

Abstract
Helicobacter pylori and Campylobacter jejuni colonize the stomach and intestinal mucus, respectively. Using a combination of mucus-secreting cells, purified mucins, and a novel mucin microarray platform, we examined the interactions of these two organisms with mucus and mucins. H. pylori and C. jejuni bound to distinctly different mucins. C. jejuni displayed a striking tropism for chicken gastrointestinal mucins compared to mucins from other animals and preferentially bound mucins from specific avian intestinal sites (in order of descending preference: the large intestine, proximal small intestine, and cecum). H. pylori bound to a number of animal mucins, including porcine stomach mucin, but with less avidity than that of C. jejuni for chicken mucin. The strengths of interaction of various wild-type strains of H. pylori with different animal mucins were comparable, even though they did not all express the same adhesins. The production of mucus by HT29-MTX-E12 cells promoted higher levels of infection by C. jejuni and H. pylori than those for the non-mucus-producing parental cell lines. Both C. jejuni and H. pylori bound to HT29-MTX-E12 mucus, and while both organisms bound to glycosylated epitopes in the glycolipid fraction of the mucus, only C. jejuni bound to purified mucin. This study highlights the role of mucus in promoting bacterial infection and emphasizes the potential for even closely related bacteria to interact with mucus in different ways to establish successful infections.
INTRODUCTION
The identification of two layers of mucus in the gastrointestinal tract (1, 2) and the profiling of bacterial species that inhabit different mucosal sites throughout the body (3–5) have evoked intense interest in how bacteria colonize mucosal surfaces. Mucus is part of the innate host defense systems of humans and animals. An important function is to create a physical barrier that limits infection by pathogenic organisms (6). Many organisms have developed strategies to overcome this barrier, and most infections occur at mucosal surfaces. Understanding the molecular mechanisms that pathogenic organisms use to colonize the mucus layer is clearly important, since such knowledge may suggest novel approaches for the prevention of colonization by pathogens.
Helicobacter pylori and Campylobacter jejuni are two closely related but distinct mucosal pathogens that have adapted to infect different niches in the mucus layer of the human gastrointestinal tract. H. pylori infection, one of the most common infections of humans, causes gastritis, duodenal ulceration, and an increased risk of developing gastric cancer (7). This organism colonizes the gastric mucosae of humans and some other primates but does not colonize other hosts naturally. H. pylori predominantly colonizes the supramucosal gel of the stomach (8). The mucus gel acts as an infectious reservoir providing a steady supply of organisms that can interact with the underlying epithelium and cause disease. The binding of H. pylori to gastric mucin has been shown to be mediated via the bacterial adhesin BabA (9), which binds to the Lewisb blood group antigen (9). H. pylori also binds to sialyl-Lewisx via the SabA outer membrane protein (OMP) (10). H. pylori exhibits high genomic plasticity due to high rates of mutation and homologous recombination (11, 12). Individual strains have been shown to modulate both their own adhesin expression and host glycan expression following infection (13, 14). The dynamic modulation of H. pylori attachment is thought to confer an advantage on the bacterium, enabling it to respond rapidly to changes in its environment.
C. jejuni is a highly motile intestinal pathogen and the leading cause of bacterial gastroenteritis in humans in the developed world. This organism colonizes chickens and other poultry naturally, without causing pathology, and chickens are an important reservoir of infection for humans. The ability of the organism to colonize intestinal mucus is thought to be an important virulence factor, and exposure of C. jejuni to the human intestinal mucin Muc2 has been shown to have an effect on virulence gene expression (15). Glycans present on chicken intestinal mucin have been shown to inhibit C. jejuni invasion of HCT-8 cells (16, 17). C. jejuni can interact with blood group antigens present in human milk, and binding to epithelial cells is inhibited by human milk-derived fucosyl oligosaccharides (18). Recent work has profiled the binding of C. jejuni to glycans at 42°C, the body temperature of avian species, and 37°C, and differences have been noted (19). However, very little information exists on the carbohydrate-lectin pairs involved in mediating infections by C. jejuni.
