

Abstract
In the bacterial degradation of steroid compounds, the enzymes initiating the breakdown of the steroid rings are well known, while the reactions for degrading steroid side chains attached to C-17 are largely unknown. A recent in vitro analysis with Pseudomonas sp. strain Chol1 has shown that the degradation of the C5 acyl side chain of the C24 steroid compound cholate involves the C22 intermediate 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20S-carbaldehyde (DHOPDCA) with a terminal aldehyde group. In the present study, candidate genes with plausible functions in the formation and degradation of this aldehyde were identified. All deletion mutants were defective in growth with cholate but could transform it into dead-end metabolites. A mutant with a deletion of the shy gene, encoding a putative enoyl coenzyme A (CoA) hydratase, accumulated the C24 steroid (22E)-7α,12α-dihydroxy-3-oxochola-1,4,22-triene-24-oate (DHOCTO). Deletion of the sal gene, formerly annotated as the steroid ketothiolase gene skt, resulted in the accumulation of 7α,12α,22-trihydroxy-3-oxochola-1,4-diene-24-oate (THOCDO). In cell extracts of strain Chol1, THOCDO was converted into DHOPDCA in a coenzyme A- and ATP-dependent reaction. A sad deletion mutant accumulated DHOPDCA, and expression in Escherichia coli revealed that sad encodes an aldehyde dehydrogenase for oxidizing DHOPDCA to the corresponding acid 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20-carboxylate (DHOPDC) with NAD+ as the electron acceptor. These results clearly show that the degradation of the acyl side chain of cholate proceeds via an aldolytic cleavage of an acetyl residue; they exclude a thiolytic cleavage for this reaction step. Based on these results and on sequence alignments with predicted aldolases from other bacteria, we conclude that the enzyme encoded by sal catalyzes this aldolytic cleavage.
INTRODUCTION
Bacteria from different phylogenetic groups are able to transform steroid compounds or to use them as growth substrates. For more than 60 years, bacterial transformation of natural steroid molecules has played an essential role in the biotechnological production of steroid-based pharmaceuticals, such as cortisol derivatives or sex hormones (1–3). Bacterial steroid metabolism is also very important for the degradation of synthetic steroid pharmaceuticals, which are thought to influence the fertility of animals and humans (4).
Despite this relevance for biotechnology and environmental microbiology, knowledge about the physiology, genetics, and biochemistry of steroid-degrading bacteria is still very limited. To date, only one degradation pathway, the so-called 9,10-seco pathway, has been described in detail (5–8). The 9,10-seco pathway is characterized by the formation of androsta-Δ1,4-diene-3,17-diones (ADDs) as central intermediates of steroid degradation. ADDs are formed by the oxidation of the steroidal A ring to the Δ1,4-3-keto structure (9–12) and the degradation of the side chain attached to C-17. ADDs are further transformed by 9α-hydroxylation, leading to an opening of the B ring and aromatization of the A ring (13, 14); in actinobacteria, 9α-hydroxylation can precede the degradation of the steroid side chain (13, 15). The resulting 9,10-seco-steroids are further degraded to acidic perhydroindane derivatives consisting of the former rings C and D (7, 16, 17). While the reactions leading to the breakdown of the A and B rings are well characterized, the reactions involved in side chain degradation and in further degradation of the perhydroindane derivatives are still largely unknown.
We have studied the degradation of steroid side chains by investigating the reactions involved in the removal of the C5 carboxylic side chain of the bile salt cholate (compound I in Fig. 1) with Pseudomonas sp. strain Chol1 as a model organism. Bile salts are surface-active steroid compounds that are produced and released by vertebrates to aid the digestion of lipophilic nutrients (18–20); in addition, bile salts can also act as pheromones in some aquatic vertebrates (21, 22). The characterization of two transposon mutants of strain Chol1, which are unable to grow with cholate, showed that the degradation of the C5 acyl side chain of cholate proceeds via the stepwise removal of an acetyl and a propionyl residue. The first mutant, strain G12, has a defect in the skt gene, encoding a putative thiolase of the SCP-x-type family; this mutant transforms cholate into two compounds with modified C5 side chains as dead-end products, namely, (22E)-7α,12α-dihydroxy-3-oxochola-1,4,22-triene-24-oate (DHOCTO, the free acid of compound III), with a double bond between C-22 and C-23, and traces of 7α,12α,22-trihydroxy-3-oxochola-1,4-diene-24-oate (THOCDO, the free acid of compound IV), with a hydroxyl group at C-22 (23). The second mutant, strain R1, has a defect in a gene encoding a putative acyl coenzyme A (acyl-CoA) dehydrogenase (ACAD); this mutant transforms cholate into a compound with a C3 acyl side chain as a dead-end product, namely, 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20-carboxylate (DHOPDC; compound VI) (24).
Fig 1.
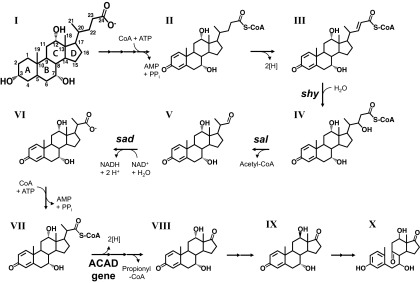
Proposed pathway for the degradation of the acyl side chain of cholate (compound I) in Pseudomonas sp. strain Chol1. Degradation proceeds via the following intermediates: compound II, Δ1,4-3-ketocholyl-CoA; compound III, (22E)7α,12α-dihydroxy-3-oxochola-1,4,22E-triene-24-oyl-CoA (DHOCTO-CoA); compound IV, 7α,12α,22-trihydroxy-3-oxochola-1,4-diene-24-oyl-CoA (THOCDO-CoA); compound V, 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20S-carbaldehyde (DHOPDCA); compound VI, 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20-carboxylate (DHOPDC); compound VII, DHOPDC-CoA; compound VIII, 7α,12α-dihydroxy-androsta-1,4-diene-9,17-dione (12α-DHADD); compound IX, 12β-DHADD; compound X, 3,7,12-trihydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione (THSATD). The following genes have been identified in strain Chol1: shy, coding for a steroid hydratase; sal, coding for a steroid aldolase (formerly annotated as skt [23]); sad, coding for a steroid aldehyde dehydrogenase; and the ACAD gene, coding for an acyl-CoA dehydrogenase (24). For legibility, only the most important reactants and cofactors are shown.
For detailed analysis of the reaction sequence of acyl side chain degradation, we used the dead-end products DHOCTO and DHOPDC as substrates for enzymes in extracts from cells of strain Chol1. With this approach, we were able to reconstitute the removal of the complete acyl side chain in vitro (25). We found that with CoA, ATP, and NAD+ as cofactors, DHOCTO is completely transformed into 7α,12α-dihydroxy-androsta-1,4-diene-3,17-dione (12α-DHADD; compound VIII) via the CoA ester of DHOPDC (compound VII). We found further that 12α-DHADD is epimerized to 12β-DHADD (compound IX) via a 12-keto intermediate. 12β-DHADD is then further degraded to the seco steroid 3,7,12-trihydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione (THSATD; compound X).
