

Abstract
The Francisella tularensis pathogenicity island (FPI) encodes many proteins that are required for virulence. Expression of these genes depends upon the FevR (PigR) regulator and its interactions with the MglA/SspA and RNA polymerase transcriptional complex. Experiments to identify how transcription of the FPI genes is activated have led to identification of mutations within the migR, trmE, and cphA genes that decrease FPI expression. Recent data demonstrated that the small alarmone ppGpp, produced by RelA and SpoT, is important for stabilizing MglA/SspA and FevR (PigR) interactions in Francisella. Production of ppGpp is commonly known to be activated by cellular and nutritional stress in bacteria, which indicates that cellular and nutritional stresses act as important signals for FPI activation. In this work, we demonstrate that mutations in migR, trmE, or cphA significantly reduce ppGpp accumulation. The reduction in ppGpp levels was similar for each of the mutants and correlated with a corresponding reduction in iglA reporter expression. In addition, we observed that there were differences in the ability of each of these mutants to replicate within various mammalian cells, indicating that the migR, trmE, and cphA genes are likely parts of different cellular stress response pathways in Francisella. These results also indicate that different nutritional and cellular stresses exist in different mammalian cells. This work provides new information to help understand how Francisella regulates its virulence genes in response to host cell environments, and it contributes to our growing knowledge of this highly successful bacterial pathogen.
INTRODUCTION
Francisella tularensis is a Gram-negative facultative intracellular pathogen that causes the human disease tularemia. F. tularensis infects a wide variety of hosts, including mammals, birds, fish, and arthropods (1–3). Although the most common route of infection for humans is cutaneous, inhalation of as few as 10 organisms can cause mortality (4–6). Due to a low infective dose, ease of aerosolization, and high mortality rate for untreated infections of F. tularensis subsp. tularensis, the bacterium has been classified as a tier 1 (category A) select agent (7–9).
F. tularensis has been identified within several environmental niches, including water, wet soil, animal carcasses, insects, and amoebas (10, 11). In order to grow within a mammalian host, F. tularensis must adapt as the organism moves from environmental sites to the extracellular space, phagosome, and, finally, the cytosol within the host. Successful infection also requires that the organism adapt to several cell types, including lung epithelia, endothelia, neutrophils, and macrophages (12–15). Each of these microenvironments poses different challenges to the survival of the organism, such as from complement proteins, antimicrobial peptides, cell-damaging reactive oxygen species, extremes of pH, and even competition for nutrients in the cytosol of the cell (16–18). The success of F. tularensis as a pathogen is dependent upon its ability to respond to these various stresses and challenges.
F. tularensis has several important virulence factors that equip it to deal with these growth and survival challenges. One critical set of genes is encoded in duplicate by the 30-kb Francisella pathogenicity island (FPI), which consists of 17 conserved open reading frames (19). Using mutational analysis, the FPI genes have been shown to be required for the escape of Francisella from the phagosome of the infected cell, which is essential for virulence (19, 20). Expression of FPI genes is regulated transcriptionally by the interactions of MglA, SspA, and FevR with RNA polymerase (21). The small molecule ppGpp, a stress alarmone that is synthesized by both RelA and SpoT, promotes the interaction of FevR with the MglA-SspA-RNA polymerase complex to increase gene expression (22, 23). In addition to the FPI genes, this regulatory complex also activates ∼80 other genes that have a wide array of physiological functions (24). Genetic screens by our group (25) and others (22) demonstrated that migR, trmE, and cphA play a role in FPI gene expression. Genetic disruption of migR, trmE, or cphA reduces, but does not completely eliminate, FPI gene expression, which contrasts with single mutations in mglA, sspA, or fevR, which virtually eliminate all expression of the FPI genes.
In general, sparse information is available for the Francisella migR, trmE, and cphA genes. The F. tularensis CphA protein is ∼50% similar to the CphA protein of cyanobacteria, which is involved in making the nitrogen reserve molecule cyanophycin (i.e., granules of arginine, lysine, or asparagine) (26). It is unknown if Francisella produces cyanophycin; however, the genome of Francisella does not appear to contain cphB, which is a cyanophycinase (27). The F. tularensis trmE gene product is ∼50% similar to the Escherichia coli TrmE protein. TrmE is a GTPase that has previously been identified in other bacteria to be involved in tRNA modification, which is important for maintenance of the reading frame during translation (28, 29). TrmE has also been shown to be involved in GadE-dependent acid resistance during growth in the presence of glucose (30–32), although its function in Francisella is currently unknown. BLAST searches with the Francisella migR gene indicate that the gene encodes a protein of unknown function, although it possesses two putative domains: an AMP-binding domain and a domain involved in fatty acid transport. Although these gene products modulate gene expression, they do not possess any detectable characteristics of transcription factors that could be identified by homology searches. While many questions as to how the products of the migR, trmE, and cphA genes process signals important to Francisella virulence and physiology remain, a picture is beginning to emerge that F. tularensis uses a stringent response network that includes these genes to respond to nutritional stresses.
It has been shown in several bacteria that responses to nutritional stresses include production of ppGpp via both RelA and SpoT (33, 34). In E. coli, both fatty acid starvation and glucose starvation are signals that activate SpoT to synthesize ppGpp (35, 36). RelA is known to respond specifically to the ribosome stalling caused by amino acid starvation by synthesizing ppGpp (37). Since the small alarmone ppGpp has been implicated in virulence regulation (38) and the bioinformatic data suggest that migR, trmE, and cphA are possibly involved in nutritional signaling, we hypothesized that the migR, trmE, and cphA genes are part of the Francisella system that senses and responds to stresses. To this end, we examined whether these genes are involved in increasing the levels of ppGpp that lead to activation of virulence gene transcription so that the organisms adapt to and survive within various cellular environments. We present data that suggest that a successful Francisella tularensis infection relies on the ability to adapt to various stresses to grow within host cells and to thrive within the host environment. This information should be useful to develop therapeutics for tularemia by disrupting ppGpp synthesis or to develop a vaccine with mutants that are able to replicate in some cells types, but not others, so that infection is limited.
MATERIALS AND METHODS
Bacterial strains, plasmid construction, and growth conditions.
F. tularensis LVS and F. tularensis Schu S4 were routinely cultured on modified Mueller-Hinton (MMH) plates (Acumedia) or in Mueller-Hinton broth supplemented with 1% (wt/vol) glucose, 0.025% ferric pyrophosphate, and 2% IsoVitaleX or in Chamberlain's defined medium (CDM), which was prepared as described previously and adjusted to the desired pH (39). All strains were grown with aeration at 37°C. When needed, kanamycin or spectinomycin was added to the growth medium at a concentration of 25 μg per ml. Serine hydroxamate was used at a 10-μg/ml concentration, when needed. Plasmid pMF108, derived from pJC84, carries the sacB gene and both kanamycin and spectinomycin resistance genes (40). Complementation vectors were generated by PCR of the target gene and its corresponding promoter and cloning of the fragments into the F. tularensis-replicating plasmid pBB103. All work with F. tularensis Schu S4 was performed within the Carver College of Medicine Biosafety Level 3 (BSL3) Core Facility, and all experimental protocols were reviewed for safety by the BSL3 Oversight Committee of the University of Iowa Carver College of Medicine. Recombinant DNA work with F. tularensis Schu S4 was approved by the Institutional Biosafety Committee.
Deletion constructs and strains.
