

Abstract
β-arrestin-2 (β-arr2) is a scaffolding protein of the arrestin family with a wide variety of cellular functions. Recent studies have demonstrated differential roles for β-arr2 in inflammation following endotoxemia and cecal ligation and puncture (CLP) models of sepsis. Because CLP-induced inflammation involves response to fecal contents and necrotic cecum in addition to microbial challenge, in this study, we examined the role of β-arr2 in an exclusively polymicrobial infection (PMI) model. In addition, we examined the role of gene dosage of β-arr2 in polymicrobial sepsis. Our studies demonstrate that β-arr2 is a negative regulator of systemic inflammation in response to polymicrobial infection and that one allele is sufficient for this process. Our results further reveal that loss of β-arr2 leads to increased neutrophil sequestration and overt inflammation specifically in the lungs following polymicrobial infection. Consistent with this, specific NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways were differentially activated in the β-arr2 knockout (KO) mice lungs compared to the wild type (WT) following PMI. Associated with enhanced inflammation in the KO mice, PMI-induced mortality was also significantly higher in KO mice than in WT mice. To understand the differential role of β-arr2 in different sepsis models, we used cell culture systems to evaluate inflammatory cytokine production following endotoxin and polymicrobial stimulation. Our results demonstrate cell-type- as well as stimulus-specific roles for β-arr2 in inflammation. Taken together, our results reveal a negative regulatory role for β-arr2 in polymicrobial infection-induced inflammation and further demonstrate that one allele of β-arr2 is sufficient to mediate most of these effects.
INTRODUCTION
Arrestins are members of a family of scaffolding proteins that include α- and β-arrestins. β-Arrestins (1 and 2) were originally discovered for their role in G protein-coupled receptor (GPCR) desensitization (1). However, recent studies have demonstrated that in addition to receptor desensitization, β-arrestins are also involved in receptor endocytosis and downstream signaling (2). In fact, the latter even has G protein-independent and arrestin-dependent components (3). Furthermore, β-arrestins can regulate signaling downstream of non-GPCRs by virtue of acting as scaffolds for major signaling molecules (4–6). This places arrestins as critical regulators of various cellular and physiological processes important in maintenance of homeostasis. It is thus not surprising that β-arrestins have been implicated in the pathogenesis of many different diseases, including arthritis (7), colorectal cancer (8), myeloid leukemia (9), multiple sclerosis (10), sepsis (11, 12), and colitis (13). In addition to mammals, β-arrestins have been shown to control unique physiological processes in other species, including Caenorhabditis elegans (14), drosophila (15), and zebra fish (16). Furthermore, β-arrestins are critical for embryonic development in mammals, as evidenced by the embryonically lethal phenotype of β-arrestin-1/2 double-knockout (KO) mice (17).
The role of β-arrestins in regulating inflammation stems from their “traditional” role in modulating GPCRs, such as C5aR (18), C3aR (19), PAR, and chemokine receptors (20–23). Furthermore, β-arrestins have been shown to act as scaffolding proteins for various signaling molecules important in mediating inflammatory responses, including TRAF6 (24), NF-κB1p105 (25), IκBα (21, 26, 27), and mitogen-activated protein kinases (MAPKs) (6, 20, 28, 29). This role as a critical scaffolding molecule extends β-arrestins' capability in modulating inflammation beyond GPCRs to non-GPCRs, such as Toll-like receptors (24, 25, 30). Studies have shown that the role of β-arrestins in inflammation is highly context dependent and that, depending on the stimulus and disease model, β-arrestins can either mediate or inhibit inflammation (11, 12, 31, 32). In this regard, we recently demonstrated that β-arrestin-2 (β-arr2) promotes an increase in systemic levels of gamma interferon (IFN-γ) and other cytokines in the endotoxemia model (11) whereas it inhibits adenovirus-induced innate responses (33). Additionally, recent studies have suggested that β-arr2 is a negative regulator of polymicrobial sepsis-induced inflammation in the cecal ligation and puncture (CLP) model (12).
Sepsis is a complex pathophysiological disease process that involves an integrative response of the host to various pathogenic stimuli, including surgery, necrosis, abscess, and polymicrobial infections. While the CLP model of polymicrobial sepsis is a gold standard model, the pathogenesis of inflammation and mortality depends on multiple aspects, including necrotic cecum and polymicrobial infection (24, 34). In fact, studies have shown that removal of the necrotic cecum in animals subjected to CLP can significantly prevent mortality (24). Given the differential roles for β-arr2 in endotoxemia and CLP models, we hypothesized that the difference is due to the latter causing a polymicrobial infection and not due to the effects of necrotic cecum and surgery. To test this hypothesis, we examined the role of β-arrestin-2 in a polymicrobial infection model (35) without involving a necrotic tissue. Additionally, in this study, we determined the gene dosage effect of β-arrestin-2 in mediating these events.
MATERIALS AND METHODS
Animals.
β-Arrestin-2 knockout mice were kindly provided by Robert Lefkowitz and bred at Michigan State University (36). Wild-type (WT) C57BL/6 mice were purchased from NCI and bred in the same facility. Animals were housed in rooms maintained at 22 to 24°C with 50% humidity and a 12-h light-dark cycle. Mouse chow and water were provided ad libitum to all animals. All experiments were performed with age- and sex-matched mice between 8 and 12 weeks of age. Animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee (IACUC) and conformed to NIH guidelines.
Preparation of the polymicrobial culture.
Polymicrobial culture was obtained as described previously (35). Briefly, cecal contents collected from WT mice were inoculated in sterile medium (brain heart infusion; BD Bacto) and cultured at 37°C with 220-rpm shaking for 18 h. The contents were then centrifuged at 432 × g for 10 min, and the bacterial pellet was resuspended in 40% glycerol and stored at −80°C. For CFU measurements, 100 μl of the polymicrobial culture was inoculated in 100 ml medium and grown for 14 h, washed with phosphate-buffered saline (PBS), and plated on Muller-Hinton agar (BD Bacto) plates using serial dilution. Once the number of CFU/ml for the culture was determined, the culture was diluted to obtain the required CFU count for the experiments. The culture stock was determined to be consistent in terms of CFU count and was confirmed to be polymicrobial based on multiple colony morphologies as well as sequencing of the microbial community. To sequence the polymicrobial culture, genomic DNA was extracted from the culture and the V3-V5 region of the 16S rRNA was amplified with barcoded primers. Amplicon sequencing was performed using the 454 GS Junior (Roche Diagnostics) platform according to the manufacturer's protocols. Sequences were analyzed using mothur (26) version 1.29.1. Sequences were aligned to the Silva reference alignment using the NAST-based aligner in mothur, trimmed to ensure that sequences overlapped, and preclustered, allowing a difference between sequences of 2 bp or less (37). Chimeric sequences were removed using the mothur implementation of UChime (38); remaining sequences were classified using RDP training set version 9 and mothur's implementation of the kmer-based Bayesian classifier. Four genera of bacteria that dominated the polymicrobial culture were identified as Bacillus, Enterococcus, Planococcus, and Streptococcus (data not shown).
Polymicrobial sepsis.