Further dissection of the reaction steps from DHOCTO to DHOPDC-CoA in vitro showed that upon NAD+ limitation, DHOCTO is completely transformed into 7α,12α-dihydroxy-3-oxopregna-1,4-diene-20S-carbaldehyde (20S-DHOPDCA; compound V), a compound with a shortened C3 side chain and a terminal aldehyde group. This conversion is dependent on CoA and ATP, indicating that the CoA ester of DHOCTO (compound III) is involved. 20S-DHOPDCA (compound V) can then be oxidized to the corresponding acid DHOPDC (compound VI), which can be activated with CoA (25). The reaction steps from DHOPDC-CoA (compound VII) to 12α-DHADD (compound VIII) are still hypothetical but most probably proceed via the ACAD-catalyzed dehydrogenation of the C3 acyl side chain, followed by its hydration at C-17 and aldolytic cleavage of propionyl-CoA from the D ring of the steroid skeleton (26, 27).
The unusual formation of an aldehyde, in this case DHOPDCA (compound V), as an intermediate of the degradation of a carboxylic acid raised the hypothesis that the reaction sequence leading from DHOCTO to DHOPDC proceeds via the cleavage of an acetyl residue by an aldolytic rather than a thiolytic reaction, which would be expected in classical β-oxidation. The goal of our present study was to investigate this hypothesis with a genetic approach based on the recently published sequence of the genomic DNA of strain Chol1 (28).
The putative first step in the degradation of DHOCTO-CoA (compound III) should be the hydration of the Δ22 double bond, leading to the formation of THOCDO-CoA (compound IV). Thus, the first goal of this study was to identify the gene for the putative hydratase catalyzing this reaction. Identification of this gene might also provide a means of generating larger amounts of THOCDO, which could be used as a substrate for further analysis of the degradation pathway in vitro. THOCDO or its CoA ester would be a plausible substrate for aldolytic cleavage leading to the aldehyde DHOPDCA and acetyl-CoA. Because THOCDO is a dead-end metabolite of the skt mutant strain G12, we had hypothesized that Skt may act as an aldolase rather than a thiolase (25).
The further degradation of the aldehyde 20S-DHOPDCA (compound V) should proceed via its oxidation to DHOPDC (compound VI). While we detected a NAD+-dependent aldehyde dehydrogenase activity in extracts of strain Chol1 cells, we do not know whether this is a specific activity or not. Thus, the second goal of this study was to identify a gene encoding an aldehyde dehydrogenase that could be responsible for the oxidation of DHOPDCA to DHOPDC. If a deletion of such a gene would affect the growth of strain Chol1 with cholate, this would be very strong evidence of an aldolytic pathway in the degradation of DHOCTO to DHOPDC.
Based on these goals, we initiated our study by searching for genes encoding enoyl-CoA hydratases and aldehyde dehydrogenases within a 79-kb gene cluster of strain Chol1 that harbors many genes with known and putative functions in steroid degradation (28).
MATERIALS AND METHODS
Cultivation of bacteria.
Pseudomonas sp. strain Chol1 and all its mutants were grown in the phosphate-buffered mineral medium MMChol as described previously (29). Strain Chol1 was grown with 2 mM cholate, and deletion mutants, which were defective in growth with cholate, were grown with 12 mM succinate in the presence of 2 mM cholate. To obtain higher quantities of the respective dead-end metabolites, mutants were transferred to a second growth passage as described previously (25). Escherichia coli strains DH5α and ST18 were grown in LB medium, in the latter case supplemented with 5-aminolevulinic acid (50 μg ml−1), at 37°C. Strains harboring plasmid pUCP18 or pEX18Ap were grown in the presence of 100 μg ml−1 carbenicillin (for Pseudomonas sp. strains) or 100 μg ml−1 ampicillin (for E. coli strains). The transposon mutant strains G12 and R1 were grown in the presence of 10 μg ml−1 kanamycin.
Cloning techniques and construction of unmarked gene deletions.
DNA was cloned, and plasmids were prepared, according to standard methods. Oligonucleotides were synthesized by Eurofins MWG Operon (Ebersberg, Germany). Genomic DNA of strain Chol1 was purified with Puregene tissue core kit B (Qiagen). For the construction of unmarked gene deletions, a splicing-by-overlap-extension (SOE) PCR-based procedure (30) was used.
For amplification of the up- and downstream flanking regions of the genes to be deleted, primer pairs A/B and C/D (sal; C211_11482), E/F and G/H (shy; C211_11487), and I/J and K/L (sad; C211_11282), described in Table 1, were used for the first PCR with genomic DNA from strain Chol1. The resulting upstream and downstream fragments of the respective genes were purified (with a QIAquick PCR purification kit) and were used as the templates for the second overlapping PCR using primer pairs A/D, E/H, and I/L, respectively.
Table 1.
Oligonucleotides used in this study
| Primer designation | Description | Sequence |
|---|---|---|
| A | Δ11482 up fw | TTTTCTAGCGGCAACGCGATGTTCTTCAGC |
| B | Δ11482 up rev | CATGGGTTTGTTCGACATGGGTGGAACATGCGCGGCGCCG |
| C | Δ11482 dn fw | CCATGTCGAACAAACCCATG |
| D | Δ11482 dn rev | TTTAAGCTTAGAGAGCACAGCACACTGTC |
| E | Δ11487 up fw | TTTTCTAGATCACAGCACGAACTATGGCG |
| F | Δ11487 up rev | AGAGAGCACAGCACACTGTCGGTCAGGCTCCCTGGAGAATCA |
| G | Δ11487 dn fw | GACAGTGTGCTGTGCTCTCT |
| H | Δ11487 dn rev | TTTAAGCTTCCTCGGTCATGTTCTCCACC |
| I | Δ11282 up fw | TTTTCTAGAGGCGATCTGTTCCAGCGATA |
| J | Δ11282 up rev | GTGGAACACCCGCTCAATCATTCGCATTCATACGGCCTCC |
| K | Δ11282 dn fw | TGATTGAGCGGGTGTTCCAC |
| L | Δ11282 dn rev | TTTAAGCTTGATGAAACCCTCGCTTTCGC |
| M | 11482 fw | TTTTCTAGAAGGCCTTATGGACGATGCTG |
| N | 11482 rev | TTTAAGCTTGCGGGTAGAAATTCCAGGCT |
| O | 11487 fw | TTTTCTAGATTGATTCTCCAGGGAGCCTGAC |
| P | 11487 rev | TTTAAGCTTCATGAGACGGTCAAAGAGAGCA |
| Q | 11282 fw | TTTTCTAGATCTGCCTCACAAATCGGGTC |
| R | 11282 rev | TTTAAGCTTAGCTGTGCGCTGTACTTTCT |
Plasmid construction, transformation, and conjugation.