New tools that enable us to study the interaction of bacteria with mucin oligosaccharides have become available recently. These include cell lines that produce adherent mucus layers (20, 21) and novel mucin microarrays that contain natural mucins from different animal species (22). This study aimed to examine the interactions of both H. pylori and C. jejuni with mucus and mucins by harnessing these novel techniques. Our results show that these two closely related bacteria interact with mucus in very different ways, and they highlight the role of mucus in promoting bacterial infection. This study also emphasizes that glycans present on host mucins and other glycosylated structures in mucus likely play a key role in the specific species and tissue tropism displayed by bacteria. These data underline the importance of elucidating how bacteria interact with mucus in order to overcome this important protective barrier and establish successful infections.
MATERIALS AND METHODS
Bacterial strains, routine maintenance, and culture.
The strains of C. jejuni used in this study included the well-characterized strains 81-176 (23, 24) and 11168 (25), two chicken isolates, CC18 and CC19 (a gift from Seamus Fanning, University College Dublin [UCD], Dublin, Ireland), and two human isolates, H1 and H3 (a gift from Margaret Byrne, Our Lady's Children's Hospital, Dublin, Ireland). C. jejuni strains were routinely cultured at 37°C on Mueller-Hinton agar (Oxoid) under microaerophilic conditions generated using CampyGen gas packs (Oxoid).
The strains of H. pylori used in this study included the sequenced strains 26695 (26), J99 (27), and G27 (28), as well as a babA knockout mutant of G27, strain G27ΔbabA (a gift from Steffen Backert). H. pylori was routinely cultured at 37°C on Columbia blood agar base (Oxoid) containing 7% (vol/vol) defibrinated horse blood under microaerophilic conditions generated using CampyGen gas packs (Oxoid). Strain G27ΔbabA was cultured on plates containing 10 μg/ml of kanamycin.
Cell culture.
The LS174T (29) and HT29 (30) human colonic carcinoma cell lines, methotrexate (MTX)-adapted HT29 cells (HT29-MTX) (31), and a mucus-secreting subclone, the HT29-MTX-E12 cell line (32), were used. The HT29-MTX-E12 cell line is a subclone of HT29-MTX cells that expresses the gastric mucin MUC5AC (21), selected on the basis of tight-junction formation and the production of an adherent mucus layer (32). HT29 cells were maintained in McCoy's medium (Lonza) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 1% (vol/vol) nonessential amino acids (Sigma), 2 mM l-GlutaMAX (Invitrogen), 100 U/ml penicillin (Sigma), 100 μg/ml streptomycin (Sigma), and 125 μg/ml amphotericin B (Sigma). LS174T, HT29-MTX, and HT29-MTX-E12 cells were maintained in Dulbecco's modified Eagle medium (DMEM; Lonza) supplemented with 10% (vol/vol) FBS, 1% (vol/vol) nonessential amino acids (Sigma), 2 mM l-GlutaMAX (Invitrogen), 100 U/ml penicillin (Sigma), 100 μg/ml streptomycin (Sigma), and 125 μg/ml amphotericin B (Sigma).
Mucus harvesting and mucin purification.
HT29-MTX-E12 cells were grown for 21 days on Transwell filters. The mucus layer was removed from the cells by treatment with N-acetyl cysteine, as described previously (16, 20). LS174T cells were cultured as described above, and once they were 60 to 80% confluent, the supernatant containing secreted mucus was collected. Mucus samples from either the reproductive tracts or digestive tracts of cows, chickens, deer, horses, mice, sheep, pigs, and rats were obtained either postmortem from healthy animals killed at commercial abattoirs or after humane euthanasia by an intravenous barbiturate overdose. Generally, mucosal surfaces were scraped with a scalpel blade to harvest secreted mucus and epithelial cells. These experimental procedures were licensed by the Department of Health and Children, Ireland, in accordance with the Cruelty to Animals Act (Ireland, 1897) and European Community Directive 86/609/EC and were sanctioned by the Animals Research Ethics Committee, University College Dublin, Dublin, Ireland. Mucins were isolated from the collected mucus and were purified as described previously (16, 22). In brief, mucus was solubilized with guanidine hydrochloride (final concentration, 4 M). Samples were reduced with dithiothreitol (DTT) (Sigma-Aldrich) at a final concentration of 0.01 M for 5 h at 37°C and were alkylated with iodoacetamide (0.025 M) (Sigma-Aldrich). Mucin was purified using CsCl density gradient separation followed by size exclusion chromatography (16, 22). Non-mucin-containing fractions from the CsCl gradient were retained for subsequent analysis.