Allelic replacement vectors were constructed to create in-frame deletion mutations of each coding region: pMF108 for trmE and pMF109 for cphA. Primers were designed to amplify 1 kb of DNA sequence both upstream and downstream of the gene being targeted for deletion. The upstream and downstream DNA fragments were cloned so that they flanked a spectinomycin resistance gene. Plasmids were introduced into F. tularensis LVS by electroporation at 2.5 kV, 25 μF, and 600 Ω before outgrowing the strains for 3 h at 37°C with aeration. Bacteria were plated on plates with kanamycin to select for transformed clones that contained the plasmid integrated at the targeted gene. Colonies were passaged twice in MMH medium with no antibiotic selection, and then dilutions were plated onto MMH agar with 8% sucrose and spectinomycin to select for the second crossover event that yielded a gene replacement mutant.
F. tularensis β-galactosidase assays.
Quantitation of β-galactosidase activity was performed using the method of Miller (41). Strains were grown to mid-log phase (optical density at 600 nm [OD600], ∼0.5 to 0.6) or late log phase (OD600, ∼0.9 to 1.2), and β-galactosidase assays were performed in triplicate for each culture.
ppGpp assay and TLC.
Intracellular levels of ppGpp were determined as previously described (42). Briefly, strains were grown in MMH broth with aeration at 37°C overnight, diluted to an OD600 of ∼0.2, and then allowed to grow for 1 h. Approximately 109 cells were pelleted by centrifugation and resuspended in 250 μl MOPS (morpholinepropanesulfonic acid) containing 55 mM mannose, 1 mM MgSO4, 0.25 mM CaCl2, 19 mM glutamic acid, and 0.0004 mM biotin (43). [32P]KH2PO4 (25 μCi; PerkinElmer) was added, and intracellular ppGpp was labeled with 32P by incubating for 1 h with aeration at 37°C. Nucleotides were extracted from the bacteria with 2 M formic acid at 4°C for 30 min. The samples were run on a polyethyleneimine cellulose thin-layer chromatography (TLC) plate (Sigma-Aldrich) using 1.5 M KH2PO4 (pH 3.4) buffer, and samples were imaged with a Packard electronic autoradiograph instant imager.
Quantitative mammalian cell growth assays.
Monocyte-derived macrophages (MDMs) were isolated from blood and grown in RPMI 1640 (Lonza). Approximately 105 macrophages were seeded into each well of 24-well dishes and infected with Francisella strains at a multiplicity of infection (MOI) of 20:1 for 1 h in the presence of 10% heat-inactivated human serum. After 1 h, MDMs were washed three times with phosphate-buffered saline (PBS) before adding RPMI 1640 for the remainder of the infection. Cell monolayers were lysed at 1 h or 24 h with 1% saponin, followed by dilution plating to quantitate the number of bacteria within each monolayer. For experiments with HEp-2 and A549 tissue culture cells, cells were seeded at ∼3 × 105 cells before they were infected at an MOI of 100:1 for 3 h in RPMI 1640 (Invitrogen) with 10% fetal bovine serum (FBS) and minimal essential medium (Invitrogen) with 10% FBS, respectively. Francisella organisms were allowed to enter these cells for 3 h, since uptake was relatively low compared to uptake by phagocytic MDMs. At 3 h postinfection, cells were washed 3 times with PBS, incubated with tissue culture medium with 30 μg/ml of gentamicin for 1 h, and then washed 3 times with PBS to remove the gentamicin. Infected cells were lysed at 4, 12, 16, or 24 h with 1% saponin, and dilution plating was performed to enumerate the bacteria.
Immunofluorescence staining and microscopy.
MDM, A549, and HEp-2 cells were seeded onto coverslips in 24-well dishes and then infected following the protocol described above. Cells were placed in blocking buffer overnight. On the following day, the coverslips were fixed using 10% formalin for 15 min, permeabilized with acetone-methanol (1:1) for 5 min, washed with PBS, and then stored in blocking buffer until immunostaining was performed. Immunostaining was performed using rabbit anti-F. tularensis serum diluted 1:20,000 (Becton, Dickinson) and mouse anti-human LAMP-1 hybridoma supernatants (clone H4A3, 1:20) obtained from the Developmental Studies Hybridoma Bank at the University of Iowa. Secondary antibodies were acquired from the University of Iowa Central Microscopy Research Facility and used at a dilution of 1:500. Samples were viewed with a Zeiss LSM 710 confocal microscope.
Murine infections.
BALB/c female mice 6 to 8 weeks of age were purchased from the National Cancer Institute (NCI). To evaluate the virulence of the F. tularensis LVS mutant strains, groups of five mice were infected intranasally with a 50-μl inoculum volume and were monitored for up to 14 days postinfection for signs of disease. Doses of 103 CFU for wild-type LVS and ∼104 and ∼105 CFU for mutant strains were estimated from OD600 readings and were confirmed by dilution and plating on agar plates. All mouse experiments were performed according to procedures approved by The University of Iowa Institutional Animal Care and Use committee.
RESULTS
Mutation of the F. tularensis migR, trmE, or cphA gene decreases igl gene expression.
Our group has previously demonstrated that a mutation in the migR gene reduces expression of an iglB-lacZ reporter as well as iglC transcript levels, as measured by reverse transcription-PCR (25). Charity et al. have also shown that disruption of the Francisella trmE or cphA gene significantly reduces expression of an F. tularensis LVS iglA-lacZ reporter (22). To examine how these genes (migR, trmE, and cphA) participate in Francisella virulence regulation, we employed homologous recombination to create trmE and cphA mutations in F. tularensis LVS (LVS ΔtrmE, LVS ΔcphA). These strains, in addition to the LVS migR::TrgTn mutant (where TrgTn represents the TargeTron intron insertion) previously created in the Jones lab, are the subject of the experiments presented here. No difference in growth in either MMH broth or CDM broth was shown between the three mutant strains and the wild-type strain (data not shown).
To demonstrate that the LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA strains had decreased levels of FPI gene expression, a reporter plasmid (pBB125) containing iglA-lacZ was introduced into each mutant strain. As previously published, the LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA strains had significantly decreased levels of expression from the iglA promoter, as measured from an iglA-lacZ plasmid reporter (Fig. 1). While the migR, trmE, and cphA mutants each had significantly reduced levels of iglA-lacZ expression (10 to 15% of that of the wild type), the level of expression of the iglA-lacZ plasmid reporter in negative-control mutant LVS fevR::TrgTn approached zero (<5% of that of the wild type).
Fig 1.

The LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA mutants have decreased iglA reporter expression. Mutations in migR and fevR were constructed by TargeTron (TrgTn) insertion (Sigma). Mutations in trmE and cphA were generated by homologous recombination. An iglA-lacZ reporter plasmid was cryotransformed into each of the F. tularensis LVS mutants, and β-galactosidase assays were performed to compare gene expression in each of the mutant strains to that in the wild type (Wt). The difference between iglA-lacZ expression in each mutant strain and the LVS parent was statistically significant (P < 0.001).
Mutation of the F. tularensis migR, trmE, or cphA gene decreases the levels of ppGpp.
Recent work has demonstrated that the Francisella relA and spoT genes play an important role in activating FPI gene expression (22, 23). RelA and SpoT together are responsible for producing the pools of the signaling molecule ppGpp in bacteria (33). In Francisella, intracellular ppGpp functions by strengthening the interactions of the FevR transcriptional activator with the MglA-SspA heterodimers and the α subunits of RNA polymerase to activate Francisella FPI gene expression (22, 23). Mutation of migR, trmE, and cphA reduces FPI gene expression, similar to the FPI gene disruption observed in an LVS relA spoT double mutant (22), which is also defective for ppGpp production. Accordingly, we examined whether the LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA strains had altered ppGpp levels using a TLC assay. The parent F. tularensis LVS strain produced large amounts of ppGpp, while LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA had very low levels of production of ppGpp (Fig. 2). In comparison to the quantity of ppGpp synthesized by the wild-type strain, which was arbitrarily set to 100%, LVS migR::TrgTn produced 5.9%, LVS ΔtrmE had 22%, and LVS ΔcphA had 3.3% of the radiolabeled counts (Fig. 2). As expected, the LVS fevR::TrgTn mutant had essentially the same amount of ppGpp as the LVS parent strain, since the FevR protein is believed to interact with ppGpp but not modulate intracellular pools of the alarmone (Fig. 2). F. tularensis mutant strains carrying plasmids with the appropriate intact gene for complementation displayed significant increases in ppGpp production (50 to 70% of that observed in the parent strain). Together, these data indicate that the LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA strains most likely have decreased levels of FPI gene expression because mutations in these genes affect sensory pathways in F. tularensis that lead to synthesis of ppGpp.