The 1,980-bp (with sal [C211_11482] deleted), 1,750-bp (with shy [C211_11487] deleted), and 2,158-bp (with sad [C211_11282] deleted) PCR products were extracted from agarose gels (with a QIAquick gel extraction kit), digested with XbaI and HindIII, and finally ligated into the corresponding site of the pEX18Ap vector (31). The resulting plasmids, pEX18Ap[Δ11482], pEX18Ap[Δ11487], and pEX18Ap[Δ11282], were transformed into E. coli strain ST18 (32). Positive clones were identified by colony PCR using primer pairs A/D, E/H, and I/L, respectively. The plasmids were mobilized into strain Chol1 by biparental mating with the respective plasmid harboring E. coli strain ST18, which is auxotrophic for 5-aminolevulinic acid (32), as the donor. For this purpose, the strains were grown in LB medium at 30°C and 200 rpm. For the growth of plasmids harboring strain ST18, the medium was supplemented with 5-aminolevulinic acid and ampicillin. A total of 3 × 109 cells of the recipient and 1 × 109 cells of the donor were harvested by centrifugation at 8,000 × g for 3 min, washed with 500 μl of prewarmed LB medium, and finally resuspended in 50 μl of LB medium. Donor and recipient were carefully mixed by pipetting and were spread onto sterile membrane filters (Durapore; 0.22 μm GV; Millipore) that were placed on prewarmed LB plates. After incubation for 4 h at 30°C, the filters were transferred to a 12-ml plastic tube containing 2 ml of 0.9% NaCl. After vortexing, the cell suspensions were transferred to 2-ml plastic tubes, centrifuged at 8,000 × g for 2 min, and resuspended in 600 μl of 0.9% NaCl. The cell suspensions were serially diluted, and aliquots were spread onto LB plates containing 100 μg ml−1 carbenicillin without additional 5-aminolevulinic acid. Markerless gene deletion by recombination was finally enforced by subsequent sacB counterselection as described previously (33). For this purpose, cells were transferred to LB plates containing 7% sucrose. Deletion of the sal, shy, and sad genes was confirmed by PCR using primer pairs A/D, E/H, and I/L, respectively, with genomic DNA from the wild type as a control.
The mutant strains were complemented by amplifying the deleted sal, shy, or sad gene, including flanking regions, from genomic DNA of the wild type using primer pair M/N, O/P, or Q/R, respectively. After digestion with XbaI and HindIII, the amplicons were ligated into the corresponding site of the pUCP18 vector (34), and the construct was transformed into E. coli strain DH5α. Positive clones were selected by colony PCR using primer pairs M/N, O/P, and Q/R. Plasmids purified with the QIAprep spin miniprep kit were transformed into competent cells (35) of strain Chol1Δsal, Chol1Δshy, or Chol1Δsad. Plasmid harboring clones were selected on MMChol plates containing ampicillin (100 μg ml−1) with cholate as the only substrate.
HPLC.
All steroid compounds were analyzed with a high-performance liquid chromatography (HPLC) system equipped with a UV/visible light diode array detector as described previously (29, 36) using a reversed-phase column (150 by 3 mm; Eurosphere II 100-5, C18 H; Knauer). K-Na-phosphate buffer (10 mM; pH 7.1) (eluent A) and acetonitrile (eluent B) were used as eluents with a total flow rate of 0.4 ml min−1. For purification of steroid compounds, a semipreparative reversed-phase column was used (250 by 8 mm; Eurosphere II, 100-5, C18 H; Knauer) with a flow rate of 2 ml min−1. For the detection and purification of cholate and its degradation intermediates, a gradient method was used starting with 20% eluent B for 2 min, rising to 70% eluent B within 9 min, and returning to 20% eluent B within 1 min, followed by an equilibration of 6 min. For improved separation of DHOCTO from Δ1/4- or Δ1,4-3-ketocholate, a gradient method was used starting with 20% eluent B for 2 min, rising to 34% eluent B within 9 min, and returning to 20% eluent B within 1 min, followed by an equilibration of 6 min. For analysis of the CoA activation assays, a gradient method was used, starting with 10% eluent B for 2 min, increasing to 56% eluent B within 23 min, and returning to 10% eluent B within 1 min, followed by an equilibration of 6 min.
Purification of steroid compounds.
Steroid compounds from culture supernatants were purified either by organic extraction with dichloromethane followed by semipreparative HPLC (24) using the HPLC gradient methods described above or by solid-phase extraction (25). DHOPDC (compound VI) and DHOCTO (the free acid of compound III) were produced and purified as described previously from supernatants of the transposon mutant strains R1 and G12, respectively (25). DHOCTO, THOCDO (the free acid of compound IV), and DHOPDCA (compound V) produced by the deletion mutant strains generated in this study were purified in the same way. For the production and purification of 12β-DHADD (compound IX), strain Chol1 was grown anaerobically with nitrate as the electron acceptor as described previously (29).
The purity of steroid compounds was assessed by HPLC analysis and mass spectrometry (MS), and concentrations of purified degradation intermediates were estimated by UV spectroscopy as described previously (25). For growth experiments, DHOPDCA, DHOPDC, and 12β-DHADD were supplied at final concentrations of approximately 0.5 mM.
Mass spectrometry.
Mass spectra of purified compounds were obtained on an LXQ tandem MS (MS-MS) instrument (Thermo Scientific) using an electrospray in the negative or positive mode. The scan range was 50 to 1,300 Da. In the negative or positive mode of ionization, the needle voltage was set to 5,430 or 4,000 V, the capillary voltage to −7 or 2 V, the tube lens to −5.1 or 109.9 V, and the capillary temperature to 300 or 250°C, respectively.
Enzyme assays.
Cell extracts of strain Chol1, the knockout mutant strains, and a plasmid harboring E. coli strain DH5α (pUCP18::C211_11282 or pUCP18) were prepared as described previously (25). For the preparation of E. coli cell extracts, overnight cultures grown in LB medium containing 100 μg ml−1 ampicillin were used. All enzyme assays were performed at 30°C. Enzyme assays for carboxylate-CoA ligase activities contained 50 mM morpholinepropanesulfonic acid (MOPS) buffer (pH 7.8), 2 mM CoA, 2.5 mM ATP, 2.5 mM MgCl2, the cell extract (1 to 1.5 mg protein ml−1), and 200 to 300 μM THOCDO (the free acid of compound IV). Assays for aldehyde dehydrogenase activity contained 50 mM MOPS buffer (pH 7.8), the cell extract (1 to 1.5 mg protein ml−1), and 200 to 300 μM DHOPDCA (compound V). NAD+ or NADP+ was added to a final concentration of 1 mM. Samples were withdrawn immediately after the reactions were started and at defined time intervals thereafter and were subsequently analyzed by HPLC.
RESULTS
Identification of a candidate gene for the hydration of DHOCTO-CoA.
Within the 79-kb steroid degradation gene cluster, we found a gene (C211_11487) encoding a putative enoyl-CoA hydratase that forms a putative operon with the skt gene (C211_11482), mentioned above. Sequence analysis of the predicted protein encoded by C211_11487 reveals a C-terminal hot-dog-fold domain that is also present in a variety of enzymes, including R-specific enoyl-CoA hydratases as well as enoyl-CoA hydratases of the MaoC-like protein family (37, 38). Additionally, the protein harbors an N-terminal DUF35/DUF35N domain that contains zinc ribbon structures as well as oligonucleotide/oligosaccharide-binding (OB) folds and has been proposed to bind to acyl-CoA moieties (39). Orthologs of this putative operon were found in the genomes of two cholate-degrading members of the Proteobacteria as well, namely, Comamonas testosteroni strain KF-1 and Pseudoalteromonas haloplanktis strain TAC125 (Fig. 2). The proteins encoded by the orthologous genes in strains KF-1 and TAC125 show 53% and 58% identities, respectively, to the gene product of C211_11487 (Fig. 3) and are predicted to act as acyl dehydratases. Based on the predicted function of C211_11487 and its vicinity to skt, we judged that this gene was a plausible target and constructed a deletion mutant.