Interrogation of NGC and natural mucin microarrays for bacterial binding.
Purified animal mucins and mucins from HT29-MTX-E12 and LS174T cells were printed onto microarray slides as described previously (22). Neoglycoconjugate (NGC) arrays were designed and printed as described previously (33). For incubations at 37°C, C. jejuni and H. pylori were cultured on agar as described above, harvested, and resuspended in Mueller-Hinton broth or in brain heart infusion (BHI) broth supplemented with 10% FBS, respectively, to an optical density at 600 nm (OD600) of 0.2. Cultures were incubated for 4 h, at which time bacteria were harvested, washed twice in phosphate-buffered saline (PBS), and resuspended in PBS to an OD600 of 1.0.
To assess the effect of increased temperature on the binding of C. jejuni, bacteria were grown in Mueller-Hinton broth for 24 h at 37°C or 42°C prior to incubation with the array. Following incubation with Syto 82 (Invitrogen) for 1 h at 37°C or 42°C, bacteria were washed 7 times in low-salt Tris-buffered saline (TBS) (22) to remove excess staining and were resuspended to an OD600 of 1.0 in low-salt TBS with 0.05% Tween 20 (TBS-T). The microarrays were incubated with fluorescently labeled bacteria using an 8-well gasket slide and incubation cassette system (Agilent Technologies) at 37°C or 42°C with gentle rotation in the dark. The slides were washed 3 times with low-salt TBS-T and once in TBS. The microarrays were dried by centrifugation and were scanned immediately.
Imaging, data extraction, and analysis.
Microarray slides were imaged in a GenePix 4000b microarray scanner (532-nm laser, 100% laser power, 70% photomultiplier tube [PMT] setting, tetramethyl rhodamine isocyanate [TRITC] emission filter) (Molecular Devices). Data from the resultant image files were extracted using GenePix Pro, version 5.1, and were exported as text to Excel, where all data analysis was performed. Local background was subtracted, and background-corrected median feature intensity was used for each feature intensity value. For statistical use, the median of six replicate spots per subarray was treated as a single data point. Data were normalized to the mean for three replicate microarray slides.
Subcellular fractionation of H. pylori and characterization of mucin binding.
Bacterial outer membrane proteins from H. pylori were lysed and isolated using sodium lauryl sarcosine as described previously (34). Briefly, H. pylori was harvested from blood agar plates, washed in sterile PBS, harvested by centrifugation at 12,000 × g for 20 min at 4°C, and frozen at −20°C. The pellet was thawed in 20 mM Tris buffer, pH 7.5, containing protease inhibitor cocktail tablets (Roche). Following 6 rounds of pulse sonication, DNase (0.1 mg/ml) and RNase (0.5 mg/ml) were added to the sonicated material, which was then incubated for 30 min at room temperature (RT). Unbroken cells were removed (2,000 × g, 20 min, 4°C), and the membranes were isolated by centrifugation of the supernatant at 40,000 × g for 30 min at 4°C. The resulting pellet was resuspended in 20 mM Tris buffer, pH 7.5, containing 0.2% sodium lauryl sarcosine and was incubated at RT for 30 min. The outer membrane fraction was pelleted by centrifugation at 40,000 × g for 30 min at 4°C and was washed 3 times in ice-cold distilled water (dH2O).
Fractions were separated by electrophoresis on 10% polyacrylamide minigels and were transferred to polyvinylidene difluoride (PVDF) membranes overnight in a wet blotter (Bio-Rad, Hercules, CA) at 10 V. Membranes were blocked in 5% gelatin for 2 h at 37°C. Purified porcine stomach mucin (13) was biotinylated by using the EZ-Link sulfo-N-hydroxysuccinimide (NHS) biotinylation kit (Pierce) according to the manufacturer's instructions. Blocked membranes were incubated with 0.25 mg/ml biotinylated mucin for 4 h at RT. Membranes were washed for 1 h with TBS-Tween (0.05% [vol/vol]) and were then incubated with streptavidin-peroxidase (1:50,000) (Sigma) for 1 h at RT. Membranes were washed overnight in TBS-Tween (0.05%), and bands were detected using enhanced chemiluminescence.