Fig 2.

LVS mutants with mutations in migR, trmE, and cphA have reduced ppGpp levels. Radiolabeled phosphate was added to each of the F. tularensis strains for 1 h, and cell lysates were run on a TLC plate to visualize ppGpp. A graphical representation of ppGpp radioactive counts after being normalized to LVS ppGpp counts, which were arbitrarily set to 100, is shown. All mutants had decreased ppGpp accumulation compared to that of wild-type strain LVS (P < 0.001). Complemented strains (designated LVS migRc, LVS trmEc, and LVS cphAc) produced significantly increased amounts of ppGpp compared to their mutant counterparts.
F. tularensis LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA have altered growth rates in mammalian cells.
Previous work from our lab characterizing the LVS migR::TrgTn mutant identified a significant growth defect within MDMs, but growth of the strain was the same as that of the LVS parent in the HEp-2 epidermoid carcinoma (larynx) tissue culture line (25). Since the F. tularensis LVS ΔtrmE and LVS ΔcphA strains are similar to the LVS migR::TrgTn mutant with respect to gene expression and modulation of ppGpp levels, we wondered whether these strains would have intracellular growth patterns similar to the pattern for the LVS migR::TrgTn strain or growth patterns that were unique to each mutant and the cell line in which it was growing. As expected, wild-type LVS grew approximately 100-fold in MDMs over the course of 24 h, while LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA had growth defects, reaching only 20%, 37%, and 14% of wild-type growth, respectively (Fig. 3A and B). The differences in intracellular growth are unlikely due to innate differences in the growth rates of the mutant strains, since in vitro growth experiments revealed no differences between these mutants and the LVS parent (data not shown). The LVS fevR::TrgTn mutant was used as a negative control and did not replicate within these host cells, as it was unable to escape the phagolysosome (Fig. 3A and B). Fold growth was calculated for each strain by dividing the number of organisms of each strain within cells at 24 h by the number of organisms that had gained entry into the cells by 1 h. The growth of the LVS parent was standardized to 100%, and the ratio of the growth of each mutant to that of the wild type is reported. In general, for these host cells and all others presented in this work, there were no significant differences in the uptake of the strains examined.
Fig 3.

LVS migR, trmE, and cphA mutants have altered growth rates in MDMs, HEp-2 cells, and A549 cells compared to the LVS wild type. Cells were infected for either 1 h (MDMs) or 4 h (HEp-2 and A549 cells), and gentamicin treatment was performed to kill extracellular bacteria. Cells were lysed with 1% saponin and serially diluted to enumerate intracellular bacteria. (A, B) Infection of MDMs (MOI, 20:1) measured at 1 h and 24 h; (C, D) infection of HEp-2 cells (MOI, 100:1) measured at 4 h and 24 h; (E, F) infection of A549 cells (MOI, 100:1) measured at 4 h and 24 h. — · — · —, LVS ΔcphA.
In addition to human MDMs, we examined the ability of the Francisella mutants to grow in the epithelial HEp-2 cell line. We observed a different pattern of growth of these strains in HEp-2 cells than in MDMs. The LVS parent strain replicated approximately 500-fold in HEp-2 cells over the 24-h infection period. Both the F. tularensis LVS migR::TrgTn and the F. tularensis LVS ΔtrmE strains replicated significantly (75% to 81% of the replication of the wild type) (Fig. 3C and D). However, different results were obtained with the LVS ΔcphA mutant. The LVS ΔcphA strain replicated to a level only 12% of that observed with the LVS parent (Fig. 3B and C), indicating that the cphA mutation attenuated the ability of the strain to respond to conditions within HEp-2 cells, significantly reducing its growth. The negative-control strain LVS fevR::TrgTn essentially failed to replicate within the HEp-2 cells.
Since the phenotypes of the F. tularensis LVS mutants varied between MDM and HEp-2 cells, we examined their growth phenotypes in another epithelial cell type, A549. A549 is an adenocarcinomic human alveolar basal epithelial cell line (44). Similar to growth in MDMs and HEp-2 cells, the LVS parent had strong growth over 24 h in A549 cells (∼350-fold) (Fig. 3E and F). Similar to the growth in the HEp-2 cells, the LVS ΔtrmE mutant had 76% of the replication of the wild type. However, both the LVS migR::TrgTn and LVS ΔcphA strains had poor replication (20% to 23%) in the A549 cells (Fig. 3E and F). The growth pattern of the LVS migR::TrgTn mutant in the A549 cells in this study differed from that which we published previously (25). Previously, we observed no growth defect with this strain in A549 cells (25), while in this study we saw a significant growth defect. Previously, we grew the bacteria in MMH broth to late log phase (25), while in this study we grew the bacteria in CDM to an early log phase. We believe that the differences in growth conditions are the most likely explanation for the variation in the intracellular growth of this strain that we observed in A549 cells. The growth pattern of the three mutants in A549 cells is different from that observed in either MDMs or HEp-2 cells. According to these results, mutation of either migR or cphA significantly reduces the ability of LVS to grow in A549 epithelial cells, establishing that these genes are directly or indirectly important for the growth of F. tularensis in these cells. As a negative control, the LVS fevR::TrgTn strain failed to replicate within A549 cells.
Microscopic analysis reveals that the LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA strains grow within different host cells with altered kinetics.
In order to gain more insight into the interactions of these mutants with host cells, confocal microscopy experiments were employed. Francisella-infected cells (MDMs, HEp-2 cells, and A549 cells) were fixed at 4 and 24 h postinfection and probed with antibodies to visualize the position of the organisms within the cells. As shown previously (45, 46), at 4 h after LVS infection, MDMs contained 1 to 2 bacteria per cell that had escaped the phagosome into the cytosol, where replication was already beginning (Fig. 4) (45, 46). By 24 h, the wild-type strain was replicating robustly in these cells. A similar pattern was observed for LVS in epithelial cell lines (HEp-2 and A549 cells). LVS organisms gained access to the cytosol by 4 h and were growing rapidly, nearly filling the cytosolic space by 24 h (Fig. 4). As a control for bacteria that were retained within the phagosome, infection of MDMs with LVS fevR::TrgTn demonstrated that these organisms were surrounded by LAMP-1 at 4 and 24 h and did not appear to replicate (Fig. 4).
Fig 4.