Fig 2.
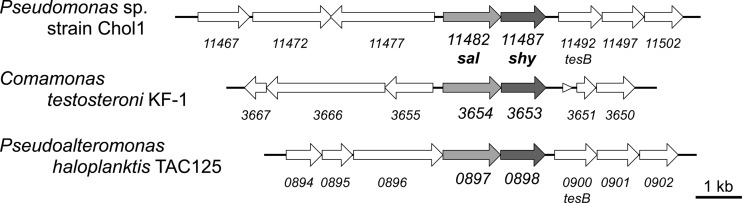
Part of the 79-kb gene cluster coding for steroid-degrading enzymes in Pseudomonas sp. strain Chol1 (GenBank accession no. NZ_AMSL00000000.1), including a putative operon containing the sal (C211_11482; formerly designated skt) and shy (C211_11487) genes. Shown is a comparison of the genetic organization of this cluster to those of analogous clusters in the cholate-degrading bacteria Pseudoalteromonas haloplanktis strain TAC125 (GenBank accession no. CR954246.1) and Comamonas testosteroni strain KF-1 (GenBank accession no. AAUJ02000001.1). Light shading, genes orthologous to sal; dark shading, genes orthologous to shy.
Fig 3.

Alignment of the amino acid sequence of the putative steroid hydratase Shy (C211_11487) in Pseudomonas sp. strain Chol1 with the predicted acyl dehydratases encoded by PD3653 (53% identity) in Comamonas testosteroni strain KF-1 and by PSHAa0898 (58% identity) in Pseudoalteromonas haloplanktis strain TAC125. Alignment was performed using Clustal W2 software. Identical residues are indicated by asterisks, conserved residues by periods, and semiconserved residues by colons.
Characterization of the deletion mutant strain Chol1Δshy.
A mutant strain with the C211_11487 gene deleted did not grow with cholate as the sole carbon source. The mutant grew with succinate in the presence of cholate (Fig. 4A), and HPLC analysis of supernatants of these cultures showed that this strain accumulated DHOCTO (the free acid of compound III) along with Δ1,4-3-ketocholate (the free acid of compound II), tailing off from the DHOCTO peak, as well as Δ1/4-3-ketocholate. The identities of DHOCTO, Δ1,4-3-ketocholate, and Δ1/4-3-ketocholate were confirmed by coelution with a culture supernatant of the skt transposon mutant strain G12 (23) containing a mixture of all three compounds and by their respective UV spectra. When these cultures were transferred to a second growth passage, DHOCTO remained as the sole end product (Fig. 4B), a finding confirmed by MS analysis of the purified compound revealing an ion ([M-H]−) with m/z = 399.11 (C24H32O5) (23). The deletion mutant strain grew with purified DHOPDC (compound VI) and 12β-DHADD (compound IX) as the only carbon sources, and HPLC analysis showed that both compounds were completely degraded (data not shown). Growth with cholate was restored when an intact copy of C211_11487 was provided in trans on the pUCP18 vector (Fig. 4A), and HPLC analysis revealed intermediary formation of DHOPDC and 12β-DHADD in supernatants of the complemented strain (data not shown). These results clearly show that the C211_11487 gene encodes an enzyme that is essential for the degradation of DHOCTO. The fact that the corresponding mutant strain does not produce THOCDO (the free acid of compound IV) and the predicted function of the product constitute strong evidence that this gene encodes an enoyl-CoA hydratase catalyzing the conversion of DHOCTO into THOCDO by hydration of the Δ22 double bond. Therefore, we named this gene shy, for steroid hydratase.
Fig 4.

(A) Growth of the deletion mutant Pseudomonas sp. strain Chol1Δshy with 12 mM succinate in the presence of 2 mM cholate (filled diamonds) or with 2 mM cholate alone (open squares) and growth of the complemented strain Chol1Δshy[pUCP18::C211_11487] with 2 mM cholate (open triangles). Error bars indicate standard deviations (n = 3). (B) HPLC chromatogram of a culture supernatant of Pseudomonas sp. strain Chol1Δshy grown with succinate (12 mM) in the presence of cholate (2 mM) after one growth passage (I) and after a second growth passage (II) where an aliquot of the first culture was diluted 1:10 into fresh medium with 12 mM succinate. The analysis wavelength was 245 nm.
Expression of shy.
THOCDO (the free acid of compound IV) would be a useful substrate for further elucidation of the reaction sequence of side chain degradation by using in vitro assays, but it is formed only in trace amounts by the skt mutant strain G12 (23). Therefore, we tested whether overexpression of shy could be used for producing THOCDO in larger amounts. Additional expression of shy on the pUCP18 vector in the wild-type strain Chol1 had no influence on the formation of THOCDO during growth with cholate. HPLC analysis of supernatants from cultures of strain Chol1[pUCP18::shy] showed that the pattern of formation and the respective amounts of cholate degradation intermediates did not differ from those for strain Chol1 (data not shown).
In the next step, we tested whether overexpression of shy would lead to a higher level of THOCDO production in the skt mutant, which is able to produce THOCDO but is unable to degrade it. For this purpose, we constructed a chromosomal skt deletion mutant. This mutant has a phenotype similar to that of the transposon mutant strain G12 but does not require previous growth with DHADD for the induction of cholate transformation (23). As expected, strain Chol1Δskt was not able to grow with cholate alone but transformed it when grown with succinate in the presence of cholate. In supernatants of these cultures, a mixture of Δ1,4-3-ketocholate and Δ1/4-3-ketocholate, as well as DHOCTO and THOCDO, accumulated. THOCDO was identified by mass spectrometry of the purified compound, revealing an ion ([M-H]−) with m/z = 317.11 (C24H34O6) (23). After a second growth passage, the amounts of DHOCTO and THOCDO increased while the amounts of Δ1,4-3-ketocholate and Δ1/4-3-ketocholate in the culture supernatants decreased (data not shown).
When strain CHol1Δskt[pUCP18::shy] was grown with succinate in the presence of cholate, the amounts of DHOCTO and THOCDO formed after the first and second growth passages were approximately twice those formed without the plasmid. Thus, culture supernatants of strain Chol1Δskt[pUCP18::shy] were used to purify THOCDO (Fig. 5).
Fig 5.