Infection assays.
Cells were seeded on Transwell filters (diameter, 12 mm; pore size, 0.4 μm; Millipore) at a density of 1 × 105 cells/filter and were grown for 21 days. Cells were fed every second day and were grown in antibiotic-free medium for 24 h prior to infection.
To determine the role of mucus in colonization, bacterial strains were grown under microaerophilic conditions, and C. jejuni was prepared for infection of the cells as described previously (20). Briefly, C. jejuni strains were grown for 24 h on Mueller-Hinton agar plates and were then transferred to a biphasic medium consisting of Mueller-Hinton agar and DMEM, supplemented with 2% FBS without antibiotics, for a further 21 to 24 h. Bacteria were harvested from the biphasic medium, washed once in DMEM, and then diluted to an OD600 of 0.2. H. pylori grown for 48 h on Columbia blood agar plates was harvested into BHI broth at pH 5.0, and the OD600 was adjusted to 0.4.
The medium in the lower chamber of the Transwell was replaced with 1.5 ml of sterile antibiotic-free McCoy's 5A medium or DMEM, each supplemented with 10% (vol/vol) FBS. For C. jejuni infections, 150 μl of a C. jejuni suspension at an OD600 of 0.2 was added to the upper chamber. H. pylori cannot survive in DMEM (35), so 100 μl of BHI broth at pH 5.0 and 50 μl of the H. pylori suspension (OD600, 0.4) were added to the upper chamber. The number of organisms added to cells was approximately 1 × 108 CFU/ml, which corresponded to a multiplicity of infection (MOI) of 50 organisms per cell.
Infected cell cultures were incubated under microaerophilic conditions generated using CampyGen gas packs (Oxoid) at 37°C for 24 h. Following incubation, infected cells were washed with sterile PBS and were then harvested using trypsin-EDTA as described previously (36). Serial dilutions of the trypsinized cells were plated out in triplicate onto Mueller-Hinton agar plates or Columbia blood agar plates for assessment of C. jejuni or H. pylori organisms associated with the cells and were incubated under microaerophilic conditions at 37°C. Colonies were enumerated after 4 to 5 days of incubation.
To determine the effect of temperature on infection and invasion by C. jejuni, strains were grown for 24 h at 37°C on Mueller-Hinton agar plates and were then transferred to Mueller-Hinton broth for a further 24 h at either 37°C or 42°C. Bacteria were harvested from the broth, washed once in DMEM, and then diluted to an OD600 of 0.2. Cell cultures were infected as described above and were incubated for 4 h at 37°C or 42°C. Following the incubation period, one set of wells was used to determine total association as described above. The second set of wells was washed once in DMEM and was then incubated with 400 μg/ml gentamicin sulfate (Sigma-Aldrich) in DMEM for 2 h to kill extracellular bacteria. Cells were then washed once in PBS and were lysed with 0.1% Triton X-100 in PBS. Serial dilutions of the cell lysate were plated out in triplicate onto Mueller-Hinton agar plates for assessment of internalized C. jejuni organisms and were incubated under microaerophilic conditions at 37°C. Colonies were enumerated after 3 to 4 days of incubation.
Probing of mucus and mucin of E12 cells with C. jejuni and H. pylori.
Crude mucus or 10 μg of purified mucin from E12 cells was immobilized onto a PVDF membrane with a pore size of 0.2 μm (Millipore). Membranes were washed in PBS and were then blocked overnight in TBS containing 5% skim milk. C. jejuni or H. pylori was cultured on agar plates as described above, harvested in PBS, washed twice, and resuspended in TBS to a final OD600 of 0.4. A suspension of either C. jejuni or H. pylori was then overlaid onto the membrane and was incubated at 37°C for 1 h. The overlays were washed three times at room temperature for 10 min on a rotary shaker in TBS containing 0.05% Tween 20. Binding of C. jejuni was detected using a 1:5,000 dilution of a rabbit anti-C. jejuni antiserum (37) and a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (Santa Cruz). H. pylori binding was detected using an anti-H. pylori polyclonal antibody (Dako) and an HRP -conjugated anti-rabbit secondary antibody (Santa Cruz). Reactive slots were detected using enhanced chemiluminescence.