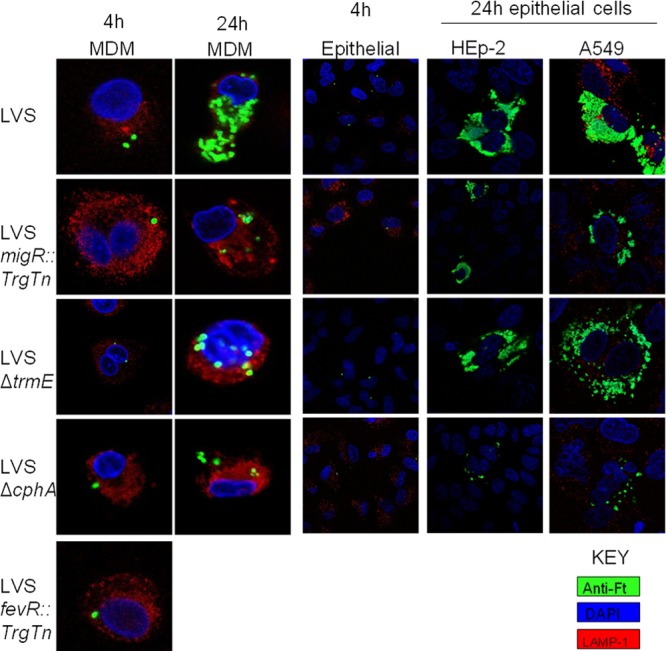
Confocal microscopy reveals that LVS mutants with mutations in migR, trmE, and cphA have altered phagosomal escape properties and cytosolic growth differences. Cells grown on coverslips were infected for either 4 h or 24 h, followed by immunofluorescence with rabbit anti-Francisella (anti-Ft) antibody, mouse anti-human LAMP-1 hybridoma supernatants, and DAPI (4′,6-diamidino-2-phenylindole). Coverslips were imaged on a Zeiss 710 confocal microscope.
The LVS migR::TrgTn mutant was frequently observed to colocalize with LAMP-1 in MDMs at 4 h postinfection, indicating that the bacteria were still within the phagolysosome (Fig. 4). At 24 h, the LVS migR::TrgTn mutant had a mixed population, with some free within the cytosol and others residing within LAMP-1-positive (LAMP-1+) phagolysosomes (Fig. 4). For epithelial cells, uptake of LVS migR::TrgTn was comparable to that for the LVS parent, in that 1 to 2 bacteria were observed in each cell after 4 h of infection. At 24 h, the organisms had multiplied significantly within the cytosol of HEp-2 cells but much less well in A549 cells (Fig. 4).
We performed a similar set of experiments with the LVS ΔtrmE mutant. At 4 h postinfection, there were 1 to 2 LVS ΔtrmE organisms per cell in all three cell types. The LVS ΔtrmE organisms observed in MDMs were still in phagosomes, as they were LAMP-1+, while at 24 h postinfection, some cytosolic replication had occurred (Fig. 4). However, at this time point, many of the bacteria within individual cells were still within a phagolysosome (Fig. 4). Within HEp-2 and A549 epithelial cells, LVS ΔtrmE was observed to replicate in the cytosol to levels nearly indistinguishable from those in wild-type LVS.
The intracellular growth data shown in Fig. 3 indicated that the LVS ΔcphA strain had the most severe growth defect within all three cell types examined, and this was supported by the confocal images of LVS ΔcphA organisms within each cell type. At 4 h postinfection, there were 1 to 2 bacteria per cell, and these were contained within phagolysosomes (Fig. 4). At 24 h postinfection, the LVS ΔcphA organisms appeared to have replicated very little or not at all in each of the cell types (Fig. 4), indicating that the cphA mutation significantly disrupts the ability of Francisella to escape the phagolysosome environment in multiple cells.
Quantitative analysis reveals that each F. tularensis mutant displays host cell growth defects unique to the mutant.
While examining the various Francisella strains by confocal microscopy, we observed that the mutant strains had a wide spectrum of growth within individual cells (MDMs, HEp-2 cells, or A549 cells). To more carefully characterize the replication abilities of each of these strains, the bacteria within at least 100 infected cells were enumerated at 24 h postinfection and compared to the number in the wild type. In all cell types that were examined, >70% of the cells infected with the wild-type strain had more than 20 organisms (for MDMs) or 36 organisms (for HEp-2 and A549 cells) organisms (Fig. 5). The remaining wild-type-infected cells mostly contained only 1 to 5 organisms. In contrast, when the mutant strains (LVS migR::TrgTn, LVS ΔtrmE, LVS ΔcphA) were examined within MDMs, 40 to 50% of the host cells contained 1 to 5 organisms per cell and approximately 30% had 10 to 20 organisms per cell (Fig. 5). Only 10 to 20% of the MDMs infected with the mutant strains had more than 20 organisms per cell, in contrast to the findings for the wild-type strain, for which >70% of the cells had more than 20 organisms. The growth patterns of the LVS migR::TrgTn and LVS ΔtrmE mutants in HEp-2 cells were generally similar to those observed for the wild-type strain, although ∼95% of the HEp-2 cells infected with the wild-type strain had >36 organisms per cell, while 70 to 80% of HEp-2 cells infected with the migR and trmE mutants had >36 organisms per cell. The other ∼20 to 25% of HEp-2 cells infected with either of these mutants had fewer organisms per cell (6 to 36 organisms), which accounts for the overall lower growth of the migR and trmE mutant strains in HEp-2 cells. In contrast, the cphA mutant was significantly reduced in its ability to grow in HEp-2 cells, with approximately 30% of the cells containing 1 to 5 organisms, 40% of the cells containing 6 to 36 organisms, and 30% of the cells containing >36 organisms. This indicates that the cphA mutant has a significant growth defect in HEp-2 cells, even though the migR and trmE mutants were relatively unaffected in their ability to grow in these cells. Interestingly, the quantitative data indicated that the mutants had a different pattern in the A549 cells. In these cells, the migR and cphA mutants were virtually unable to grow, as 75 to 90% of the cells infected with these strains had only 1 to 5 organisms, with only ∼8% of ΔcphA-infected cells or ∼20% of migR::TrgTn-infected cells containing more than 5 organisms. In contrast, the wild type and trmE mutant had very similar growth phenotypes in A549 cells, where 15 to 20% of the cells had 1 to 5 organisms and 50 to 70% of the cells had >36 organisms.
Fig 5.

Quantitative analysis of the cellular growth of F. tularensis mutants demonstrates that each strain has a unique pattern. MDMs, HEp-2 cells, and A549 cells were infected with LVS, LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA using the standard intracellular growth assay. At 24 h of growth, cells were fixed and the samples were prepared for confocal microscopy. The number of bacteria per infected cell was determined for each of the three cell lines via confocal microscopy, and the data are presented in the graphs. Greater than 100 infected cells were counted per strain and cell type. (A) MDMs; (B) HEp-2 cells; (C) A549 cells.
Since the LVS mutants have reduced FPI gene expression, a likely explanation for the reduced intracellular growth rates of these strains is a reduced efficiency in escaping the phagosome, as phagosome escape is the best-characterized function of the FPI genes. However, it is also possible that the strains are altered in their ability to grow in the cytosol. To gain insights into the growth defect in host cells, we performed a set of experiments in HEp-2 cells with LVS, LVS fevR::TrgTn, LVS migR::TrgTn, LVS ΔtrmE, and LVS ΔcphA and determined how many organisms were present in the cells at 12 h and 16 h, in addition to the numbers at the 4-h and 24-h time points at which measurements are typically made. Interestingly, each of the LVS mutants had a substantial delay before they began to grow significantly (∼12 h for the mutants versus 4 h for the wild type), but following this delay in growth, each of the mutants appeared to be able to grow as fast as the parent LVS strain, as the slopes of the growth curves were essentially the same (Fig. 6). We believe that these data are most consistent with the F. tularensis LVS mutants being delayed in their ability to escape the phagolysosome, although a phagosome integrity assay could be performed to determine more precisely when these mutants escape the phagosome. The LVS fevR::TrgTn strain did not replicate appreciably at any of the time points examined.
Fig 6.

LVS migR, trmE, and cphA mutants have delayed replication in HEp-2 cells compared to that in the LVS parent strain. HEp-2 cells were infected at an MOI of 100:1 for 4 h, followed by gentamicin treatment for 1 h to kill extracellular bacteria. Infected cells were then lysed at either 4, 12, or 16 h with 1% saponin and serially diluted to enumerate intracellular bacteria.