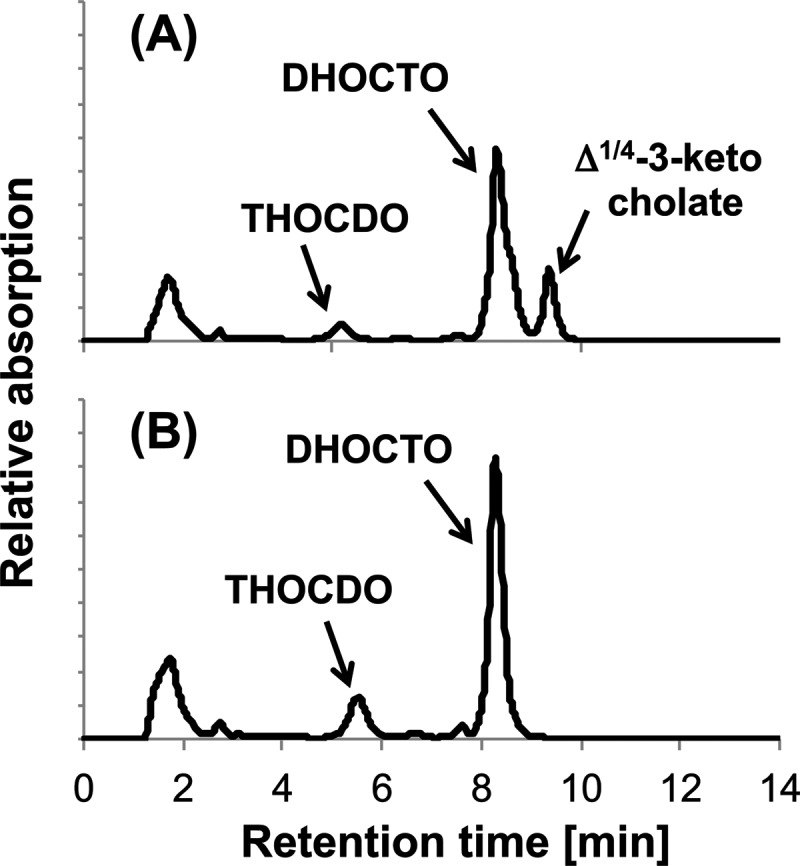
HPLC chromatograms of culture supernatants of Pseudomonas sp. strain Chol1Δsal with no additional plasmid (A) and with plasmid pUCP18::shy (B) grown with succinate (12 mM) in the presence of cholate (2 mM) after the second growth passage. The analysis wavelength was 245 nm.
Transformation of THOCDO in vitro.
To investigate the fate of THOCDO (the free acid of compound IV) in vitro, we used desalted cell extracts of the wild-type strain Chol1 for CoA activation assays with THOCDO as the substrate. HPLC analysis of the results of these assays showed CoA-, ATP-, and cell extract-dependent consumption of THOCDO and the concomitant formation of DHOPDCA (compound V). In the absence of CoA or ATP, DHOPDCA was not formed (Fig. 6). Although the CoA ester of THOCDO was never observed in our assays, these results strongly suggest that THOCDO-CoA (compound IV) is the precursor for the formation of DHOPDCA.
Fig 6.

Transformation of THOCDO (diamonds) into DHOPDCA (circles) in CoA activation assays containing desalted cell extracts of Pseudomonas sp. strain Chol1. Controls without CoA (solid gray line) or without ATP (dashed gray line) showed no transformation of THOCDO and no formation of DHOPDCA.
When desalted or nondesalted cell extracts of the deletion mutant strain Chol1Δshy or Chol1Δskt were used in THOCDO activation assays, the concentration of THOCDO did not decrease under any condition applied.
Identification of a candidate gene for the oxidation of DHOPDCA to DHOPDC.
The oxidation of DHOPDCA (compound V) to DHOPDC (compound VI) should be catalyzed by an aldehyde dehydrogenase. Within the 79-kb steroid degradation gene cluster we identified a gene (C211_11282) coding for a putative aldehyde dehydrogenase. BLAST analysis of the protein product of C211_11282 revealed high similarity to predicted aldehyde dehydrogenases from known cholate-degrading bacteria, including Comamonas testosteroni strain KF-1 (58% similarity) and Pseudoalteromonas haloplanktis strain TAC125 (53% similarity) (Fig. 7). Sequence analysis revealed the presence of conserved residues of a putative NAD(P)+ binding site within the protein product of C211_11282. Based on this information, we judged C211_11282 to be a plausible target and constructed a deletion mutant.
Fig 7.

Alignment of the amino acid sequence of the gene product of sad (C211_11282) in Pseudomonas sp. strain Chol1 with those of the predicted aldehyde dehydrogenases encoded by PD3690 (58% identity) in Comamonas testosteroni strain KF-1 and PSHAa2139 (53% identity) in Pseudoalteromonas haloplanktis strain TAC125. Alignment was performed using Clustal W2 software. Identical residues are indicated by asterisks, conserved residues by periods, and semiconserved residues by colons.
Characterization of the deletion mutant strain Chol1Δsad.
A mutant strain with the C211_11282 gene deleted showed a growth phenotype altered from that of the wild-type strain Chol1 when cholate was the only substrate used. The cultures showed extremely slow growth (Fig. 8A), reaching a maximal optical density at 600 nm (OD600) of approximately 0.3 after a prolonged incubation of about 48 h (data not shown). The mutant strain grew with succinate in the presence of cholate; after succinate depletion, the OD600 dropped quickly, and the culture fluid became more viscous, indicating cell lysis (Fig. 8A). HPLC analysis of supernatants from cultures grown with succinate in the presence of cholate revealed the accumulation of 20S-DHOPDCA (compound V) as the major end product of cholate transformation (Fig. 8B). The identity of 20S-DHOPDCA was confirmed by MS analysis of the purified compound, revealing an ion ([M+H]+) with m/z = 359.0 (C22H30O4) (25). A minor product eluting just after 20S-DHOPDCA was identified as the 20R diastereoisomer on the basis of its UV spectrum and chromatographic properties, and its formation could be due to a chemical transformation reaction as described previously (25). Small amounts of DHOPDC (compound VI) were also detected in those culture supernatants. When the mutant strain was incubated with cholate as the only substrate, HPLC analysis revealed the formation of 20S-DHOPDCA and 20R-DHOPDCA after 48 h. The deletion mutant grew with purified DHOPDC (compound VI) and 12β-DHADD (compound IX) as the only carbon sources, and HPLC analysis showed that both compounds were completely degraded (data not shown). Growth of the deletion mutant strain with cholate was restored when an intact copy of C211_11282 was provided in trans on the pUCP18 vector (Fig. 8A), and HPLC analysis revealed the intermediary formation of DHOPDC and 12β-DHADD in supernatants of the complemented strain (data not shown).
Fig 8.

(A) Growth of the deletion mutant Pseudomonas sp. strain Chol1Δsad with 12 mM succinate and 2 mM cholate (filled diamonds) or with 2 mM cholate alone (open squares) and growth of the complemented strain Chol1Δsad[pUCP18::C211_11282] with 2 mM cholate (open triangles). Error bars indicate standard deviations (n = 3). (B) HPLC chromatogram of a culture supernatant of Pseudomonas sp. strain Chol1Δsad grown with succinate (12 mM) in the presence of cholate (2 mM) after incubation for 23 h. The analysis wavelength was 245 nm.
