNA binding. (i) EMSA.
To further characterize the activity of the MarR variants, we purified the mutated proteins to apparent homogeneity by following the procedure described in Materials and Methods (see also Fig. S2A in the supplemental material). We used electrophoretic mobility shift assay (EMSA) to compare semiquantitatively the binding affinities of the proteins to a 35-mer duplex DNA containing site 2 (S2). Figure 4 shows representative EMSAs. G95S and D26N mutants bind DNA with a higher affinity than that of wild-type MarR. T72A and H19A mutants showed similar DNA binding activities, though lower than that of wild-type MarR, while the K44A mutant DNA binding affinity was even lower. R86A, P57A-T72A, and M74A-R77A mutants did not bind the DNA at all under the given conditions.
Fig 4.
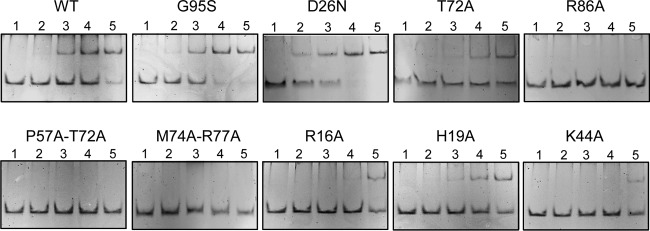
Electrophoretic mobility shift assay of wild-type (WT) MarR and mutants. Annealed DNA S2 at 50 nM was incubated with increasing concentrations of protein and resolved as described in Materials and Methods. Lanes: 1, no MarR; 2, 12.5 nM MarR dimer; 3, 25 nM MarR dimer; 4, 50 nM MarR dimer; 5, 100 nM MarR dimer.
(ii) Thermal stabilization of MarR by DNA.
To characterize the DNA binding activity of the MarR variants using an independent method, we examined the thermal denaturation of MarR in the presence of DNA duplex S2. First, we monitored the fluorescence of SYPRO orange while denaturing native, globular MarR. The low fluorescence signal observed at low temperature indicated the presence of a globular protein (Fig. 5B, “No DNA”). As the temperature rose, the fluorescence signal increased as the protein melted. The temperature at the midpoint of this transition is the apparent melting temperature (Tm) (Fig. 5B, “No DNA”). For all the proteins studied, Tms were similar, indicating comparable protein stabilities (see Fig. S2B in the supplemental material). Incubation of wild-type MarR with increasing concentrations of DNA duplex S2 (see Fig. 5A for sequence) led to a shift of the Tm toward higher temperature, which indicated protein stabilization upon DNA binding (Fig. 5B and C). As expected, OmpR, the negative control, was not stabilized by the DNA used (Fig. 5C). Indeed, OmpR, the cytoplasmic osmoregulator of the EnvZ/OmpR two-component regulatory system, recognizes specific DNA sequence not found in marO promoter (36, 37). To assess the specificity of the binding, we also used two modified complementary oligonucleotides carrying two base substitutions (C→A and G→T) located in the palindromic recognition region of S2 DNA (M2A; see Fig. 5A for sequence). These base substitutions affected MarR DNA binding, as shown by EMSA in Fig. 5D. Figure 5C shows that M2A was not able to stabilize MarR such as S2, validating the method to compare DNA binding activities. DNA binding activity of the mutants was then assessed by comparing the protein stabilization in the presence of 3 μM DNA. The stabilization of D26N and G95S superrepressors by S2 DNA was greater than that of wild-type MarR, confirming the higher affinity for DNA for these superrepressors (Fig. 5E). Our results also showed that T72A, R16A, and H19A mutants were able to bind S2, as shown by the stabilization of the protein in the presence of DNA, though the stabilization was inferior to that of wild-type MarR (Fig. 5E). Low protein stabilization was observed for K44A, P57A-T72A, R86A, and M74A-R77A mutants, indicating a large defect in binding to DNA (Fig. 5E).
Thermal stabilization of MarR variants by ligand.