Age-matched male mice were intraperitoneally injected with 10 × 106 CFU polymicrobial culture in 200 μl PBS. Six hours later, mice were euthanized, peritoneal fluid, spleen, and plasma were collected, and lung and liver tissue were harvested and snap frozen in liquid nitrogen for further analysis as described previously (11). Briefly, the peritoneal cavity was lavaged with 4 ml 1640 RPMI (Gibco) medium with 5% fetal bovine serum (FBS) (Gibco) and 55 μM β-mercaptoethanol (Gibco), and supernatant from this initial wash was stored at −80°C for cytokine analysis. The cavity was further washed with another 20 ml medium, and the cells from all washes were pooled for cellular analysis. Blood was collected by cardiac puncture; 100 μl was subjected to red blood cell (RBC) lysis and a subsequent wash in medium to get cells for flow cytometry analysis. The rest of it was centrifuged at 5,000 rpm for 2 min to obtain plasma that was stored at −80°C for cytokine analysis. Spleen was crushed, subjected to RBC lysis, and filtered through 40-μm nylon mesh. The cells were counted, resuspended in RPMI 1640 (with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 55 μM β-mercaptoethanol), and plated at a concentration of 10 × 106 cells/ml. Twenty-four hours later, the supernatant was collected for cytokine determination and stored at −80°C.
Survival study.
Male mice were administered an intraperitoneal injection of polymicrobial culture (40 × 106 CFU in 200 μl PBS) and monitored for survival for 24 h. The dose for survival studies was based on the 50% lethal dose (LD50) determined through a pilot experiment done on wild-type mice.
Cytokine/chemokine measurements.
Cytokines were measured from plasma, splenic culture supernatant, and peritoneal fluid using enzyme-linked immunosorbent assay (ELISA) kits from eBiosciences, Inc., according to the manufacturer's protocol and as described before (39). In order to pool the data from different experiments, the raw values were converted to fold change over average WT concentrations for each experiment.
Flow cytometry.
Peritoneal and blood cells collected from septic mice 6 hours after polymicrobial injection were processed as described above. They were then stained with antibody cocktail made in 2.4G2 supernatant (fcγR blocking antibody) to block nonspecific binding and washed with staining buffer (PBS with sodium azide and bovine calf serum [BCS]). The antibodies against the cell surface markers CD11b, F4/80, Gr-1, CD3, CD19, and CD11c were obtained from eBiosciences and used according to the manufacturer's instructions.
MPO assay.
Tissue myeloperoxidase (MPO) activity was performed as described before (39). Briefly, snap-frozen lung and liver tissues were homogenized in 50 mM potassium phosphate (pH 6.0) buffer. After centrifugation, the pellets were resuspended and vortexed in 50 mM potassium phosphate (pH 6.0) buffer containing 0.5% hexadecyltrimethylammonium bromide to release MPO. An aliquot of the supernatant was incubated at 25°C in 50 mM potassium phosphate (pH 6.0) buffer containing 0.0005% H2O2 and 167 μg/ml o-dianisidine hydrochloride. MPO activity was determined spectrophotometrically by measuring the change in absorbance at 450 nm over time using a 96-well plate reader. It was then normalized to total protein from the tissue initially homogenized, determined by the Bradford method.
Quantitative RT-PCR.
To determine the relative levels of a specific RNA transcript, RNA was isolated from snap-frozen tissue using the Qiagen RNeasy minikit according to the manufacturer's protocol and as described earlier (40). Reverse transcription (RT) was carried out with 1 μg of RNA using the Promega cDNA synthesis kit. Quantitative RT-PCR (qRT-PCR) was performed with ABI Fast 7500 (Applied Biosystems), and all genes were normalized to hypoxanthine phosphoribosyltransferase (HPRT) as previously described (40). The following primers were used for the respective genes: tumor necrosis factor alpha (TNF-α) forward, TCTCATCAGTTCTATGGCCC-3; TNF-α reverse, GGGAGTAGACAAGCTACAAC; IκBα forward, TGG CCA GTG TAG CAG TCT TG; IκBα reverse, GAC ACG TGT GGC CAT TGT AG; interleukin 6 (IL-6) forward, ACA AGT CGG AGG CTT AAT TAC ACA T; IL-6 reverse, TTG CCA TTG CAC AAC TCT TTT C; keratinocyte chemoattractant (KC) forward, CTTGAAGGTGTTGCCCTGAG; KC reverse, TGGGGACACCTTTTAGCATC; macrophage inflammatory protein 2 (MIP2) forward, GGCAAGGCTAACTGACCTGGAAAGG; MIP2 reverse, ACAGCGAGGCACATGAGGTACGA; HPRT forward, AAG CCT AAG ATG AGC GCA AG; HPRT reverse, TTA CTA GGC AGA TGG CCA CA. Primer sets for β-arrestin-1 and β-arrestin-2 were obtained from Qiagen and were used as described earlier (41).
Western blotting.
Snap-frozen liver tissue was homogenized in lysis buffer (20 mM Tris-HCl [pH 7.4], 1 mM EDTA, 150 mM NaCl) containing 1% Triton X-100 and protease and phosphatase inhibitors. Homogenized tissue was spun at 13,000 rpm for 10 min at 4°C. The protein concentration of the supernatant was determined using the Bradford method. Western blots for pERK1/2, extracellular signal-regulated kinase 2 (ERK2), IκBα, pJNK1/2, Jun N-terminal kinase 1/2 (JNK1/2), pP38, pP105, and tubulin were performed as previously described (25). Briefly, equivalent concentrations of protein samples were run on polyacrylamide gels and transferred to nitrocellulose membranes. Blots were then probed with primary and fluorescent secondary antibody as described previously. Blots were scanned and bands were quantified using a Li-COR Odyssey scanner. For data analysis, pERK1/2 was normalized to ERK2, pIκBα was normalized to IκBα, and pJNK, pP38, and pP105 were normalized to actin/tubulin as loading controls.
Cell culture.
All cells were cultured in RPMI 1640 containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 55 μM β mercaptoethanol and incubated at 37°C and 5% CO2. In the case of polymicrobial stimulation, initial stimulus was done in antibiotic-free medium for an hour, following which an antibiotic mixture (100 U/ml penicillin, 100 μg/ml streptomycin, and 10 μg/ml gentamicin) was added for the rest of the duration.
Basal peritoneal cells were obtained by collecting peritoneal washes from naive mice. Briefly, 30 ml medium was used to lavage the peritoneal cavity. Cells were subjected to RBC lysis and plated at 0.4 million cells/well in 500 μl medium.
Splenocytes were harvested and processed to obtain single-cell suspensions. Briefly, spleen was crushed, subjected to RBC lysis, filtered through a 40-μm filter, and resuspended to give a final concentration of 5 million cells/well in 1 ml medium.
Bone marrow-derived macrophages (BMDM) were obtained by culturing bone marrow cells in the presence of L929 cell-conditioned medium (LCCM) as described previously (42). Briefly, tibia and femur were collected and flushed using a 27-gauge needle and cells were passed through the needle twice for single-cell suspensions. Following RBC lysis, cells were filtered and cultured in 30% LCCM for 7 days to generate BMDMs. The cells were finally plated at 0.5 million cells/ml in 1 ml medium for stimulation.
To obtain neutrophils, 1 ml thioglycolate was injected into the peritoneal cavity and cells were harvested from the cavity 6 h later as described above. We confirmed that the cells harvested from the peritoneal cavity 6 h after thioglycolate injection were predominantly neutrophils (∼70%), with macrophages comprising only a minor population (∼10%) (flow cytometry data not shown). Even though the cellular proportion was unaffected by loss of β-arr2, the total cell count was marginally (not statistically) decreased in the KO mice (2.24 ± 0.6 × 106) and unaffected in the HET mice (5.34 ± 0.4 × 106) compared to the WT mice (5.15 ± 0.9 × 106). The cells were eventually plated at 1 million cells/well in 1 ml medium for stimulation.