To test whether 20S-DHOPDCA and 20R-DHOPDCA are intermediates of cholate degradation in vivo, a mixture of these two compounds was purified and was used as the substrate for growth experiments with strain Chol1 and the mutant strains Chol1Δshy and Chol1Δsal. All strains were able to utilize 20S-DHOPDCA as a growth substrate, and HPLC analysis revealed that it was depleted from the culture supernatants (data not shown). The amount of 20R-DHOPDCA also decreased in all cultures, but it was never depleted completely (data not shown). During growth, the downstream intermediates DHOPDC and 12β-DHADD accumulated transiently in the culture supernatants.
In summary, these results clearly indicate that 20S-DHOPDCA (compound V) is a true intermediate in the metabolic pathway for cholate degradation and that the C211_11282 gene codes for an enzyme that is responsible for the degradation of this aldehyde. Therefore, this gene was named sad, for steroid aldehyde dehydrogenase.
Heterologous expression of sad and activity in vitro.
To further confirm the activity of Sad in vitro, we expressed the gene in E. coli strain DH5α on plasmid pUCP18::sad and performed enzymatic tests with 20S-DHOPDCA (compound V) as the substrate. HPLC analysis of the results of these assays showed that 20S-DHOPDCA was readily oxidized to DHOPDC (compound VI) by desalted cell extracts with NAD+ as the electron acceptor (Fig. 9). When NADP+ was added as the electron acceptor, 20S-DHOPDCA was still oxidized to DHOPDC, but the reaction rate was significantly lower than that with NAD+ (data not shown). In control experiments with cell extracts of the E. coli vector control, 20S-DHOPDCA was not degraded.
Fig 9.
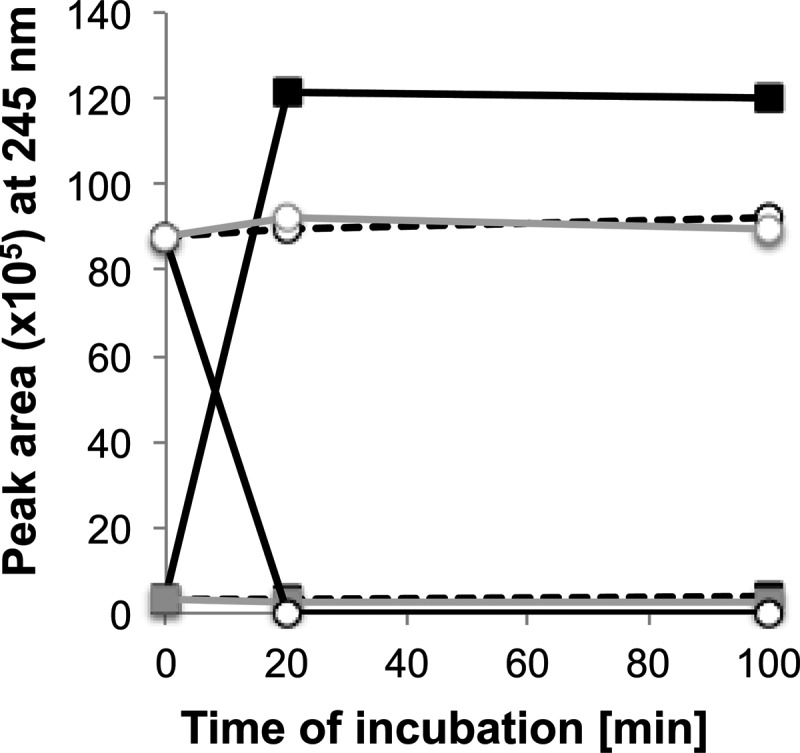
Activity of the steroid aldehyde dehydrogenase Sad from Pseudomonas sp. strain Chol1 expressed in E. coli strain DH5α[pUCP18::sad]. Shown is the oxidation of 20S-DHOPDCA (circles) to DHOPDC (squares) in the presence of NAD+ (solid black lines) in desalted cell extracts of strain DH5α[pUCP18::sad]. Without the addition of NAD+ (dashed black lines) or with desalted cell extracts of strain DH5α[pUCP18] (vector control) (solid gray lines, parallel to dashed lines), 20S-DHOPDCA remained stable and no DHOPDC was formed.
DISCUSSION
The deacetylation of the acyl side chains of steroid compounds generating C22 steroids from C24 steroids has been thought to proceed via a thiolytic reaction step, as in classical β-oxidation of fatty acids (5, 6, 8, 40). In contrast, our recent finding that the degradation of the C5 acyl side chain of the C24 steroid compound cholate involves the formation of the C22 aldehyde intermediate 20S-DHOPDCA (compound V in Fig. 1) in Pseudomonas sp. strain Chol1 suggested that this deacetylation proceeds via an aldolytic reaction step (25). In the present study, we identified two genes in strain Chol1, shy and sad, that are required for the formation of 20S-DHOPDCA from the C24 compound DHOCTO (the free acid of compound III) and for its further degradation to the C22 compound DHOPDC (compound VI), respectively. To our knowledge, these are the first genes shown to encode an acyl-CoA hydratase and an aldehyde dehydrogenase with essential functions in the bacterial degradation of a steroid side chain. The characterization of the respective mutants provided compelling evidence that the removal of an acetyl residue from the acyl side chain of cholate proceeds via an aldolytic and not a β-ketothiolytic splitting of a carbon-carbon bond.
First, the sad deletion mutant showed strongly impaired growth with cholate and accumulated the aldehyde 20S-DHOPDCA (compound V) in the culture supernatant. Heterologous expression of Sad showed unambiguously that this enzyme functions as an aldehyde dehydrogenase catalyzing the NAD+-dependent oxidation of 20S-DHOPDCA to DHOPDC (compound VI). If the degradation of DHOCTO to DHOPDC proceeded via a β-ketothiolytic reaction, the inactivation of the sad gene should not affect cholate degradation. Thus, the phenotype of the sad mutant and the activity of the encoded enzyme clearly show that the cleavage of an acetyl residue from the acyl side chain of DHOCTO proceeds via aldolytic cleavage. The fact that the sad deletion mutant showed very slow growth with cholate and formed traces of DHOPDC indicates that at least one further aldehyde dehydrogenase located in the genome of strain Chol1 could catalyze the oxidation of 20S-DHOPDCA to DHOPDC. However, the fact that 20S-DHOPDCA accumulated strongly during the growth of the sad deletion mutant with succinate in the presence of cholate clearly shows that the alternative aldehyde dehydrogenases cannot compensate for the loss of Sad, indicating that the alternative aldehyde dehydrogenases are not specific for 20S-DHOPDCA. In addition, the fact that 20R-DHOPDCA is not completely degraded in cultures of strain Chol1 growing with a mixture of both isomers shows that none of the available aldehyde dehydrogenases can oxidize this diastereomer. The rapid cell lysis in outgrown cultures of strain Chol1Δsad accumulating DHOPDCA indicated that the aldehyde is toxic to the cells. No such strong cell lysis is observed with the ACAD mutant strain R1, which accumulates DHOPDC (24). Thus, Sad also has a detoxifying function for strain Chol1 during growth with cholate.