As an assay of the ability of different MarR mutants to bind ligand, we measured thermal stability using SYPRO orange in the manner used to study the MarR-DNA interaction. We first monitored the effect of salicylate on the thermal stability of wild-type MarR. The titration curve showed a concentration-dependent stabilization of wild-type MarR, although the high salicylate concentration used to obtain saturation indicated possible nonspecific binding (Fig. 6A). We then compared the thermal stabilization of each mutant in the presence of 1 mM salicylate, the concentration used in our in vivo assays. Salicylate at 1 mM raised the wild-type MarR Tm by ∼2°C (Fig. 6B). Similar thermal stabilization was observed for T72A, P57A-T72A, R86A, M74A-R77A, G95S, and H19A mutants (Fig. 6B), indicating wild-type salicylate binding activity for these proteins. For D26N, R16A, and K44A mutants, thermal stabilization was smaller than that of wild-type MarR, with decreases of 1.6-, 2.6-, and 2.3-fold, respectively. We also tested the thermal stabilization of these proteins in the presence of 50 μM DNP, a concentration that induced LacZ expression in vivo ∼10-fold for SPC107/marRwt (data not shown). While levels of thermal stabilization by DNP were similar for wild-type MarR and T72A, P57A-T72A, R86A, M74A-R77A, and G95S mutants, it was greatly reduced for D26N, H19A, and K44A mutants, i.e., 12-, 4.3-, and 6-fold, respectively. For the R16A mutant, the thermal stabilization by DNP was reduced 1.5-fold (Fig. 6C). These results suggest that D26N, R16A and K44A mutants have reduced binding to both salicylate and DNP, while for the H19A mutant, only DNP binding is decreased. The results also suggest that binding of both ligands to the other mutants tested, including those with changes involving residues comprising the SAL-1 and SAL-2 sites, as well as the G95S mutant, is similar to that of wild-type MarR.
Fig 6.

Thermal stabilization of MarR upon ligand binding. (A) Specific thermal stabilization of MarR upon binding to salicylate. Dimeric protein at 1.5 μM was denatured in the presence of increasing concentrations of salicylate. Change in protein melting temperature (ΔTm) was plotted as a function of salicylate concentration. OmpR was used as a negative control and, as expected, was not stabilized by salicylate. (B) Change in Tm in the presence of 1 mM sodium salicylate. (C) Change in Tm in the presence of 50 μM DNP. Salicylate and DNP did not display any change in fluorescence above background at any of the temperatures tested (data not shown). The data represent the means ± standard deviations of at least 3 independent experiments. Statistically significant differences for a mutant compared to the wild type (WT) are shown as double asterisks (P < 0.01).
DISCUSSION
Spontaneous mutations inactivating MarR are often recovered in vivo and result in resistance to a range of antibiotics and disinfectants via MarA (25, 38–40). In the MarR structure, residues interacting with the salicylates in sites SAL-A and SAL-B are located within the DNA binding domain of the protein (9). Not surprising, therefore, are our findings that most mutants carrying a substitution in one or two of these residues presented a repression defect in vivo. In accordance with these observations, a mutation in P57, R86, M74, or R77 corresponded to a large decrease in antibiotic susceptibility.