O-glycan analysis of purified mucin from HT29-MTX-E12 cells.
Mucins were purified as described above and were freeze-dried. Then 28% NH3·H2O saturated with (NH4)2CO3 was added to a final mucin concentration of 1 mg/ml. The mixture was incubated at 65°C for 16 h in order to release the O-glycans. For glycan enrichment after release, Hypercarb MicroSPE tips were conditioned twice with 200 μl of methanol, twice with 200 μl of Milli-Q water, and finally twice with 200 μl of 10 mM ammonium carbonate before the sample (in an aqueous solution; final volume, 200 μl) was applied. After the sample had passed through the tip, the chromatographic bed was washed 5 times with 200 μl of Milli-Q water in order to remove excess release reagents. The retained glycans were eluted from the Hypercarb bed 5 times, using 100 μl of 10 mM ammonium carbonate solution in 80% (vol/vol) acetonitrile each time, and the eluates from all these elution steps were collected in a single microcentrifuge tube and were dried down in a SpeedVac concentrator. Once dry, the samples were treated with formic acid to convert them back to the aldose form and were derivatized with 2-aminobenzamide (2-AB) as described previously (38). 2-AB-labeled glycans were analyzed by hydrophilic interaction chromatography (HILIC) (38, 39), and the glucose unit (GU) values were compared to those in the O-glycan database (http://glycobase.nibrt.ie/glycobase/show_nibrt.action). Further confirmation of the structures proposed was obtained by exoglycosidase digestion.
Exoglycosidase digestion.
Arrays of exoglycosidases were used in combination with HILIC–high-performance liquid chromatography (HPLC) to determine the sequence, monosaccharide type, and linkage of sugar residues. Exoglycosidase sequencing was performed on 2-AB-labeled O-glycans in 10 μl of a solution containing enzymes at standard concentrations in the manufacturer's recommended buffers for 16 h at 37°C. The enzymes used were as follows: Arthrobacter ureafaciens sialidase (ABS) (EC 3.2.1.18), 1 to 2 U/ml; Streptococcus pneumoniae sialidase recombinant in Escherichia coli (NAN1) (EC 3.2.1.18), 1 U/ml; bovine kidney α-fucosidase (BKF) (EC 3.2.1.51), 1 U/ml; almond meal α-fucosidase (AMF) (EC 3.2.1.111), 3 mU/ml; bovine testis β-galactosidase (BTG) (EC 3.2.1.23), 2 U/ml; Streptococcus pneumoniae β-galactosidase (SPG) (EC 3.2.1.23), 80 mU/ml; and β-N-acetylglucosaminidase cloned from S. pneumoniae, expressed in Escherichia coli (GUH) (EC 3.2.1.30), 10 mU/ml. After digestion, samples were separated from the exoglycosidases before HPLC analysis by centrifugation in a Nanosep 10K Omega microcentrifuge filter (Pall, VWR).
WAX chromatography.
Neutral and acidic oligosaccharides were separated by weak anion-exchange (WAX)-HPLC using a Vydac 301VHP575 column (7.5 by 50 mm; Anachem) on a 2695 Alliance Separations module with a 474 fluorescence detector set (excitation and emission wavelengths, 330 and 420 nm, respectively) (Waters). Solvent A consisted of 0.1 M ammonium acetate buffer, pH 7.0, in 20% (vol/vol) acetonitrile, and solvent B was 20% acetonitrile. Gradient conditions were as follows: a linear gradient of 0 to 5% solvent A over 12 min at a flow rate of 1 ml/min, followed by 5 to 21% solvent A over 13 min, 21 to 50% solvent A over 25 min, 80 to 100% solvent A over 5 min, and 5 min at 100% solvent A. Samples were injected in water, and a fetuin N-glycan standard was used for calibration.
Immunofluorescent staining of HT29-MTX-E12 cells.
HT29-MTX-E12 cells growing on Transwell filters were washed with PBS, removed from their plastic supports, and sandwiched between two thin pieces of chicken liver prior to mounting in optimal cutting temperature (OCT) medium (BDH) as described previously, (40). Sections (20 μm) were cut using a cryostat, collected on polylysine-coated microscope slides, allowed to air dry, and then either used immediately or stored at −20°C until use.