Collectively, these data provide insights into the growth properties of Francisella mutants that have altered FPI expression and indicate that intact sensing pathways that modulate ppGpp levels are critical for the ability of Francisella to lyse the phagosome and subsequently grow in the cytosolic environment.
Addition of serine hydroxamate and/or pH variation in the growth medium modulates expression of the iglA promoter in both F. tularensis LVS and Schu S4.
To provide additional evidence that ppGpp is involved in stimulating expression of the genes under the control of FevR, MglA, and SspA and to further understand FPI regulation, we examined if exogenous methods of inducing cellular and nutritional stress in Francisella could activate the iglA promoter. Both F. tularensis LVS and Schu S4 were grown in CDM to test the effects of stresses (amino acid starvation and pH variations) on iglA-lacZ expression. For LVS, when the medium pH was acidified to pH 5.5 from pH 7.0, there was a 2-fold reduction in iglA-lacZ expression, while increasing the medium pH to 8.5 induced iglA-lacZ expression 2-fold (Fig. 7A). Thus, we can alter iglA gene expression 4-fold by changing the pH of the medium. We also manipulated ppGpp levels by adding to the growth medium serine hydroxamate, which simulates amino acid starvation (47). As shown in Fig. 7A, the presence of serine hydroxamate induced iglA expression ∼2.5-fold, providing additional indirect evidence that an increase in ppGpp levels due to amino acid starvation stress induces FPI gene expression. Interestingly, we found that the effects of alkaline pH and serine hydroxamate were additive and together increased FPI transcription 8-fold or more. To date, this is the highest induction of Francisella FPI gene expression reported. Under all conditions tested, no differences were observed using a control plasmid carrying a non-FevR-regulated lacZ reporter (data not shown). Furthermore, we determined if either pH or serine hydroxamate acted as a signal for the LVS migR::TrgTn, LVS ΔtrmE, or LVS ΔcphA mutant. Strains carrying iglA-lacZ reporters were grown in media with various pHs or medium with or without serine hydroxamate before performing Miller assays. Each mutant still exhibited a response to various pHs and serine hydroxamate, although with a significantly reduced response compared to that of the parent strain (data not shown).
Fig 7.

Alkaline pH and serine hydroxamate (SHX) increase iglA-lacZ reporter expression and modulate ppGpp levels in F. tularensis. Triplicates of cultures of either LVS or Schu S4 carrying a plasmid with iglA-lacZ were grown to early log phase, and β-galactosidase assays were performed. (A) LVS was grown in CDM poised at pH 5.5, 7.0, or 8.5 with or without serine hydroxamate, and the level of β-galactosidase activity was compared to the level in CDM at pH 5.5. (B) F. tularensis LVS was grown in CDM poised at either pH 5.5, 7.0, or 8.5, bacterial samples were radiolabeled, and ppGpp was separated by TLC and quantitated. The amount of ppGpp produced by LVS at pH 5.5 was arbitrarily set to 1, and the other samples were compared against this sample as the standard. (C) F. tularensis LVS was grown in CDM at pH 7.0 with and without serine hydroxamate, and ppGpp accumulation was separated by TLC and quantitated. The amount of ppGpp produced by LVS without serine hydroxamate was set to 1, and the sample with serine hydroxamate was compared against this as the standard. (D) Schu S4 with the iglA-lacZ reporter plasmid was grown in CDM poised at pH 5.5, 7.0, or 8.5 with or without serine hydroxamate. The level of β-galactosidase activity under each growth condition was compared to the level of β-galactosidase activity in CDM at pH 5.5.
We would predict that growth in basic pH or in the presence of serine hydroxamate would create growth stress for the bacteria, which would be manifested by increased ppGpp production. In order to test this hypothesis, TLC was performed with extracts of bacteria that were grown in CDM poised at pH 5.5, 7.0, and 8.5 and in the presence of serine hydroxamate at pH 7.0. As the pH became more basic, an increase in ppGpp which correlated with an increase in iglA-lacZ expression was observed (Fig. 7B). As the pH increased from 5.5 to 7.0, there was an ∼2.5-fold increase in ppGpp, and in a comparison of pH 5.5 to 8.5, there was an ∼3.5-fold increase in ppGpp production. Similarly, in the presence of serine hydroxamate, ppGpp increased ∼3.0-fold (Fig. 7C). These data further demonstrate the role of ppGpp in regulation of the FPI.
As we are most interested in F. tularensis Schu S4 virulence and pathogenesis, we introduced the iglA-lacZ reporter into F. tularensis Schu S4 and performed a set of experiments similar to those performed with the LVS strain. Importantly, the stress signals that modulated LVS iglA-lacZ expression also activated Schu S4 iglA-lacZ expression. The F. tularensis Schu S4 strain with the iglA-lacZ reporter, grown in CDM at pH 5.5, gave the lowest expression, while growth of Schu S4 in CDM at pH 8.5 increased iglA-lacZ expression ∼3-fold above that seen at pH 5.5 (Fig. 7B). Similar to LVS, addition of serine hydroxamate to the Schu S4 growth medium (pH 8.5) increased reporter expression 2- to 2.5-fold (Fig. 7B). The induction of the lacZ reporter was dependent upon the iglA promoter, as the lacZ vector, without the iglA promoter, did not respond to either pH variation or serine hydroxamate (data not shown). As with the LVS experiments, the effect of pH 8.5 and serine hydroxamate was to induce the iglA promoter in an additive fashion (Fig. 7B). These data demonstrate that nutritional and cellular stresses significantly induce virulence gene expression in both the LVS and virulent Schu S4 F. tularensis strains.
Quantitation of virulence in murine infections.
As we have shown, F. tularensis LVS strains with mutations in trmE, migR, or cphA display significant growth defects in different cell types in vitro. We extended our characterization of these strains by examining the ability of each mutant to cause disease in a mouse model of tularemia. Groups of BALB/c mice were infected intranasally with either wild-type LVS (3 × 103 CFU), LVS migR::TrgTn (2.1 × 104 and 2.1 × 105 CFU), LVS ΔtrmE (1.4 × 104 and 1.4 × 105 CFU), or LVS ΔcphA (2.1 × 104 and 2.1 × 105 CFU). Following inoculation, the health of the mice was followed for 14 days postinfection. All mice infected with wild-type strain LVS succumbed to death on day 6 postinfection (Fig. 8). Mice infected with LVS migR::TrgTn or LVS ΔcphA did not die, nor was inflammation (fur ruffling or conjunctivitis) observed, indicating that the 50% lethal dose (LD50) for both of these strains is above 1.4 × 105 CFU. Mice infected with either dose of the LVS ΔtrmE mutant displayed ruffled fur and minor conjunctivitis, which resolved by day 10 postinfection. However, at the highest dose administered (1.4 × 105 CFU) for the LVS ΔtrmE-infected mice, three of the five mice succumbed to the infection (Fig. 8). Using these data, the LD50 for the LVS ΔtrmE strain was calculated to be 9.5 ×104 CFU (48).
Fig 8.

Evaluation of murine virulence of F. tularensis migR, trmE, and cphA mutants. Mice were infected with 3,000 CFU of wild-type strain LVS or with LVS migR::TrgTn, LVS ΔtrmE, or LVS ΔcphA mutant strains with two separate doses to evaluate the effect of each mutation on the virulence of the strain. As can be seen, all mice infected with 3,000 CFU of LVS died by day 6 postinfection. The LD50 values of the migR and cphA mutants were >1 × 105 CFU, since no mice died at the highest dose. The calculated LD50 value for the trmE mutant was 9.5 × 104 CFU, as no mice died at a dose of 14,000 CFU but 60% of mice died at the 140,000-CFU dose.