Second, the shy deletion mutant could not grow with cholate and accumulated DHOCTO as a dead-end metabolite, indicating that Shy acts as a hydratase catalyzing the hydration of the Δ22 double bond of DHOCTO-CoA (compound III), with THOCDO-CoA (compound IV) as a reaction product. In our in vitro assays, we had no indication that THOCDO (the free acid of compound IV) was converted into a C22 keto-acyl-CoA steroid compound, which would be the required intermediate for the splitting of the carbon-carbon bond via thiolytic cleavage. This result is in agreement with the fact that no genes encoding putative β-hydroxyacyl-CoA dehydrogenases were identified in the 79-kb steroid degradation gene cluster (28). Instead, THOCDO was readily converted into 20S-DHOPDCA in vitro in a CoA- and ATP-dependent reaction, suggesting that the CoA ester of THOCDO (compound IV) is the direct precursor of 20S-DHOPDCA (compound V). Such a conversion of a β-hydroxyacyl compound to an aldehyde is in full agreement with aldolytic cleavage of a carbon-carbon bond and, thus, strongly supports our hypothesis.
In accordance with our previous results with the transposon mutant strain G12 (23, 25), the newly constructed skt deletion mutant strain also accumulated THOCDO as an end product of cholate transformation. This phenotype, combined with the new findings of the present study, strongly reinforces our previous hypothesis that this gene codes for an enzyme that catalyzes this postulated aldolytic cleavage. Therefore, we rename the C211_11482 gene sal, for steroid aldolase.
This assignment is strongly supported by alignments with other proteins with similarities to the SCP-x-type thiolase family. Genes for three further members of this family, namely, ltp3 and ltp4 from Rhodococcus rhodochrous (41) and ipfD from Sphingomonas sp. strain Ibu-2 (42), were recently revealed to be essential for the acyl side chain degradation of C-24 branched phytosterols and the phenylacetic acid ibuprofen, respectively. The respective substrates of Ltp3, Ltp4, and IpfD contain tertiary β-hydroxyl groups, which mechanistically preclude oxidation to the corresponding β-keto groups and subsequent thiolytic cleavage. This led to the conclusion that these enzymes catalyze aldolytic cleavage reactions (41). Just like Sal, all three proteins lack the catalytically active N-terminal cysteine, which is highly conserved in catabolic thiolases (43) such as BbsB, which catalyzes a thiolytic cleavage reaction during anaerobic toluene degradation in the denitrifying bacterium Thauera aromatica strain K172 (44), or FadA in E. coli strain K-12 (Fig. 10). Although THOCDO-CoA (compound IV), the putative substrate of Sal, contains a secondary β-hydroxyl group, our biochemical results and the sequence similarity to Ltp3, Ltp4, and IpfD strongly support the notion that Sal belongs to this proposed new group of SCP-x-type aldolases.
Fig 10.

Alignment of a section of the amino acid sequence of Sal in Pseudomonas sp. strain Chol1 with those of the predicted aldolases Ltp3 from Rhodococcus rhodochrous DSM 43269 and IpfD from Sphingomonas sp. strain Ibu-2, the thiolase BbsB from Thauera aromatica strain K172, and the β-keto-thiolase FadA from E. coli K-12. Alignment was performed using Clustal W2 software. Identical residues are indicated by asterisks, conserved residues by periods, and semiconserved residues by colons. The catalytic N-terminal cysteine residue (highlighted) is conserved in thiolases, such as BbsB and FadA, but is not present in SalA, Ltp3, and IpfD.
Surprisingly, the sal deletion mutant accumulated mainly DHOCTO and only small amounts of THOCDO, even when Shy was expressed on a plasmid. In addition, THOCDO was not converted at all in cell extracts of the shy deletion mutant, despite the presence of a functional sal gene. A possible mechanistic explanation for these effects could be that Shy and Sal may require mutual protein-protein interactions for their full activity. This interaction may occur via the N-terminal DUF35/DUF35N domain of Shy, which is also present in the predicted proteins encoded by ipfE and bbsA in Sphingomonas sp. strain Ibu-2 and T. aromatica strain K172, respectively (42, 44). Interestingly, in both cases, these proteins are thought to be required for the activity of the SCP-x-type enzyme (IbfD or BbsB).
Regarding the high similarity of Shy, Sal, and Sad to predicted proteins and regarding the similar genetic organization of shy and sal in Comamonas testosteroni strain KF-1 and Pseudoalteromonas haloplanktis strain TAC125, it is likely that the same reaction steps are used for the degradation of the acyl side chains in these cholate-degrading bacteria as well. To analyze the conversion of DHOCTO into DHOPDC in biochemical detail, we are working on the isolation of the enzymes involved from strain Chol1 for the reconstitution of these reaction steps in a completely defined system.
ACKNOWLEDGMENTS
We thank Stefanie Krahn for experimental support. Mass spectrometric measurements by Marc Schürmann (Münster, Germany) are gratefully acknowledged.
This work was funded by a grant from the Deutsche Forschungsgemeinschaft to B.P. (DFG, PH71/3-1).
Footnotes
Published ahead of print 24 May 2013
REFERENCES
- 1. Hogg JA. 1992. Steroids, the steroid community, and Upjohn in perspective: a profile of innovation. Steroids 57:593–616 [DOI] [PubMed] [Google Scholar]
- 2. Fernandes P, Cruz A, Angelova B, Pinheiro H, Cabral J. 2003. Microbial conversion of steroid compounds: recent developments. Enzyme Microb. Technol. 32:688–705 [Google Scholar]
- 3. Donova MV, Egorova OV. 2012. Microbial steroid transformations: current state and prospects. Appl. Microbiol. Biotechnol. 94:1423–1447 [DOI] [PubMed] [Google Scholar]
- 4. Silva CP, Otero M, Esteves V. 2012. Processes for the elimination of estrogenic steroid hormones from water: a review. Environ. Pollut. 165:38–58 [DOI] [PubMed] [Google Scholar]
- 5. Hayakawa S. 1982. Microbial transformation of bile acids. A unified scheme for bile acid degradation, and hydroxylation of bile acids. Z. Allg. Mikrobiol. 22:309–326 [DOI] [PubMed] [Google Scholar]
- 6. Philipp B. 2011. Bacterial degradation of bile salts. Appl. Microbiol. Biotechnol. 89:903–915 [DOI] [PubMed] [Google Scholar]
- 7. Yam KC, Okamoto S, Roberts JN, Eltis LD. 2011. Adventures in Rhodococcus—from steroids to explosives. Can. J. Microbiol. 57:155–168 [DOI] [PubMed] [Google Scholar]
- 8. Horinouchi M, Hayashi T, Kudo T. 2012. Steroid degradation in Comamonas testosteroni. J. Steroid Biochem. Mol. Biol. 129:4–14 [DOI] [PubMed] [Google Scholar]
- 9. Grimm C, Maser E, Möbus E, Klebe G, Reuter K, Ficner R. 2000. The crystal structure of 3α-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J. Biol. Chem. 275:41333–41339 [DOI] [PubMed] [Google Scholar]