For stimulation, ultrapure lipopolysaccharide (LPS) (Invivogen) and polymicrobial culture (obtained as described above) were added at specified concentrations and multiplicities of infection (cells/bacteria), respectively. Supernatant was collected 18 h later and stored at −80°C for further analysis.
Statistical analysis.
All experimental data in the figures are expressed as means and standard errors of the means (SEM) and analyzed using GraphPad Prism software. Experiments were performed 3 times, and total n represents the number of mice used in all 3 experiments combined. Student's t test (for comparing groups with equal variances) or a Mann-Whitney test (for comparing groups with unequal variances) was used to compare two experimental groups, and analysis of variance (ANOVA) (with a Bonferroni posttest) was used for more than two groups. Differences in the survival were determined using the log-rank test. P values of <0.05 were considered significant.
RESULTS
Gene dosage-dependent effect of β-arrestin-2 on PMI-induced cytokine production.
To induce polymicrobial sepsis, we injected the polymicrobial culture intraperitoneally into the 3 groups of mice (wild type [WT], β-arr2−/− [KO], and β-arr2+/− [HET]) and assessed cytokine production as a measure of inflammation at different sites (systemic and local) at 6 h following infection (35, 43, 44). Uninfected mice from the three genotypes were used as controls. As shown in Fig. 1A, plasma IL-6 and IL-10 levels were significantly enhanced in KO mice compared to the WT mice. Note that the plasma IL-6 and IL-10 were below detection limits in uninfected mice and that TNF-α was not detectable at this time point in the infected or uninfected mice (data not shown). Similar to the plasma cytokine levels, splenic and peritoneal IL-6 and IL-10, except peritoneal IL-6 (P = 0.09), were upregulated following microbial challenge and significantly enhanced in KO mice compared to the WT mice (Fig. 1B and C). Interestingly, cytokine levels in these different sites were similar between the HET and WT mice, suggesting that expression from one allele is sufficient to inhibit polymicrobial infection-induced cytokine production (Fig. 1B). The difference in cytokine levels between the genotypes was not due to differential bacterial load since bacterial counts in blood and spleen were similar between the three groups (Fig. 1D). Thus, β-arr2 acts as a negative regulator of cytokine production following PMI even though there appears to be no difference in bacterial dissemination.
Fig 1.
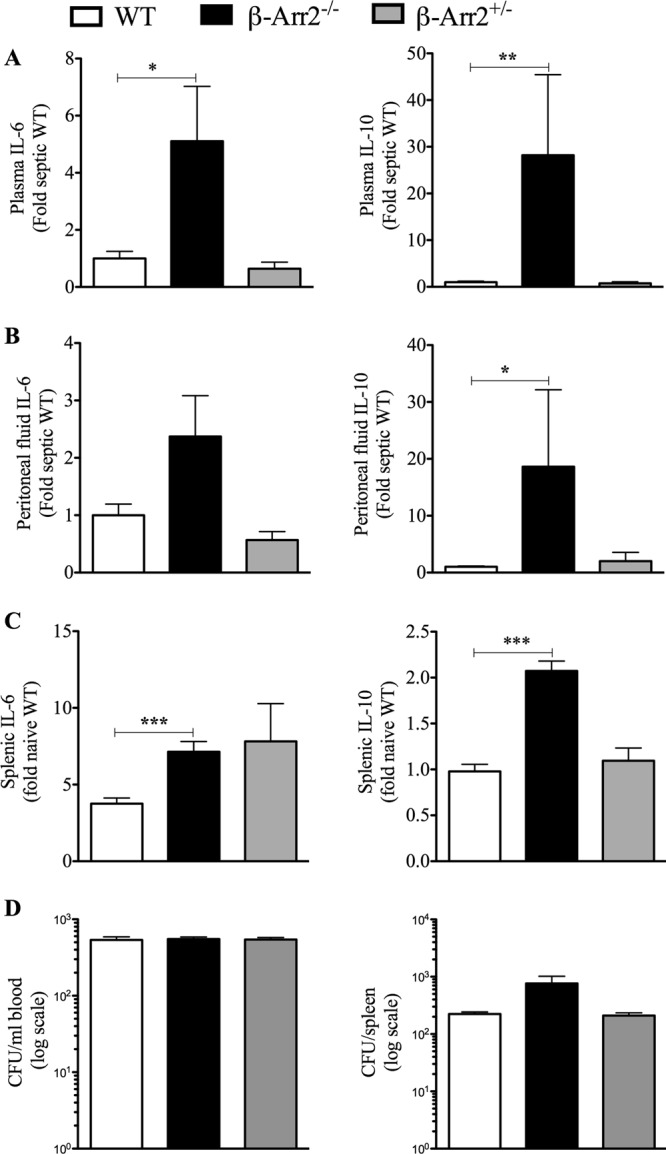
Cytokine production induced by polymicrobial injection is enhanced in β-arrestin-2 knockout mice. Wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice were intraperitoneally injected with polymicrobial culture. Six hours later, mice were euthanized and samples were collected as described in Materials and Methods. (A) Plasma cytokine. (B) Cytokines in splenic culture supernatant. Splenic cells were cultured at 5 × 106 cells/ml, and 24 h later, the supernatant was collected and assayed for the cytokine concentration. (C) Cytokines in peritoneal fluid. All concentrations were converted to fold change over septic WT for plasma and peritoneal fluid and over naive WT for spleen. Uninfected naive animals had undetectable levels of cytokines in plasma and the peritoneum. (D) Bacterial load in blood and spleen of infected mice, represented on a log scale. Uninfected naive mice had no bacterial load. *, P < 0.05; **, P < 0.01; ***, P < 0.001; compared to the WT using a t test or Mann-Whitney test. n = 3 for naive mice and 8 to 9 for septic mice for each genotype.
Differential regulation of immune cell infiltration by β-arrestin-2.
We also assessed local (peritoneal) and systemic (blood) infiltration of neutrophils and macrophages following PMI as another measure of inflammatory response following microbial challenge. The numbers of neutrophils and macrophages in blood and the peritoneal cavity were comparable in naive mice from the three genotypes (data not shown) and increased following polymicrobial injection. At the local site of infection (i.e., the peritoneal cavity), the number of macrophages but not the number of neutrophils was significantly lower in KO mice than in WT mice (Fig. 2A). Interestingly, infiltration of these cells was similar between the peritoneal cavities of HET and WT mice. In contrast to the peritoneal cavity, blood neutrophil and macrophage numbers were significantly increased in KO (but not HET) mice compared to the WT (Fig. 2B). These results suggest that in the complete absence of β-arr2 protein, there is an increased influx of innate immune cells into blood but not to the site of infection.
Fig 2.