DISCUSSION
While genes on the Francisella pathogenicity island were identified as having an essential virulence function some 10 years ago, unraveling the details of the regulation of the FPI has been difficult. Francisella is unusual in that analysis of the genome reveals few, if any, typical bacterial regulators. The chromosome of Francisella appears to encode only one alternate sigma factor, RpoH, and no complete two-component systems. Portions of two two-component systems, PmrA and QseC, are present in virulent Francisella strains, but neither of these has a partner that can be identified via homology searches. Published work has demonstrated that the PmrA protein of F. tularensis subsp. novicida plays a role in activating FPI expression (49). However, the PmrA protein does not regulate all of the genes in the FevR/MglA/SspA regulon, and it regulates other genes not part of the FevR/MglA/SspA regulon (49). Mutation of the qseC gene has been shown to alter the F. tularensis lipopolysaccharide, which makes the organism sensitive to complement and reduces the ability of the organism to survive in macrophages (50). However, no link to FPI gene expression has been described for the qseC gene.
Recent observations, however, have begun to provide insights into how F. tularensis induces expression of the critical FPI virulence genes. Work by Charity et al. (22) has demonstrated that an F. tularensis LVS ΔrelA ΔspoT double mutant has significantly reduced expression of FevR-regulated genes. In addition, the F. tularensis LVS ΔrelA ΔspoT strain has both reduced growth in the J774.A macrophage cell line and a significant reduction in virulence for mice. A second study (23) with F. novicida found that a relA mutant had a slight decrease in J774.A growth but also saw a significant reduction in the virulence of the F. novicida strain for mice.
The results from our iglA-lacZ reporter experiments indicate that mutations in the Francisella genes migR, trmE, and cphA significantly decrease FPI gene expression. Further, the TLC data demonstrate that strains with mutations in these genes have reduced ppGpp production, presumably because each of the mutations has disrupted a pathway that LVS normally uses to increase production of this small alarmone to activate FPI expression. While this group of mutations has similar effects on iglA-lacZ expression and ppGpp production, we observed distinct differences in these strains when we examined their abilities individually to grow in different types of cultured cells. Perhaps not surprisingly, we observed a correlation of the lowest reporter expression with the most profound growth defect within cells. LVS ΔcphA had the largest decrease in reporter expression and also struggled to replicate in all of the cell types, whereas LVS ΔtrmE had the highest level of reporter expression and had a significant growth defect only within MDMs. Together, the data suggest that FPI expression in the wild-type strain ensures highly efficient escape from the phagosome and that a decrease in FPI gene expression reduces the ability of Francisella to efficiently escape the phagosome to grow in the host cell cytosol. Having some FPI expression may allow escape and some replication but does not allow the bacteria to optimally replicate. The microscopy data presented here support this idea because the LVS ΔcphA mutant replicated within some cells, although to low levels.
The use of primary MDMs and epithelial tissue culture cells in these experiments allowed us to make important comparisons between wild-type strain LVS and the Francisella mutants. In primary MDMs, each of the three mutants was significantly defective in the ability to escape from the phagolysosome and grow within the cells, in contrast to the well-established phenotype of LVS, which escapes from the phagosome within 4 h and grows to very high organism loads in the cytosol. The growth environment of the tissue culture cell lines is apparently less restrictive, since the LVS ΔtrmE mutant grew well in both HEp-2 and A549 cells. The LVS migR::TrgTn mutant grew well in HEp-2 cells but poorly in A549 cells; however, the LVS ΔcphA mutant grew poorly in both HEp-2 and A549 cells. These data indicate that F. tularensis LVS escapes from the phagosome and growth within MDMs requires that each of the three genes migR, trmE, and cphA be intact. The same conclusion is not true for HEp-2 cells, as only the mutation in cphA reduced growth by more than 50%, and in A549 cells, mutations in either cphA or migR reduced growth to 20% of the growth in LVS. Our results indicate that these regulatory pathways are employed individually or in concert to ensure the growth of Francisella. One possible application of these findings of the ability to reduce but not eliminate FPI expression may be in the development of F. tularensis vaccine strains. A key feature of an attenuated vaccine strain is the ability to activate the appropriate immune responses by delivering the vaccine strain to the same compartments that the fully virulent pathogen would reach during virulence (51). Reducing FPI expression could accomplish this goal while still significantly attenuating the virulence properties of the strain.
Importantly, the defects in iglA expression, ppGpp production, and growth in cells observed for each of the LVS mutants also correlated with losses in virulence for mice (Fig. 8). Previous workers have demonstrated that a trmE mutant is avirulent by an intradermal route of infection but the virulence of a cphA or migR mutant is unaffected by an intradermal route of infection (22). In contrast, the intranasal LD50 values for the migR and cphA mutants were >1 × 105 bacteria and the LD50 of a trmE mutant was calculated to be 9.5 × 104 bacteria, which represent decreases in virulence of 2 log units or more. These data provide evidence that sensory pathways may be crucial to the virulence strategy by one route of infection and are less important by another route.
Our data that growth stresses (i.e., alkaline pH and serine hydroxamate) activate FPI genes, along with the data demonstrating that migR, trmE, and cphA participate in ppGpp production, suggest that Francisella has evolved pathways to sense growth signals to activate the fevR regulon and FPI gene expression through modulation of ppGpp levels. Efforts to understand how migR, trmE, and cphA function to increase ppGpp levels by looking for similarities to other genes has yielded only a small amount of information. The MigR protein has a conserved AAS_C domain that has been shown in other bacteria to be present in proteins that are involved in fatty acid transport and that interact with acyl carrier proteins (51). In E. coli, it has been demonstrated that the acyl carrier protein interacts with SpoT and acts as a switch for ppGpp degradation or synthesis, depending upon the presence or absence of a bound fatty acid (36). On the basis of this information, a possible explanation for the migR mutant phenotype is that disruption of the F. tularensis migR gene leads to a SpoT protein that is locked into a conformation that only degrades ppGpp. This situation would lead to reduced accumulation of ppGpp. Reduced ppGpp levels would result in lower levels of expression of FevR-dependent genes. The cphA gene is implicated in creating amino acid storage molecules (equal parts aspartic acid and arginine) in cyanobacteria (26). While there is no evidence that cyanophycin is produced by Francisella strains, one hypothesis for the explanation of the phenotypes of this F. tularensis mutant, without more specific information, is that deletion of cphA increases the availability of amino acids for growth, reducing the production of ppGpp and genes activated by high levels of the alarmone. Information about the function of the trmE gene product is also scarce. In E. coli, the trmE gene has been shown to encode a GTPase that is involved in synthesizing the hypermodified nucleoside 5-methylaminomethyl-2-thiouridine, which is found in the wobble position of bacterial tRNAs specific for lysine, glutamic acid, and, possibly, glutamine (30). Disruption of the trmE gene may create a situation where amino acid limitation is not sensed well or perhaps the mutation reduces the ability of RelA to detect ribosome stalling. Either of these situations would lead to a reduced stress response and corresponding decreases in ppGpp levels. Future work will focus on determining whether RelA or SpoT mediates the activity of these genes to modulate ppGpp levels and exert regulatory effects on FPI gene expression.