- 10. Kataoka S, Nakamura S, Ohkubo T, Ueda S, Uchiyama S, Kobayashi Y, Oda M. 2006. Crystallization and preliminary X-ray analysis of the complex of NADH and 3α-hydroxysteroid dehydrogenase from Pseudomonas sp. B-0831. Acta Crystallogr. Sect. F Biol. Cryst. Commun. 62:569–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Oosterwijk N, Knol J, Dijkhuizen L, van der Geize R, Dijkstra BW. 2012. Structure and catalytic mechanism of 3-ketosteroid-Δ4-(5α)-dehydrogenase from Rhodococcus jostii RHA1 genome. J. Biol. Chem. 287:30975–30983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L. 2001. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Δ1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counterselectable marker. FEMS Microbiol. Lett. 205:197–202 [DOI] [PubMed] [Google Scholar]
- 13. Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. 2011. Activity of 3-ketosteroid 9α-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J. Biol. Chem. 286:40717–40724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petrusma M, Hessels G, Dijkhuizen L, van der Geize R. 2011. Multiplicity of 3-ketosteroid-9α-hydroxylase enzymes in Rhodococcus rhodochrous DSM43269 for specific degradation of different classes of steroids. J. Bacteriol. 193:3931–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swain K, Casabon I, Eltis LD, Mohn WW. 2012. Two transporters essential for the reassimilation of novel cholate metabolites by Rhodococcus jostii RHA1. J. Bacteriol. 194:6720–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lack NA, Yam KC, Lowe ED, Horsman GP, Owen RL, Sim E, Eltis LD. 2010. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J. Biol. Chem. 285:434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Casabon I, Crowe AM, Liu J, Eltis LD. 2013. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol. Microbiol. 87:269–283 [DOI] [PubMed] [Google Scholar]
- 18. Hofmann AF, Mysels KJ. 1987. Bile salts as biological surfactants. Colloids Surf. 30:145–173 [Google Scholar]
- 19. Ridlon J, Kang D, Hylemon P. 2006. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 47:241–259 [DOI] [PubMed] [Google Scholar]
- 20. Hagey LR, Møller PR, Hofmann AF, Krasowski MD. 2010. Diversity of bile salts in fish and amphibians: evolution of a complex biochemical pathway. Physiol. Biochem. Zool. 83:308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meredith TL, Caprio J, Kajiura SM. 2012. Sensitivity and specificity of the olfactory epithelia of two elasmobranch species to bile salts. J. Exp. Biol. 215:2660–2667 [DOI] [PubMed] [Google Scholar]
- 22. Fine JM, Sorensen PW. 2010. Production and fate of the sea lamprey migratory pheromone. Fish Physiol. Biochem. 36:1013–1020 [DOI] [PubMed] [Google Scholar]
- 23. Birkenmaier A, Möller HM, Philipp B. 2011. Identification of a thiolase gene essential for β-oxidation of the acyl side chain of the steroid compound cholate in Pseudomonas sp. strain Chol1. FEMS Microbiol. Lett. 318:123–130 [DOI] [PubMed] [Google Scholar]
- 24. Birkenmaier A, Holert J, Erdbrink H, Möller HM, Friemel A, Schoenenberger R, Suter MJ, Klebensberger J, Philipp B. 2007. Biochemical and genetic investigation of initial reactions in aerobic degradation of the bile acid cholate in Pseudomonas sp. strain Chol1. J. Bacteriol. 189:7165–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holert J, Kulić Ž, Yücel O, Suvekbala V, Suter MJ, Möller HM, Philipp B. 2013. Degradation of the acyl side chain of the steroid compound cholate in Pseudomonas sp. strain Chol1 proceeds via an aldehyde intermediate. J. Bacteriol. 195:585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kieslich K. 1985. Microbial side-chain degradation of sterols. J. Basic Microbiol. 25:461–474 [DOI] [PubMed] [Google Scholar]
- 27. Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. 2011. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J. Biol. Chem. 286:43668–43678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holert J, Alam I, Larsen M, Antunes A, Bajic VB, Stingl U, Philipp B. 2013. Genome sequence of Pseudomonas sp. strain Chol1, a model organism for the degradation of bile salts and other steroid compounds. Genome Announc. 1(1):e00014–12. 10.1128/genomeA.00014-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Philipp B, Erdbrink H, Suter MJ, Schink B. 2006. Degradation of and sensitivity to cholate in Pseudomonas sp. strain Chol1. Arch. Microbiol. 185:192–201 [DOI] [PubMed] [Google Scholar]
- 30. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 31. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 32. Thoma S, Schobert M. 2009. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 294:127–132 [DOI] [PubMed] [Google Scholar]
- 33. Choi K-H, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30. 10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schweizer HP. 1991. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97:109–121 [DOI] [PubMed] [Google Scholar]
- 35. Chuanchuen R, Narasaki CT, Schweizer HP. 2002. Benchtop and microcentrifuge preparation of Pseudomonas aeruginosa competent cells. Biotechniques 33:760–763 [DOI] [PubMed] [Google Scholar]
- 36. Jagmann N, Brachvogel H, Philipp B. 2010. Parasitic growth of Pseudomonas aeruginosa in co-culture with the chitinolytic bacterium Aeromonas hydrophila. Environ. Microbiol. 12:1787–1802 [DOI] [PubMed] [Google Scholar]
- 37. Pidugu LS, Maity K, Ramaswamy K, Surolia N, Suguna K. 2009. Analysis of proteins with the “hot dog” fold: prediction of function and identification of catalytic residues of hypothetical proteins. BMC Struct. Biol. 9:37. 10.1186/1472-6807-9-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dillon SC, Bateman A. 2004. The Hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinformatics 5:109. 10.1186/1471-2105-5-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krishna SS, Aravind L, Bakolitsa C, Caruthers J, Carlton D, Miller MD, Abdubek P, Astakhova T, Axelrod HL, Chiu HJ, Clayton T, Deller MC, Duan L, Feuerhelm J, Grant JC, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, McMullan D, Morse AT, Nigoghossian E, Okach L, Reyes R, Rife CL, van den Bedem H, Weekes D, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2010. The structure of SSO2064, the first representative of Pfam family PF01796, reveals a novel two-domain zinc-ribbon OB-fold architecture with a potential acyl-CoA-binding role. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66:1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szentirmai A. 1990. Microbial physiology of sidechain degradation of sterols. J. Ind. Microbiol. Biotechnol. 6:101–115 [Google Scholar]
- 41. Wilbrink MH, van der Geize R, Dijkhuizen L. 2012. Molecular characterization of ltp3 and ltp4, essential for C24-branched chain sterol-side-chain degradation in Rhodococcus rhodochrous DSM 43269. Microbiology 158:3054–3062 [DOI] [PubMed] [Google Scholar]
- 42. Murdoch RW, Hay AG. 2013. Genetic and chemical characterization of ibuprofen degradation by Sphingomonas Ibu-2. Microbiology 159:621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haapalainen AM, Meriläinen G, Wierenga RK. 2006. The thiolase superfamily: condensing enzymes with diverse reaction specificities. Trends Biochem. Sci. 31:64–71 [DOI] [PubMed] [Google Scholar]
- 44. Leuthner B, Heider J. 2000. Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of beta oxidation of the intermediate benzylsuccinate. J. Bacteriol. 182:272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]