Sections were fixed on slides with 2% formalin for 10 min, permeabilized with 0.2% (wt/vol) saponin (Sigma) in PBS for 10 min, blocked for 1 h in 1% (wt/vol) bovine serum albumin (BSA; Sigma) and 10% (vol/vol) serum in PBS, and probed with an anti-Lewisb antibody (SPM194; Santa Cruz) or anti-sialyl-Lewisx (Molecular Probes) and a secondary anti-mouse antibody conjugated to Alexa Fluor 488 (Invitrogen). DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen) was used to counterstain the nuclei. Coverslips were mounted on the slides using Fluorescent Mounting Medium (Dako), and the sections were examined with a fluorescence microscope.
Detection of Lewisb blood group antigen in HT29-MTX-E12 mucus fractions.
Following CsCl gradient fractionation of E12 mucus, non-mucin-containing fractions were slot blotted onto a PVDF membrane (Millipore). Membranes were washed in PBS and were then blocked overnight in TBS containing 5% skim milk. Lewisb was detected in the fractions by using an antibody against Lewisb blood group antigen (SPM194; Santa Cruz) and a secondary anti-mouse antibody conjugated to HRP. Reactive slots were detected by enhanced chemiluminescence.
Glycolipid isolation and characterization and C. jejuni and H. pylori binding.
Glycolipids were isolated from mucus by multiple rounds of chloroform-methanol extraction. Mucus was harvested as described above, and organic extraction of mucus was carried out according to the method of Muindi et al. (41). Briefly, lipids were obtained by successive extractions with chloroform-methanol (1:1 followed by 1:2 [vol/vol]) and a final extraction with chloroform-methanol-water (4.8:3.5:1). The resulting glycolipids were separated by thin-layer chromatography (TLC) using chloroform-methanol-0.25% KCl (5:4:1) as a solvent system with silica TLC plates (Fisher). Glycolipid bands were visualized with a 0.1% orcinol-H2SO4 stain and were charred using a heat gun. TLC plates containing separated glycolipids were treated with 0.1% polyisobutylmethacrylate in acetone, dried, and probed with antibodies against Lewisb and sialyl-Lewisx. The TLC plates were also overlaid with either H. pylori or C. jejuni organisms and were probed with antibodies raised against H. pylori or C. jejuni. TLC plates were probed with alkaline phosphatase-conjugated secondary antibodies (Santa Cruz), and antigen-antibody complexes were detected using the colorimetric substrate BCIP (5-bromo-4-chloro-3-indolylphosphate)-NBT (nitroblue tetrazolium) (Sigma).
Statistical analysis.
Infection assays were carried out on three separate occasions in triplicate. Microarray interrogation was conducted in duplicate per microarray slide, and data were normalized to the means for three replicate microarray slides. Graphs were drawn using GraphPad Prism. Results are presented as means ± standard deviations (indicated by error bars) for replicate experiments. The Student t test was used to estimate statistical significance; a P value of <0.01 was considered significant.
RESULTS
The interaction of C. jejuni and H. pylori with purified native mucins from different animal species reveals species tropism.
We investigated the binding of H. pylori and C. jejuni to a panel of natural mucins from different animals printed on a microarray that enables the generation of quantitative binding data. C. jejuni and H. pylori bound to distinctly different mucins, and each showed tropism for mucins from specific sites among different animal species.
C. jejuni displayed a tropism for chicken mucin in preference to mucins from other animals (Table 1). All 6 isolates of C. jejuni (81-176, 11168, 2 chicken isolates, and 2 human clinical isolates) showed a clear tropism for mucin from chickens, with the strength of the interaction dependent on the site of origin of the mucin (in descending order of interaction strength: the large intestine, proximal small intestine, and cecum) (Fig. 1). The binding of C. jejuni to chicken mucin was not influenced by growth at human or avian body temperature (Fig. 1). C. jejuni also bound to mucins from other animals, including equine, porcine, ovine, and bovine mucins. However, the strength of the interactions with these mucins was not as great as that seen with chicken large-intestine mucin (Table 1). The patterns of binding were consistent across different C. jejuni isolates and were comparable at 37°C and 42°C (see Table S1 in the supplemental material).