The data that we have presented here contribute to the current model of FPI regulation (Fig. 9). In this model, SspA and MglA interact with the α subunits of RNA polymerase to form a complex that associates at low affinity with the FevR protein and promoter sequences. The FevR protein has a helix-turn-helix motif, and the protein is proposed to bind to DNA, although its DNA-binding activity is still unproven. Under inducing conditions, ppGpp concentrations increase due to synthesis by RelA and SpoT (52). Production of ppGpp is regulated by pathways that include the migR, trmE, and/or cphA gene and possibly others. The increasing concentration of ppGpp shifts the equilibrium so that the interactions of MglA/SspA and FevR with RNA polymerase are stabilized. The data shown in Fig. 7 support this model by demonstrating that environmental and nutritional stresses significantly increase FPI (i.e., iglA) gene expression, most likely by increasing the intracellular pools of ppGpp. These data indicate that the virulence strategy that F. tularensis has evolved is aimed at escaping and/or avoiding stressful growth conditions, such as those in a phagosome or a dying host cell.
Fig 9.

Working model for Francisella tularensis FevR-dependent virulence gene regulation. Upon detecting signals from the environment, such as pH, fatty acid stress, or lack of nutrients, sensing pathways that include TrmE, CphA, or MigR act as a signal to the cell to activate ppGpp through either RelA or SpoT. ppGpp then stabilizes the regulatory complex between MglA, SspA, and FevR, which then transcribes target genes, such as those encoded on the FPI.
The activation of virulence genes by ppGpp is not a novel concept and is found in several bacteria (38, 53). It is not surprising that addition of serine hydroxamate increases FPI expression in Francisella tularensis. In other bacteria, it has been shown that serine hydroxamate binds to ribosomes and causes ribosomes to stall. These stalled ribosomes then release bound RelA, which enzymatically generates ppGpp (47). Our data are consistent with this model, as we observed increased ppGpp production after growth in medium containing serine hydroxamate (Fig. 7).
In some pathogenic bacteria, pH has been shown to be a signal that activates virulence gene expression. For example, the transcription of the Shigella flexneri virB gene, which is required for transcription of invasion genes, is decreased by acidic pH and increased by basic pH (54). This regulatory mechanism appears to be part of a larger response to multiple stress conditions that are under the control of the alternative sigma factor, RpoS. An overlapping stress response could explain the regulation of FPI genes by basic pH. For instance, under alkaline conditions iron becomes less soluble, resulting in a stressed growth state (55). Also, high-pH conditions cause indirect effects relevant to high osmolarity, heat shock, and oxidative stress conditions (56). The cumulative result of several simultaneous stresses may be the mechanism by which alkaline pH activates FPI gene expression.
This work contributes to a better understanding of the challenges that Francisella faces to initiate infection. Individual organisms enter a cell via a phagosome, an environment that challenges the bacteria with low pH, oxidative stress, and low nutrient levels as the compartment is converted into a phagolysosome. Engaging its virulence mechanisms, the bacteria escape the maturing phagolysosome to enter the cytosol, where F. tularensis not only survives but grows very rapidly. Presumably, after multiple rounds of replication, host cell nutrients are consumed and waste products produce a basic microenvironment, similar to that observed when Francisella grows in vitro (57). Published microarray data provide additional evidence that the bacteria encounter stressful conditions upon entry into host cells and again at later stages of growth (40). Genes involved in general and oxidative stress (e.g., clpP, dnaK, groEL, groES, and msrA) are significantly upregulated at both 1 and 2 h postinfection (phagosome stage), before decreasing at 4 and 8 h (early cytosolic growth stage). The same stress genes are again induced at 12 h postinfection, with transcription increasing at the 24-h-postinfection time point (40). In general, a similar pattern of regulation of many of the FPI genes was also observed in these microarray experiments (40).
We present evidence here that F. tularensis strains sense stressful environments through the use of pathways that include MigR, TrmE, and CphA. F. tularensis responds to these conditions by increasing intracellular levels of ppGpp, which, in turn, lead to increased transcription of FevR-activated genes, including FPI genes. This information contributes to our understanding of the challenges to which Francisella must respond to cause human disease. In addition, the data indicating that individual mutants defective in ppGpp production grow differently in different host cell types provide new information about how different host cells present different growth challenges to this bacterial pathogen. Finally, this work should be useful in creating potential F. tularensis vaccine strains that retain a portion of their ability to carry out productive interactions with a host, albeit at a significantly reduced level.
ACKNOWLEDGMENTS
We are grateful for use of the University of Iowa Carver College of Medicine Biosafety Level 3 Core Facility as well as the University of Iowa Central Microscopy Research Facility. We thank Simon Dove for sharing of unpublished data and Jean Sippy for technical assistance with TLC experiments and analysis.
Financial support for this work was provided by NIH grants 2PO1 AI044642 (to B.D.J.) and 2U54 AI057160, Project 9 (to B.D.J. and L.-A.H.A.), which funds the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Disease Research. L.-A.H.A. was also supported by National Institutes of Health grant 5PO1 AI044642-11.
Footnotes
Published ahead of print 28 May 2013
REFERENCES
- 1. Kuehn A, Schulze C, Kutzer P, Probst C, Hlinak A, Ochs A, Grunow R. 2012. Tularaemia seroprevalence of captured and wild animals in Germany: the fox (Vulpes vulpes) as a biological indicator. Epidemiol. Infect. 14:833–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morner T. 1992. The ecology of tularaemia. Rev. Sci. Tech. 11:1123–1130 [PubMed] [Google Scholar]
- 3. Nylund A, Ottem KF, Watanabe K, Karlsbakk E, Krossoy B. 2006. Francisella sp. (family Francisellaceae) causing mortality in Norwegian cod (Gadus morhua) farming. Arch. Microbiol. 185:383–392 [DOI] [PubMed] [Google Scholar]
- 4. Elkins KL, Cowley SC, Bosio CM. 2007. Innate and adaptive immunity to Francisella. Ann. N. Y. Acad. Sci. 1105:284–324 [DOI] [PubMed] [Google Scholar]
- 5. Rockx-Brouwer D, Chong A, Wehrly TD, Child R, Crane DD, Celli J, Bosio CM. 2012. Low dose vaccination with attenuated Francisella tularensis strain SchuS4 mutants protects against tularemia independent of the route of vaccination. PLoS One 7:e37752. 10.1371/journal.pone.0037752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McLendon MK, Apicella MA, Allen LA. 2006. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Annu. Rev. Microbiol. 60:167–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Isherwood KE, Titball RW, Davies DH, Felgner PL, Morrow WJ. 2005. Vaccination strategies for Francisella tularensis. Adv. Drug Deliv. Rev. 57:1403–1414 [DOI] [PubMed] [Google Scholar]
- 8. Lukasova E, Cermak P, Smela G, Jedlickova A. 2010. Tularaemia—an overview of the current knowledge. Klin. Mikrobiol. Infekc. Lek. 16:22–27 (In Czech.) [PubMed] [Google Scholar]
- 9. Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773 [DOI] [PubMed] [Google Scholar]
- 10. Foley JE, Nieto NC. 2010. Tularemia. Vet. Microbiol. 140:332–338 [DOI] [PubMed] [Google Scholar]
- 11. Petersen JM, Schriefer ME. 2005. Tularemia: emergence/re-emergence. Vet. Res. 36:455–467 [DOI] [PubMed] [Google Scholar]
- 12. Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. 2007. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect. Immun. 75:1034–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meibom KL, Barel M, Charbit A. 2009. Loops and networks in control of Francisella tularensis virulence. Future Microbiol. 4:713–729 [DOI] [PubMed] [Google Scholar]