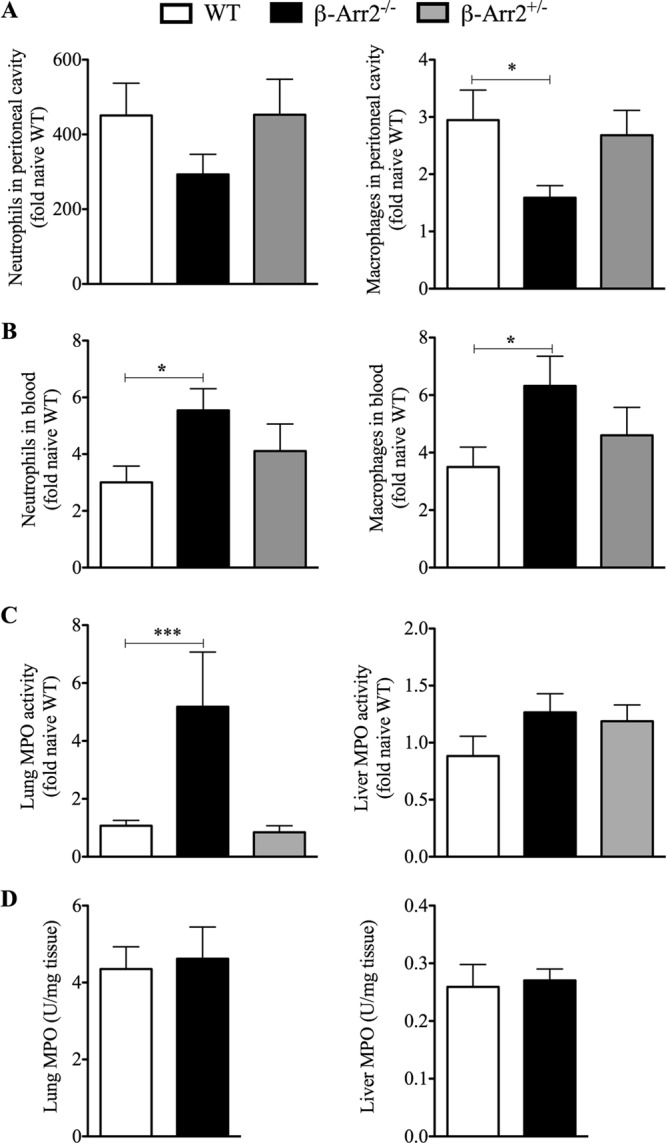
β-Arrestin-2 differentially regulates immune cell infiltration following polymicrobial infection. Wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice were intraperitoneally injected with polymicrobial culture. Six hours later, mice were euthanized and samples were collected as described in Materials and Methods. Cells were subjected to flow cytometry and identified on the basis of the cell surface markers CD11b+ Gr-1+ (polymorphonuclear cells [PMN]) and CD11b+ F4/80+ (macrophages). (A) Total number of cells harvested from the peritoneal cavity of each mouse. (B) Total number of cells/ml of blood. (C) MPO activity as a marker for neutrophil sequestration was determined in lung and liver tissue following PMI. Data were normalized as fold naive WT; basal values were similar in all three genotypes. (D) Lung and liver MPO activity following LPS injection. Wild-type and β-arrestin-2 knockout mice were intraperitoneally injected with LPS (5 μg/g of body weight), and lung and liver tissue was collected for MPO activity 6 h later. *, P < 0.05; ***, P < 0.001; compared to septic WT using a t test or Mann-Whitney test. n = 3 for naive mice and 8 or 9 for septic mice of each genotype.
To assess the fate of cells entering the bloodstream, MPO content, an indicator of neutrophil sequestration, was determined in lung and liver tissue from septic mice. Basal MPO levels were equivalent in lungs from WT, KO, and HET mice (data not shown), and microbial challenge did not cause an increase in MPO content in WT mice (Fig. 2C). However, lungs from septic KO mice had significantly enhanced MPO activity compared to that of the WT (Fig. 2C). Interestingly, MPO activity in liver was unaffected by loss of β-arr2 or microbial stimulation (Fig. 2C). The role of β-arr2 thus appears to be organ specific in terms of neutrophil sequestration following polymicrobial sepsis. More importantly, increased MPO content was not observed in lungs from septic HET mice, suggesting a gene dosage effect of β-arr2 in regulating neutrophil migration.
To determine if lipopolysaccharide (LPS; a major component of Gram-negative bacteria) following systemic infection is the likely mediator of this differential neutrophil migration in the KO, MPO content was determined in lung tissue following intraperitoneal LPS injection. In contrast to PMI stimulation, MPO content of lung tissue from KO mice was comparable to that of the WT following LPS administration (Fig. 2D), suggesting that the effect on MPO activity in the lung is specific to the PMI model.
β-arrestin-2 regulates inflammatory gene expression in lungs following PMI.
Impaired neutrophil chemotaxis in β-arrestin-2−/− mice following polymicrobial sepsis was specific for the lung and not observed in the liver. Hence, we further examined the role of β-arrestin-2 on cytokine and chemokine genes in these organs following septic peritonitis. As is evident from Fig. 3A, cytokine and chemokine expression was induced in WT lung following PMI. Interestingly, uninfected KO lung had higher expression of TNF-α and IL-10 than did uninfected WT lung. However, septic KO lung tissue demonstrated significantly enhanced mRNA for IL-6, TNF-α, IL-10, KC, and mip2 compared to that for septic WT mice (Fig. 3A). In addition, mRNA expression of IκBα (a gene tightly regulated and induced by the NF-κB pathway [45]) was significantly elevated in KO lung tissue compared to that in the WT mice. Contrary to the distinctly enhanced “inflammatory signature” in the KO mice, the HET mice had reduced expression levels of KC, mip2, and IκBα but similar levels of IL-6, TNF-α, and IL-10 compared to the WT lung.
Fig 3.

Gene dosage-dependent regulation of inflammatory genes in lung by β-arrestin-2 following polymicrobial infection. RNA was obtained from lung (A) and liver (B) tissue samples from wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice at 6 h after polymicrobial infection. mRNA expression of the indicated inflammatory genes was performed using real-time qRT-PCR as described in Materials and Methods. The values were converted to fold naive WT for each experiment. *, P < 0.05; **, P < 0.01; using a t test or Mann-Whitney test. n = 3 for naive mice and 7 to 9 for septic mice of each genotype.
Basal expression of tested inflammatory mediators was unaffected by loss of β-arrestin-2 in liver tissue. Unlike that in the lung, IL-6, TNF-α, and IκBα expression was not even induced in the liver tissue in response to bacterial challenge. Additionally, except for mip2 expression, which was higher in the KO liver tissue, all other tested inflammatory markers were comparable between WT and KO liver (Fig. 3B). Further, IL-6, TNF-α, and IL-10 expression was lower in septic HET liver tissue than in the WT. Thus, both enhanced neutrophil sequestration and increased expression of inflammatory genes appear to be specific to lung tissue in KO mice following polymicrobial sepsis. Furthermore, both these observations are dependent on loss of both β-arrestin-2 alleles since neither was enhanced in the HET mice.
Differential regulation of signaling in the lungs by β-arrestin-2 following PMI.
β-arrestin-2 has been shown to act as a scaffolding protein for NF-κB and MAPK signaling molecules (6, 21, 24, 26–28, 46). To examine the signaling mechanisms associated with hyperinflammation observed in the KO lungs, we determined the phosphorylation status of major NF-κB (IκBα, P105) and MAPK (ERK, JNK, P38) signaling molecules. Interestingly, compared to the uninfected controls, MAPK signaling was significantly downregulated in infected WT mice at this time point. In contrast, pP105 levels were significantly elevated postinfection while pIκBα showed no difference. In addition, compared to infected WT mice, KO-infected mice had significantly elevated pJNK and pIκBα levels while pP38 showed a similar trend (P = 0.06; two-tailed t test) (Fig. 4). Lung tissue from septic HET mice had MAPK and NF-κB activation similar to that of the WT. Together, these data suggest that β-arr2 is likely important for downregulating some of these specific NF-κB and MAPK pathways in the lungs following polymicrobial infection.