Table 1.
Binding of C. jejuni strain 81-176 and H. pylori strain 26695 to natural mucins on a mucin microarray
| Mucin sourcea | Level of bindingb of: |
|
|---|---|---|
| C. jejuni 81-176 | H. pylori 26695 | |
| Abomasum (B) | 2,780 | 2,158 |
| Cervicovaginal (B) | 472 | 521 |
| Cervix (B) | 497 | 505 |
| Duodenum (B) | 795 | 289 |
| Spiral colon (B) | 1,131 | 288 |
| Trachea (B) | 533 | 261 |
| Endometrium (B) | 554 | 177 |
| Cecum (C) | 3,884 | 387 |
| Proximal small intestine (C) | 8,775 | 625 |
| Large intestine (C) | 14,344 | 571 |
| Abomasum (D) | 2,021 | 2,261 |
| Duodenum (D) | 121 | 487 |
| Jejunum (D) | 144 | 653 |
| Spiral ascending colon (D) | 816 | 172 |
| HT29-MTX-E12 cells | 3,192 | 1,108 |
| Dorsal ascending colon (E) | 925 | 2,036 |
| Duodenum (E) | 1,183 | 1,794 |
| Left ventral ascending colon (E) | 1,883 | 965 |
| Right ventral ascending colon (E) | 3,255 | 1,786 |
| Jejunum (E) | 1,891 | 3,064 |
| Stomach (E) | 218 | 493 |
| Trachea (E) | 313 | 404 |
| LS174T cells | 4,298 | 14,409 |
| Cecum (M) | 483 | 1,157 |
| Large intestine (M) | 1,001 | 1,907 |
| Small Intestine (M) | 202 | 353 |
| Stomach (M) | 694 | 1,074 |
| Abomasum antrum (O) | 343 | 1,126 |
| Descending colon (O) | 866 | 1,826 |
| Duodenum (O) | 1,576 | 1,124 |
| Ileum (O) | 724 | 778 |
| Jejunum (O) | 652 | 1,393 |
| Spiral colon (O) | 2,654 | 7,759 |
| Cecum (P) | 4,221 | 6,038 |
| Descending colon (P) | 1,423 | 256 |
| Jejunum (P) | 1,055 | 2,903 |
| Spiral colon (P) | 1,211 | 3,372 |
| Stomach (P) | 2,522 | 7,915 |
| Cecum (R) | 1,031 | 4,302 |
| Colon (R) | 1,011 | 482 |
| Duodenum (R) | 902 | 5,827 |
| Ileum (R) | 501 | 1,446 |
| Stomach (R) | 1,101 | 4,688 |
B, bovine; C, chicken; D, deer; E, equine; M, mouse; O, ovine; P, porcine; R, rat.
Expressed as the mean fluorescence intensity (in relative fluorescence units) from duplicate individual microarrays across 3 different array slides. The binding temperature was 37°C.
Fig 1.

Interaction of C. jejuni with chicken mucins printed on a mucin microarray slide. Microarrays containing purified chicken mucins from different sites in the intestinal tract were probed with 6 strains of fluorescently labeled C. jejuni organisms. For incubations at 37°C, C. jejuni strains were cultured on agar as described in Materials and Methods, harvested, and resuspended in Mueller-Hinton broth supplemented with 10% FBS. Cultures were grown for 4 h, at which time bacteria were harvested, washed, and resuspended in PBS to an OD600 of 1.0. To assess the effect of temperature on the binding of C. jejuni, bacteria were grown in Mueller-Hinton broth for 24 h at 37°C or 42°C. Labeling and microarray interrogations were carried out at 37°C or 42°C. The histogram shows the mean fluorescence intensities from duplicate subarrays on three replicate microarray slides; values for each subarray are medians for six feature replicates. Error bars indicate the standard deviations of the means for three microarray slides. RFU, relative fluorescence units.