- 14. Lindemann SR, McLendon MK, Apicella MA, Jones BD. 2007. An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infect. Immun. 75:3178–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moreland JG, Hook JS, Bailey G, Ulland T, Nauseef WM. 2009. Francisella tularensis directly interacts with the endothelium and recruits neutrophils with a blunted inflammatory phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 296:L1076–L1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krause KH. 2000. Professional phagocytes: predators and prey of microorganisms. Schweiz Med. Wochenschr. 130:97–100 [PubMed] [Google Scholar]
- 17. Oyston PC. 2008. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J. Med. Microbiol. 57:921–930 [DOI] [PubMed] [Google Scholar]
- 18. Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 7:333–340 [DOI] [PubMed] [Google Scholar]
- 19. Nano FE, Schmerk C. 2007. The Francisella pathogenicity island. Ann. N. Y. Acad. Sci. 1105:122–137 [DOI] [PubMed] [Google Scholar]
- 20. Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, Klose KE. 2009. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74:1459–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauriano CM, Barker JR, Yoon SS, Nano FE, Arulanandam BP, Hassett DJ, Klose KE. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U. S. A. 101:4246–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Charity JC, Blalock LT, Costante-Hamm MM, Kasper DL, Dove SL. 2009. Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog. 5:e1000641. 10.1371/journal.ppat.1000641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dean RE, Ireland PM, Jordan JE, Titball RW, Oyston PC. 2009. RelA regulates virulence and intracellular survival of Francisella novicida. Microbiology 155:4104–4113 [DOI] [PubMed] [Google Scholar]
- 24. Brotcke A, Monack DM. 2008. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun. 76:3473–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buchan BW, McCaffrey RL, Lindemann SR, Allen LA, Jones BD. 2009. Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect. Immun. 77:2517–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hai T, Frey KM, Steinbuchel A. 2006. Engineered cyanophycin synthetase (CphA) from Nostoc ellipsosporum confers enhanced CphA activity and cyanophycin accumulation to Escherichia coli. Appl. Environ. Microbiol. 72:7652–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adames K, Euting K, Broker A, Steinbuchel A. 2013. Investigations on three genes in Ralstonia eutropha H16 encoding putative cyanophycin metabolizing enzymes. Appl. Microbiol. Biotechnol. 97:3579–3591 [DOI] [PubMed] [Google Scholar]
- 28. Armengod ME, Moukadiri I, Prado S, Ruiz-Partida R, Benitez-Paez A, Villarroya M, Lomas R, Garzon MJ, Martinez-Zamora A, Meseguer S, Navarro-Gonzalez C. 2012. Enzymology of tRNA modification in the bacterial MnmEG pathway. Biochimie 94:1510–1520 [DOI] [PubMed] [Google Scholar]
- 29. Moukadiri I, Prado S, Piera J, Velazquez-Campoy A, Bjork GR, Armengod ME. 2009. Evolutionarily conserved proteins MnmE and GidA catalyze the formation of two methyluridine derivatives at tRNA wobble positions. Nucleic Acids Res. 37:7177–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gong S, Ma Z, Foster JW. 2004. The Era-like GTPase TrmE conditionally activates gadE and glutamate-dependent acid resistance in Escherichia coli. Mol. Microbiol. 54:948–961 [DOI] [PubMed] [Google Scholar]
- 31. Sayed AK, Foster JW. 2009. A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol. Microbiol. 71:1435–1450 [DOI] [PubMed] [Google Scholar]
- 32. Sayed AK, Odom C, Foster JW. 2007. The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiology 153:2584–2592 [DOI] [PubMed] [Google Scholar]
- 33. Jain V, Kumar M, Chatterji D. 2006. ppGpp: stringent response and survival. J. Microbiol. 44:1–10 [PubMed] [Google Scholar]
- 34. Tozawa Y, Nomura Y. 2011. Signalling by the global regulatory molecule ppGpp in bacteria and chloroplasts of land plants. Plant Biol. (Stuttg.) 13:699–709 [DOI] [PubMed] [Google Scholar]
- 35. Seyfzadeh M, Keener J, Nomura M. 1993. spoT-dependent accumulation of guanosine tetraphosphate in response to fatty acid starvation in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 90:11004–11008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Battesti A, Bouveret E. 2006. Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Mol. Microbiol. 62:1048–1063 [DOI] [PubMed] [Google Scholar]
- 37. Payoe R, Fahlman RP. 2011. Dependence of RelA-mediated (p) ppGpp formation on tRNA identity. Biochemistry 50:3075–3083 [DOI] [PubMed] [Google Scholar]
- 38. Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. 2010. ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74:171–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chamberlain RE. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13:232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, Brouwer D, Nair V, Fischer ER, Wicke L, Curda AJ, Kupko JJ, III, Martens C, Crane DD, Bosio CM, Porcella SF, Celli J. 2009. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell. Microbiol. 11:1128–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 42. Schneider DA, Murray HD, Gourse RL. 2003. Measuring control of transcription initiation by changing concentrations of nucleotides and their derivatives. Methods Enzymol. 370:606–617 [DOI] [PubMed] [Google Scholar]
- 43. Mendrygal KE, Gonzalez JE. 2000. Environmental regulation of exopolysaccharide production in Sinorhizobium meliloti. J. Bacteriol. 182:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. 1976. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 17:62–70 [DOI] [PubMed] [Google Scholar]
- 45. Clemens DL, Lee BY, Horwitz MA. 2004. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72:3204–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LA, Jones BD. 2011. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect. Immun. 79:581–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tosa T, Pizer LI. 1971. Biochemical bases for the antimetabolite action of l-serine hydroxamate. J. Bacteriol. 106:972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 49. Mohapatra NP, Soni S, Bell BL, Warren R, Ernst RK, Muszynski A, Carlson RW, Gunn JS. 2007. Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect. Immun. 75:3305–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mokrievich AN, Kondakova AN, Valade E, Platonov ME, Vakhrameeva GM, Shaikhutdinova RZ, Mironova RI, Blaha D, Bakhteeva IV, Titareva GM, Kravchenko TB, Kombarova TI, Vidal D, Pavlov VM, Lindner B, Dyatlov IA, Knirel YA. 2010. Biological properties and structure of the lipopolysaccharide of a vaccine strain of Francisella tularensis generated by inactivation of a quorum sensing system gene qseC. Biochemistry (Mosc.) 75:443–451 [DOI] [PubMed] [Google Scholar]
- 51. Cooper CL, Hsu L, Jackowski S, Rock CO. 1989. 2-Acylglycerolphosphoethanolamine acyltransferase/acyl-acyl carrier protein synthetase is a membrane-associated acyl carrier protein binding protein. J. Biol. Chem. 264:7384–7389 [PubMed] [Google Scholar]
- 52. Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol. 11:100–105 [DOI] [PubMed] [Google Scholar]
- 53. Kalia D, Merey G, Nakayama S, Zheng Y, Zhou J, Luo Y, Guo M, Roembke BT, Sintim HO. 2013. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p) ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 42:305–341 [DOI] [PubMed] [Google Scholar]
- 54. Cheng F, Wang J, Peng J, Yang J, Fu H, Zhang X, Xue Y, Li W, Chu Y, Jin Q. 2007. Gene expression profiling of the pH response in Shigella flexneri 2a. FEMS Microbiol. Lett. 270:12–20 [DOI] [PubMed] [Google Scholar]
- 55. Morgan B, Lahav O. 2007. The effect of pH on the kinetics of spontaneous Fe(II) oxidation by O2 in aqueous solution—basic principles and a simple heuristic description. Chemosphere 68:2080–2084 [DOI] [PubMed] [Google Scholar]
- 56. Leyer GJ, Johnson EA. 1993. Acid adaptation induces cross-protection against environmental stresses in Salmonella typhimurium. Appl. Environ. Microbiol. 59:1842–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fuller JR, Kijek TM, Taft-Benz S, Kawula TH. 2009. Environmental and intracellular regulation of Francisella tularensis ripA. BMC Microbiol. 9:216. 10.1186/1471-2180-9-216 [DOI] [PMC free article] [PubMed] [Google Scholar]