Fig 4.

Differential regulation of MAPK and NF-κB kinase pathways by β-arrestin-2 in the lung following polymicrobial infection. Lung protein lysates from wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice at 6 h after polymicrobial infection were assessed for the phosphorylation status of major signaling molecules as described in Materials and Methods. Note that the blots were probed with multiple primary antibodies and later analyzed by Li-COR Odyssey (except pIκBα) as stated in Materials and Methods. pERK was normalized to ERK, pIκBα was normalized to IκBα, pJNK was normalized to JNK/actin, and pP38 and pP105 were normalized to actin/tubulin as loading controls. Raw values were converted to fold septic WT from each blot. Representative blots are shown in Fig. S1 in the supplemental material. *, P < 0.05; **, P < 0.01; ***, P < 0.001, using a t test or Mann-Whitney test. n = 3 to 6 for naive mice and 8 or 9 for septic mice for all genotypes.
β-arrestin-2 mRNA expression is upregulated in lungs from WT mice following PMI.
β-arrestin-1 and β-arrestin-2 have distinct as well as overlapping functions (19, 31, 33, 47). Keeping that in mind, we examined the possibility that the level of β-arr1 might be differentially regulated in KO mice to carry out compensatory functions. To test this, we determined the expression levels of both arrestins in the lung tissue of naive and septic mice. β-arr1 mRNA expression was unaffected by loss of β-arr2 under basal conditions and, furthermore, was unaltered following induction of polymicrobial sepsis in all three genotypes. β-arr2 mRNA expression, on the other hand, was significantly upregulated in the WT septic lungs following PMI (Fig. 5). Both alleles of β-arrestin-2, however, were necessary for this upregulation since there was no increase in β-arr2 levels in the HET mice following PMI. β-arr2 thus acts as an important negative regulator of pulmonary inflammation, and its expression is upregulated in the lungs following microbial challenge.
Fig 5.
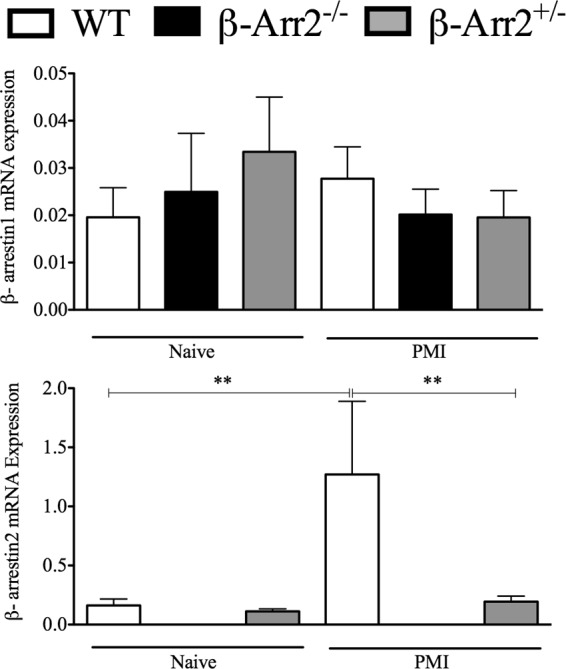
Differential regulation of β-arrestin expression in the lung following polymicrobial infection. Lung RNA samples described in Fig. 3 were subjected to real-time qRT-PCR for determining the expression levels of β-arrestin-1 (A) and β-arrestin-2 (B) in mice injected with polymicrobial culture. Untreated control mice were used as basal controls. **, P value of <0.01 using a Mann-Whitney test. n = 7 to 9 for each treatment group and genotype.
β-arrestin-2 modulates sepsis-induced mortality.
IL-6, IL-10, and other cytokines have often been used to predict early deaths in sepsis, hinting at the presence of cause-and-effect or correlative mechanisms at play for the two events (7, 43, 44). In the present study, consistent with the systemic cytokine profile, polymicrobial injection resulted in higher mortality in the β-arrestin-2 knockout mice than in WT mice (Fig. 6). HET mice, however, had mortality comparable to that of the WT following lethal PMI. Together, our results demonstrate a close association between inflammatory response and mortality, with both being higher in KO mice than in WT mice following polymicrobial infection (Fig. 6).
Fig 6.
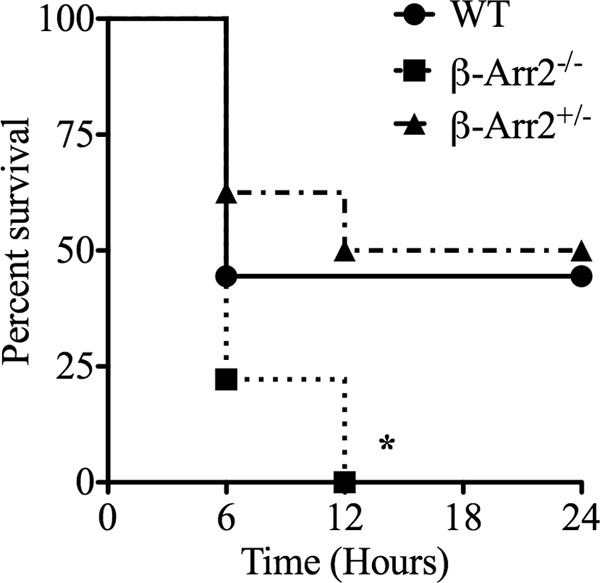
Gene dosage-dependent role for β-arrestin-2 in preventing mortality following polymicrobial infection. Wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice were intraperitoneally injected with polymicrobial culture (40 × 106 CFU) in a 200-μl volume. Mice were then monitored for survival for 24 h. *, P value of <0.05 compared to the WT by a log-rank (Mantel Cox) test. n = 7 mice for each genotype.
Role of β-arrestin-2 in regulating cytokine production in vitro.
β-arrestin-2 has been ascribed contrasting roles in regulating inflammation following different stimuli (11, 12, 24, 33). Additionally, following PMI, we observed a site-specific regulation of cytokine production by β-arr2. We hypothesized that β-arrestin-2 has a cell-type-specific and stimulus-dependent role in regulating inflammation. To test this, we used peritoneal cells from naive mice as well as splenocytes, bone marrow-derived macrophages (BMDMs), and thioglycolate-induced neutrophils as cellular models to examine the effect of LPS and polymicrobial challenge on IL-6, TNF-α, and IL-10 production. Peritoneal cells and splenocytes from KO mice demonstrated enhanced cytokine production compared to the WT (see Fig. S1 and S2 in the supplemental material). Compared to these two populations, IL-6 and TNF-α production was lower but IL-10 production was higher from KO BMDMs than WT cells in a stimulus-specific manner (Fig. 7). HET BMDMs produced lower IL-6 in response to LPS but higher IL-10 following polymicrobial stimulation. Thus, in BMDMs, in contrast to in peritoneal cells and splenocytes, β-arr2 has a diverse role in cytokine production, acting as a positive regulator of proinflammatory cytokines and negative regulator of anti-inflammatory cytokines. In contrast to BMDMs, thioglycolate-elicited neutrophils from KO mice produced significantly enhanced IL-6 and IL-10 compared to WT mice following LPS but not microbial stimulation (Fig. 8). Neutrophils from HET mice had lower IL-10 than WT mice following microbial stimulation. These results suggest that β-arr2 is a negative regulator of IL-6 and IL-10 production in these neutrophils in a stimulus-specific manner. Overall, these results suggest a stimulus- and cell-type-specific role for β-arrestin-2 in mediating cytokine production.