The structure of the MarR-salicylate complex shows two salicylate molecules per monomer, both located within the DNA binding motif and both solvent exposed. As described earlier, because of the high salicylate concentration and the importance of salicylate in crystal contacts, it was not clear whether these sites were physiologically significant. Our present findings show that none of the SAL-A or SAL-B site residues was essential for the binding of salicylate in vitro. All together, these results indicate that the presence of salicylate was necessary to obtain stable crystals but did not reveal the true regulatory salicylate binding sites. We hypothesize that the high concentration of sodium salicylate stabilized the DNA binding domain and restricted the conformational flexibility of MarR, resulting in a closed conformation which did not allow the ligand to bind at its regulatory sites. Interestingly, the G95S mutant did not respond to salicylate in vivo. Our in vitro study demonstrated that the G95S mutant was able to bind both salicylate and DNP as well as did wild-type MarR, suggesting that the nonresponder phenotype of the G95S mutant may arise simply from a greater affinity for DNA. The G95S mutation is at the tip of the wing of the winged helix-turn-helix (HTH) DNA binding motif. A serine at position 95 of MarR could generate van Der Waals contacts with the DNA like those observed for a similarly located serine in the structure of the OhrR-DNA complex (16), increasing affinity. Alternatively, Ser-95 could also generate an extra hydrogen bond with the phosphate backbone. Gly-95 in wild-type MarR would be unable to form such van Der Waals interactions, allowing a greater torsional flexibility in the protein backbone. The additional bond with the DNA due to the serine residue in G96S might disfavor the movement of the DNA binding domain upon ligand binding and the conformational change necessary for the release from the DNA.
In the structure of the MarR homologue MTH313-salicylate complex, the salicylate binding site was buried between the dimerization and DNA binding domains. This site was hypothesized to have a high potential to bind different anionic compounds due to its size and the presence of basic residues in the pocket (17). Our results demonstrated that basic residues located in a similarly conserved pocket in MarR were important for salicylate and DNP binding. An exact interpretation of our results is difficult since no structure of MarR with ligand bound in the pocket is available. By analogy with the MTH313-salicylate structure, we hypothesize that the side chains of residues Arg-16 and Lys-44 interact with the hydroxyl and carboxylate groups of the salicylate molecule. Residue Asp-26 could position a water molecule leading to additional bonds with either the hydroxyl or carboxylate group of salicylate. Our analysis showed that while not involved in salicylate binding, His-19 was important for binding DNP, indicating that MarR can bind several structurally related anionic ligands through an adaptable binding pocket. An additional possible bond between His-19 and one of the nitro groups of DNP could also explain the stronger interaction with DNP compared to salicylate. Though none was identified in this study, additional residues could interact with the two nitro groups of DNP and contribute to its higher affinity for MarR.
Mutations in residues located in helices H1 and H2 affected binding to DNA. It is plausible that residues R16, H19, and D26 located in H1 could interact with residues located on helice H2 or H5 and trigger a movement of the DNA binding domain to promote DNA binding. Interactions of these residues with a ligand could then induce a conformational change that modifies the DNA binding of MarR.
Although members of the MarR family generally have low primary sequence similarity (∼25%), all known structures show high structural homology. MarR homologues control pathways that are critical to bacterial physiology such as stress responses (41, 42), virulence (43), metabolism (10), and multiple-drug resistance (23, 44, 45). The activity of a subset of these proteins can be modified through the formation of a disulfide bond (19, 41, 46). However, most respond to specific ligands. Comparison of the structures of M. autotrophicum MTH313, Staphylococcus epidermidis TcaR, and Sulfolobus tokodaii ST1710 salicylate complexes reveals a similar salicylate binding pocket located between helices H1 and H2 (17, 20, 21). Interestingly, the carboxylate moiety of salicylate interacts with the side chain of a basic residue located on helix H1 in all cases, suggesting that these proteins have evolved to interact with anionic ligands similar to salicylate.
Supplementary Material
ACKNOWLEDGMENTS
We thank bioMérieux for providing Etests strips used in this work. We appreciated helpful discussions with Laurent Bouillaut and Jared Pitts about the protein stabilization experiments.
This work was supported by U.S. Public Health Service grant AI56021 from the National Institutes of Health.
Footnotes
Published ahead of print 17 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02224-12.