Fig 7.
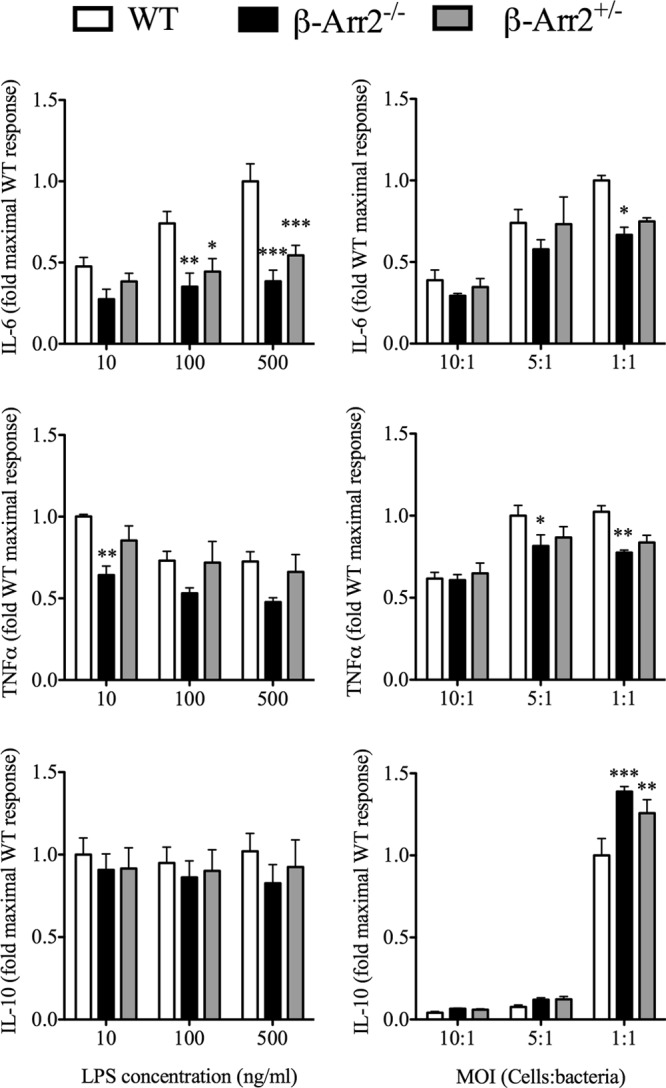
Differential regulation of IL-6, TNF-α, and IL-10 by β-arrestin-2 in bone marrow-derived macrophages. Bone marrow-derived macrophages were obtained from wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice and plated as described in Materials and Methods. LPS and polymicrobial culture were used to stimulate the cells at different concentrations and multiplicities of infection (MOI), respectively. Eighteen hours later, the supernatant was collected and assayed for IL-6, IL-10, and TNF-α concentrations. Basal cytokine production was below the detection limit of the ELISA kits. The data were transformed to fold WT maximal response. *, P < 0.05; **, P < 0.01; ***, P < 0.001; compared to the WT as determined by a 2-way ANOVA followed by a Bonferroni posttest. n = 4 or 5 mice for each genotype.
Fig 8.
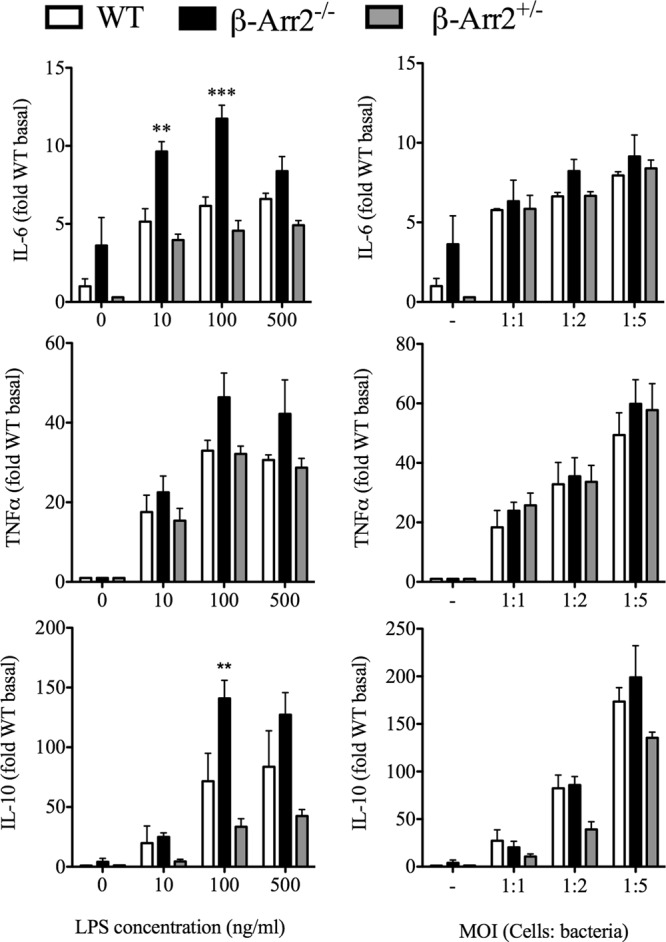
Stimulus-specific role of β-arrestin-2 in regulating cytokine production from neutrophils. Neutrophils were obtained from thioglycolate-injected wild-type (WT), β-arrestin-2 homozygous knockout (β-arr2−/−), and β-arrestin-2 heterozygous (β-arr2+/−) mice as described in Materials and Methods. Cells were plated (as described in Materials and Methods) and stimulated with LPS and polymicrobial culture at different concentrations and multiplicities of infection (MOI), respectively, for 18 h, and supernatants were assayed for IL-6, IL-10, and TNF-α concentrations. Cytokine levels were transformed as fold over WT basal. *, P < 0.05; **, P < 0.01; ***, P < 0.001; compared to the WT as determined by a 2-way ANOVA followed by a Bonferroni posttest. n = 3 for each genotype.
DISCUSSION
The role of β-arrestin-2 (β-arr2) in modulating inflammatory changes in different sepsis models has yielded contradictory results, with β-arr2 KO mice having higher mortality in the cecal ligation and puncture (CLP) model (12) but lower mortality in the endotoxemia model (11). Unlike the endotoxemia model, in CLP, the inflammatory stimuli include microbes, fecal material, and necrotic tissue (48), and β-arr2 was shown to be a negative regulator of both inflammation and mortality in this model. We therefore used the polymicrobial infection (PMI) model to determine whether β-arr2 has similar function following microbial challenge independent of the effects of necrotic tissue and surgery. Similar to the CLP-induced sepsis, but in contrast to the endotoxemia, β-arr2 KO mice exhibited exacerbated systemic inflammation and poor survival following PMI, indicating that microbial challenge is the dominant stimulus in response to which β-arr2 exerts its influence in polymicrobial sepsis models. Additionally, even though TLR4 signaling has been ascribed a critical role in pathogenesis of the PMI model (35), the role of β-arr2 in microbial infection as a negative regulator appears to be dominant over and distinct from its role as a positive regulator of inflammation in response to LPS. Distinct roles for β-arr2 in regulating cytokine production in response to LPS and microbial challenge were observed in vitro as well.
In addition to being model specific, β-arr2 appears to regulate inflammatory genes in a tissue-specific manner in the PMI model. Expression of certain inflammatory mediators (IL-10, KC) was enhanced in the lung but not liver of septic KO mice, suggesting that β-arr2's negative regulatory role in this model is tissue specific. While it is possible that there may be differences in kinetics of cytokine production in different tissues, the site-specific regulation may also be due to the cell-type-specific role of β-arr2. The latter scenario is supported by our in vitro experiments, wherein the role of β-arr2 in mediating cytokine production was found to be responder specific. This was particularly interesting given the contrasting roles ascribed to β-arr2 when using different cell models (12, 24, 31). Using the in vitro systems led us to further postulate that given the integrative nature of systemic inflammatory responses, functions observed in vitro might not be reflective/indicative of this protein's in vivo role. Even though the biochemical basis for these differences is not clear, our results underscore the importance of β-arr2 in inflammation as well as suggest that the role of β-arr2 in inflammation may be disease specific depending on the stimulus and the dominant cell type involved in disease pathogenesis.
β-arr2 has been shown to be an important regulator of chemotaxis in different models (21–23, 32). Consistent with that, our studies also reveal that β-arr2 is an important regulator of directional migration of neutrophils. Although polymicrobial infection did not cause any differential infiltration of immune cells to the site of infection (peritoneum), MPO activity in lung but not liver was significantly elevated in the β-arr2 KO mice. This neutrophil sequestration in KO lungs following microbial challenge was also observed in the surgical CLP model of polymicrobial sepsis (12, 32). This site-specific regulation in the KO mice may be due to specific “neutrophil-favoring” chemokine gradients regulating infiltration into the lungs (since KC and mip2 mRNA expression was higher in KO lung than the WT lung) or due to an inherent defect in the ability of β-arr2 KO neutrophils to carry out “directional migration” toward the site of infection. It is, however, clear that this directional migration is specific for this PMI model, since LPS-induced neutrophil sequestration in the lungs was unaffected by the loss of β-arr2.
In addition to enhanced sequestration of neutrophils in the KO lungs, inflammatory gene expression was also enhanced in the lungs from KO mice. β-Arrestin-2 has been shown to act as a scaffolding protein for NF-κB and MAPK signaling molecules (6, 21, 24, 26–28, 46). Altered activation of some of these pathways in the lung and enhanced gene expression suggest that β-arr2 likely regulates these pathways negatively, thereby affecting pulmonary inflammation. Interestingly, at the 6-hour time point that we studied, MAPK activation (pERK, pJNK, and p-p38) appears to be decreased following infection. It is possible that these pathways are activated at early time points and that regulatory mechanisms come into play to negatively regulate these pathways and suppress consequent exacerbated inflammation. This mechanism appears to be largely lost in the β-arr2 KO, suggesting that β-arr2 is important for downregulation of these specific pathways at the time point we tested. Enhanced NF-κB activation as evidenced by higher Iκbα phosphorylation may, in part, explain exacerbated inflammatory genes in the KO-infected lungs. Consistent with that, mRNA expression of IκBα (NF-κB-regulated gene) was significantly higher in the KO lung than in the WT. Interestingly, pulmonary mRNA expression of β-arr2 itself was upregulated following infection. This regulation of β-arr2 expression under inflammatory conditions has been previously observed in an arthritis model (49) and was found to have an important role to play in its pathogenesis. A similar regulation of β-arr2 expression in the sepsis model suggests an important role for β-arr2 in regulating pulmonary inflammation following sepsis.
Consistent with previous studies on CLP-induced mortality as well as the hyperinflammatory phenotype observed following PMI, β-arr2 KO mice had higher lethality than the WT mice. Again, as was observed for most other parameters, one allele of β-arr2 was sufficient in preventing PMI-induced mortality. Enhanced lethality of the β-arr2 KO mice is likely because of higher systemic and lung inflammatory cytokines and likely pulmonary damage in the KO mice. Interestingly, enhanced pulmonary neutrophil sequestration in KO mice was observed only in microbial models of sepsis and not in endotoxemia. These data, taken in conjunction with our previous observations on the decreased mortality of β-arr2 KO mice in the endotoxemia model, suggest that the enhanced lethality in the β-arr2 KO mice is specific for models of microbial challenge.
Even though β-arr2 knockout mice were shown to be susceptible to CLP-induced septic mortality (12), our studies demonstrate that the susceptibility of the KO mice is dependent solely on microbial challenge and independent of other effects of surgery or necrotic cecum. In future studies, we will determine the molecular mechanisms by which β-arrestin-2 modulates bacterial infection, which would likely result in identification of new therapeutic targets to treat bacterial sepsis.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge support from the NIH (grants HL095637, AR055726, and AR056680 [to N.P.] and AI090872 [to R.A.B.]).
We thank the university lab animal resources for taking excellent care of our animals. We are grateful to Robert J. Lefkowitz for kindly providing us the β-arrestin-2 knockout mice. We thank Kylie Farrell and Jennifer Auchtung for assistance in sequencing the polymicrobial culture.
Footnotes
Published ahead of print 10 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00653-13.
REFERENCES
- 1. Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. 1987. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. U. S. A. 84:8879–8882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Magalhaes AC, Dunn H, Ferguson SSG. 2012. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 165:1717–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajagopal S, Rajagopal K, Lefkowitz RJ. 2010. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9:373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeFea K. 2008. β-Arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br. J. Pharmacol. 153:S298–S309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. 2007. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 69:483–510 [DOI] [PubMed] [Google Scholar]
- 6. Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. 2001. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U. S. A. 98:2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barkhausen T, Tschernig T, Rosenstiel P, van Griensven M, Vonberg R-P, Dorsch M, Mueller-Heine A, Chalaris A, Scheller J, Rose-John S, Seegert D, Krettek C, Waetzig GH. 2011. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 39:1407–1413 [DOI] [PubMed] [Google Scholar]