REFERENCES
- 1. Blair JM, Piddock LJ. 2009. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr. Opin. Microbiol. 12:512–519 [DOI] [PubMed] [Google Scholar]
- 2. Cohen SP, McMurry LM, Levy SB. 1988. marA locus causes decreased expression of OmpF porin in multiple-antibiotic-resistant (Mar) mutants of Escherichia coli. J. Bacteriol. 170:5416–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S. 2011. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front. Microbiol. 2:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirai K, Aoyama H, Suzue S, Irikura T, Iyobe S, Mitsuhashi S. 1986. Isolation and characterization of norfloxacin-resistant mutants of Escherichia coli K-12. Antimicrob. Agents Chemother. 30:248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin RG, Rosner JL. 1995. Binding of purified multiple antibiotic-resistance repressor protein (MarR) to mar operator sequences. Proc. Natl. Acad. Sci. U. S. A. 92:5456–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen SP, Levy SB, Foulds J, Rosner JL. 1993. Salicylate induction of antibiotic resistance in Escherichia coli: activation of the mar operon and a mar-independent pathway. J. Bacteriol. 175:7856–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alekshun MN, Levy SB. 1999. Alteration of the repressor activity of MarR, the negative regulator of the Escherichia coli marRAB locus, by multiple chemicals in vitro. J. Bacteriol. 181:4669–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pomposiello PJ, Bennik MH, Demple B. 2001. Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J. Bacteriol. 183:3890–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. 2001. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 A resolution. Nat. Struct. Biol. 8:710–714 [DOI] [PubMed] [Google Scholar]
- 10. Chubiz LM, Rao CV. 2010. Aromatic acid metabolites of Escherichia coli K-12 can induce the marRAB operon. J. Bacteriol. 192:4786–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barbosa TM, Levy SB. 2000. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J. Bacteriol. 182:3467–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White DG, Goldman JD, Demple B, Levy SB. 1997. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J. Bacteriol. 179:6122–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coyer J, Andersen J, Forst SA, Inouye M, Delihas N. 1990. micF RNA in ompB mutants of Escherichia coli: different pathways regulate micF RNA levels in response to osmolarity and temperature change. J. Bacteriol. 172:4143–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou JH, Greenberg JT, Demple B. 1993. Posttranscriptional repression of Escherichia coli OmpF protein in response to redox stress: positive control of the micF antisense RNA by the soxRS locus. J. Bacteriol. 175:1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong M, Fuangthong M, Helmann JD, Brennan RG. 2005. Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol. Cell 20:131–141 [DOI] [PubMed] [Google Scholar]
- 17. Saridakis V, Shahinas D, Xu X, Christendat D. 2008. Structural insight on the mechanism of regulation of the MarR family of proteins: high-resolution crystal structure of a transcriptional repressor from Methanobacterium thermoautotrophicum. J. Mol. Biol. 377:655–667 [DOI] [PubMed] [Google Scholar]
- 18. Yu L, Fang J, Wei Y. 2009. Characterization of the ligand and DNA binding properties of a putative archaeal regulator ST1710. Biochemistry 48:2099–2108 [DOI] [PubMed] [Google Scholar]
- 19. Newberry KJ, Fuangthong M, Panmanee W, Mongkolsuk S, Brennan RG. 2007. Structural mechanism of organic hydroperoxide induction of the transcription regulator OhrR. Mol. Cell 28:652–664 [DOI] [PubMed] [Google Scholar]
- 20. Chang YM, Jeng WY, Ko TP, Yeh YJ, Chen CK, Wang AH. 2010. Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl. Acad. Sci. U. S. A. 107:8617–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumarevel T, Tanaka T, Umehara T, Yokoyama S. 2009. ST1710-DNA complex crystal structure reveals the DNA binding mechanism of the MarR family of regulators. Nucleic Acids Res. 37:4723–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alekshun MN, Levy SB. 1999. Characterization of MarR superrepressor mutants. J. Bacteriol. 181:3303–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seoane AS, Levy SB. 1995. Characterization of MarR, the repressor of the multiple antibiotic resistance (mar) operon in Escherichia coli. J. Bacteriol. 177:3414–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. George AM, Levy SB. 1983. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 155:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oethinger M, Podglajen I, Kern WV, Levy SB. 1998. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob. Agents Chemother. 42:2089–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duval V, Nicoloff H, Levy SB. 2009. Combined inactivation of lon and ycgE decreases multidrug susceptibility by reducing the amount of OmpF porin in Escherichia coli. Antimicrob. Agents Chemother. 53:4944–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 28. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29. Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. 2006. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U. S. A. 103:15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niesen FH, Berglund H, Vedadi M. 2007. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2:2212–2221 [DOI] [PubMed] [Google Scholar]
- 31. Fraser JA, Madhumalar A, Blackburn E, Bramham J, Walkinshaw MD, Verma C, Hupp TR. 2010. A novel p53 phosphorylation site within the MDM2 ubiquitination signal: II. A model in which phosphorylation at SER269 induces a mutant conformation to p53. J. Biol. Chem. 285:37773–37786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. 2004. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332:153–159 [DOI] [PubMed] [Google Scholar]
- 33. Phillips K, de la Pena AH. 2011. The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol. 94:10.28.1–10.28.15. 10.1002/0471142727.mb1028s94 [DOI] [PubMed] [Google Scholar]
- 34. Sulavik MC, Gambino LF, Miller PF. 1995. The MarR repressor of the multiple antibiotic resistance (mar) operon in Escherichia coli: prototypic member of a family of bacterial regulatory proteins involved in sensing phenolic compounds. Mol. Med. 1:436–446 [PMC free article] [PubMed] [Google Scholar]
- 35. Brooun A, Tomashek JJ, Lewis K. 1999. Purification and ligand binding of EmrR, a regulator of a multidrug transporter. J. Bacteriol. 181:5131–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshida T, Qin L, Egger LA, Inouye M. 2006. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J. Biol. Chem. 281:17114–17123 [DOI] [PubMed] [Google Scholar]
- 37. Mizuno T, Mizushima S. 1990. Signal transduction and gene regulation through the phosphorylation of two regulatory components: the molecular basis for the osmotic regulation of the porin genes. Mol. Microbiol. 4:1077–1082 [DOI] [PubMed] [Google Scholar]
- 38. Linde HJ, Notka F, Metz M, Kochanowski B, Heisig P, Lehn N. 2000. In vivo increase in resistance to ciprofloxacin in Escherichia coli associated with deletion of the C-terminal part of MarR. Antimicrob. Agents Chemother. 44:1865–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maneewannakul K, Levy SB. 1996. Identification for mar mutants among quinolone-resistant clinical isolates of Escherichia coli. Antimicrob. Agents Chemother. 40:1695–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warner DM, Yang Q, Duval V, Chen M, Xu Y, Levy SB. 2013. Involvement of MarR and YedS in carbapenem resistance in a clinical isolate of Escherichia coli from China. Antimicrob. Agents Chemother. 57:1935–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuangthong M, Atichartpongkul S, Mongkolsuk S, Helmann JD. 2001. OhrR is a repressor of ohrA, a key organic hydroperoxide resistance determinant in Bacillus subtilis. J. Bacteriol. 183:4134–4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perera IC, Grove A. 2010. Molecular mechanisms of ligand-mediated attenuation of DNA binding by MarR family transcriptional regulators. J. Mol. Cell Biol. 2:243–254 [DOI] [PubMed] [Google Scholar]
- 43. Dolan KT, Duguid EM, He C. 2011. Crystal structures of SlyA protein, a master virulence regulator of Salmonella, in free and DNA-bound states. J. Biol. Chem. 286:22178–22185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumaraswami M, Schuman JT, Seo SM, Kaatz GW, Brennan RG. 2009. Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 37:1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Starr LM, Fruci M, Poole K. 2012. Pentachlorophenol induction of the Pseudomonas aeruginosa mexAB-oprM efflux operon: involvement of repressors NalC and MexR and the antirepressor ArmR. PLoS One 7:e32684. 10.1371/journal.pone.0032684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soonsanga S, Lee JW, Helmann JD. 2008. Conversion of Bacillus subtilis OhrR from a 1-Cys to a 2-Cys peroxide sensor. J. Bacteriol. 190:5738–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.