- 8. Bonnans C, Flaceliere M, Grillet F, Dantec C, Desvignes J-P, Pannequin J, Severac D, Dubois E, Bibeau F, Escriou V, Crespy P, Journot L, Hollande F, Joubert D. 2012. Essential requirement for β-arrestin2 in mouse intestinal tumors with elevated Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 109:3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fereshteh M, Ito T, Kovacs JJ, Zhao C, Kwon HY, Tornini V, Konuma T, Chen M, Lefkowitz RJ, Reya T. 2012. β-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 109:12532–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forooghian F, Cheung RK, Smith WC, O'Connor P, Dosch HM. 2007. Enolase and arrestin are novel nonmyelin autoantigens in multiple sclerosis. J. Clin. Immunol. 27:388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter KJ, Gonipeta B, Parvataneni S, Appledorn DM, Patial S, Sharma D, Gangur V, Amalfitano A, Parameswaran N. 2010. Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by β-arrestins. J. Cell. Physiol. 225:406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan H, Bitto A, Zingarelli B, Luttrell LM, Borg K, Halushka PV, Cook JA. 2010. Beta-arrestin 2 negatively regulates sepsis-induced inflammation. Immunology 130:344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee T, Lee E, Irwin R, Lucas PC, McCabe LR, Parameswaran N. 2013. β-arrestin-1 deficiency protects mice from experimental colitis. Am. J. Pathol. 182:1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palmitessa A, Benovic JL. 2010. Arrestin and the multi-PDZ domain-containing protein MPZ-1 interact with phosphatase and tensin homolog (PTEN) and regulate Caenorhabditis elegans longevity. J. Biol. Chem. 285:15187–15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tipping M, Kim Y, Kyriakakis P, Tong M, Shvartsman SY, Veraksa A. 2010. β-Arrestin Kurtz inhibits MAPK and Toll signalling in Drosophila development. EMBO J. 29:3222–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilbanks AM, Fralish GB, Kirby ML, Barak LS, Li YX, Caron MG. 2004. Beta-arrestin 2 regulates zebrafish development through the hedgehog signaling pathway. Science 306:2264–2267 [DOI] [PubMed] [Google Scholar]
- 17. Li H, Sun X, LeSage G, Zhang Y, Liang Z, Chen J, Hanley G, He L, Sun S, Yin D. 2010. β-Arrestin 2 regulates toll-like receptor 4-mediated apoptotic signalling through glycogen synthase kinase-3β. Immunology 130:556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braun L, Christophe T, Boulay F. 2003. Phosphorylation of key serine residues is required for internalization of the complement 5a (C5a) anaphylatoxin receptor via a β-arrestin, dynamin, and clathrin-dependent pathway. J. Biol. Chem. 278:4277–4285 [DOI] [PubMed] [Google Scholar]
- 19. Vibhuti A, Gupta K, Subramanian H, Guo Q, Ali H. 2011. Distinct and shared roles of β-arrestin-1 and β-arrestin-2 on the regulation of C3a receptor signaling in human mast cells. PLoS One 6:e19585. 10.1371/journal.pone.0019585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao M, Wimmer A, Trieu K, DiScipio RG, Schraufstatter IU. 2004. Arrestin regulates MAPK activation and prevents NADPH oxidase-dependent death of cells expressing CXCR2. J. Biol. Chem. 279:49259. [DOI] [PubMed] [Google Scholar]
- 21. Luan B, Zhang Z, Wu Y, Pei G. 2005. β-Arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-κB activation. EMBO J. 24:4237–4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. 2002. Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 99:7478–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vroon A, Heijnen CJ, Kavelaars A. 2006. GRKs and arrestins: regulators of migration and inflammation. J. Leukoc. Biol. 80:1214–1221 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. 2006. Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 7:139–147 [DOI] [PubMed] [Google Scholar]
- 25. Parameswaran N, Pao CS, Leonhard KS, Kang DS, Kratz M, Ley SC, Benovic JL. 2006. Arrestin-2 and G protein-coupled receptor kinase 5 interact with NF B1 p105 and negatively regulate lipopolysaccharide-stimulated ERK1/2 activation in macrophages. J. Biol. Chem. 281:34159–34170 [DOI] [PubMed] [Google Scholar]
- 26. Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. 2004. β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc. Natl. Acad. Sci. U. S. A. 101:8603–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. 2004. Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 14:303–317 [DOI] [PubMed] [Google Scholar]
- 28. McDonald PH, Chow C-W, Miller WE, Laporte SA, Field ME, Lin F-T, Davis RJ, Lefkowitz RJ. 2000. β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290:1574–1577 [DOI] [PubMed] [Google Scholar]
- 29. Ahn S, Wei H, Garrison TR, Lefkowitz RJ. 2004. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J. Biol. Chem. 279:7807–7811 [DOI] [PubMed] [Google Scholar]
- 30. Barratt-Due A, Thorgersen EB, Lindstad JK, Pharo A, Brekke O-L, Christiansen D, Lambris JD, Mollnes TE. 2010. Selective inhibition of TNF-α or IL-1β does not affect E. coli-induced inflammation in human whole blood. Mol. Immunol. 47:1774–1782 [DOI] [PubMed] [Google Scholar]
- 31. Fan H, Luttrell LM, Tempel GE, Senn JJ, Halushka PV, Cook JA. 2007. β-Arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol. Immunol. 44:3092–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Basher F, Fan H, Zingarelli B, Borg KT, Luttrell LM, Tempel GE, Halushka PV, Cook JA. 2008. β-Arrestin 2: a negative regulator of inflammatory responses in polymorphonuclear leukocytes. Int. J. Clin Exp. Med. 1:32–41 [PMC free article] [PubMed] [Google Scholar]
- 33. Seregin SS, Appledorn DM, Patial S, Bujold M, Nance W, Godbehere S, Parameswaran N, Amalfitano A. 2010. β-Arrestins modulate adenovirus-vector-induced innate immune responses: differential regulation by β-arrestin-1 and β-arrestin-2. Virus Res. 147:123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ayala A, Song GY, Chung CS, Redmond KM, Chaudry IH. 2000. Immune depression in polymicrobial sepsis: the role of necrotic (injured) tissue and endotoxin. Crit. Care Med. 28:2949–2955 [DOI] [PubMed] [Google Scholar]
- 35. Alves-Filho JC, de Freitas A, Russo M, Cunha FQ. 2006. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit. Care Med. 34:461–470 [DOI] [PubMed] [Google Scholar]
- 36. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. 1999. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286:2495–2498 [DOI] [PubMed] [Google Scholar]
- 37. Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. 10.1371/journal.pone.0027310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patial S, Shahi S, Saini Y, Lee T, Packiriswamy N, Appledorn DM, LaPres JJ, Amalfitano A, Parameswaran N. 2011. G-protein coupled receptor kinase 5 mediates lipopolysaccharide-induced NFκB activation in primary macrophages and modulates inflammation in vivo in mice. J. Cell. Physiol. 226:1323–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Packiriswamy N, Lee T, Raghavendra PB, Durairaj H, Wang H, Parameswaran N. 2013. G-protein-coupled receptor kinase-5 mediates inflammation in a polymicrobial sepsis model in mice. J. Innate Immun. 10.1159/000347002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loniewski K, Shi Y, Pestka J, Parameswaran N. 2008. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol. Immunol. 45:2312–2322 [DOI] [PubMed] [Google Scholar]
- 42. Zhang X, Goncalves R, Mosser DM. 2008. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter ]?>14:Unit 14.1. 10.1002/0471142735.im1401s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giannoudis PV, Harwood PJ, Loughenbury P, Van Griensven M, Krettek C, Pape HC. 2008. Correlation between IL-6 levels and the systemic inflammatory response score: can an IL-6 cutoff predict a SIRS state? J. Trauma 65:646–652 [DOI] [PubMed] [Google Scholar]
- 44. Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. 2002. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17:463–467 [DOI] [PubMed] [Google Scholar]
- 45. Oeckinghaus A, Ghosh S. 2009. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1:a000034. 10.1101/cshperspect.a000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kizaki T, Izawa T, Sakurai T, Haga S, Taniguchi N, Tajiri H, Watanabe K, Day NK, Toba K, Ohno H. 2008. β2-Adrenergic receptor regulates Toll-like receptor-4-induced nuclear factor-κB activation through β-arrestin 2. Immunology 124:348–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeFea KA. 2007. Stop that cell! β-Arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annu. Rev. Physiol. 69:535–560 [DOI] [PubMed] [Google Scholar]
- 48. Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, III, Bland KI, Chaudry IH. 2005. Cecal ligation and puncture. Shock 24:52. [DOI] [PubMed] [Google Scholar]
- 49. Li P, Cook JA, Gilkeson GS, Luttrell LM, Wang L, Borg KT, Halushka PV, Fan H. 2011. Increased expression of beta-arrestin 1 and 2 in murine models of rheumatoid arthritis: isoform specific regulation of inflammation. Mol. Immunol. 49:64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.