H. pylori bound to a distinctly different subset of mucins than that seen with C. jejuni (Table 1). Three wild-type strains of H. pylori and a BabA knockout strain were used to probe the array. The wild-type organisms all displayed appreciable binding to porcine stomach mucin, although the intensity of the interaction was substantially less than the intensity of the binding of C. jejuni to chicken large-intestine mucin. H. pylori also bound to other mucins, including rat gastric and duodenal mucins (Fig. 2; Table 1). The wild-type strains all displayed similar binding profiles. However, strain G27ΔbabA exhibited reduced binding to a number of mucins, including those of porcine and rat origins, relative to binding by the parental strain (Fig. 2), suggesting that the BabA outer membrane protein (OMP) was playing a role in mediating the interaction between H. pylori G27 and a number of mucins.
Fig 2.

Interaction of H. pylori with porcine and rat mucins. Mucin microarrays were probed with fluorescently labeled H. pylori organisms. Three wild-type strains and strain G27ΔbabA were used. G27ΔbabA does not express the BabA outer membrane protein, which mediates binding to the Lewisb blood group antigen. H. pylori was cultured on agar as described in Materials and Methods, harvested, and resuspended in brain heart infusion broth supplemented with 10% FBS. Cultures were incubated at 37°C for 4 h, at which time bacteria were harvested, washed, and resuspended in PBS to an OD600 of 1.0. Strain G27ΔbabA was cultured in the presence of 10 μg/ml of kanamycin. The histogram shows the mean fluorescence intensities from duplicate subarrays on three replicate microarray slides; values for each subarray are medians for six feature replicates. Error bars indicate the standard deviations of the means for three microarray slides.
Role of OMPs in mediating the binding of H. pylori to mucin.
In order to investigate the potential contribution of OMPs to mucin binding, we probed OMPs from each of the H. pylori strains with biotinylated porcine stomach mucin. Mucin bound specifically to an 80-kDa OMP in strains G27 and J99 but not in G27ΔbabA or 26695 (Fig. 3A). Direct binding of these H. pylori isolates to Lewis blood group antigen structures on an NGC array confirmed that strain 26695 demonstrated lower levels of binding to both Lewisb and sialyl-Lewisx structures than strains J99 and G27 (Fig. 3B). The results suggest that BabA, when expressed, can mediate the binding of H. pylori to porcine stomach mucin. However, in the case of strain 26695, for which no OMP could be detected to mediate binding, this interaction may be mediated by a non-protein-based bacterial adhesin.
Fig 3.

Role of OMPs in mediating the binding of H. pylori to mucins. (A) Detection of H. pylori surface adhesins involved in mucin binding. Outer membrane protein fractions of H. pylori were prepared and were probed with biotinylated porcine stomach mucin. A protein of ∼80 kDa, corresponding to the expected size of the surface adhesin BabA, was detected in strains J99 and G27 but was absent from 26695 and from a babA knockout mutant of G27. (B) Binding of H. pylori strains to Lewis blood group antigen structures. Microarrays containing neoglyconjugate Lewis blood group antigen structures were probed with fluorescently labeled H. pylori organisms. The structures probed were Lewisx-BSA (LexBSA), di-Lewisx-aminophenylethyl-human serum albumin (DiLexHSA), tri-Lewisx-HSA (TriLexHSA), 3′-sialyl-Lewisx-BSA (SLexBSA), 3-sulfo-Lewisx-BSA (3SuLexBSA), 6-sulfo-Lewisx-BSA (6SuLexBSA), and lacto-N-difucohexaose I-BSA, (LNDHIBSA), which consists of Lewisb tetrasaccharide and lactose and is used as a Lewisb active structure in immunochemical and functional inhibition studies. Shown are histograms representing the mean fluorescence intensities from three replicate microarray slides of H. pylori strains binding to printed neoglycoconjugates. The dashed line marks the level of marginal or background binding. Error bars represent the standard deviations of the means for three replicate microarray slides.
Mucus promotes infection by both H. pylori and C. jejuni.
These experiments point to a fundamental role of mucus in C. jejuni and H. pylori infection. In order to explore this further, we examined the interactions of the organisms with HT29-MTX-E12 cells, which harbor an adherent mucus layer, and compared colonization efficiencies with parental HT29-MTX (mucus-secreting) and HT29 (non-mucus-secreting) cells. To determine the effects of secreted mucins and an adherent mucus layer on the interactions between host cells and C. jejuni or H. pylori, a comparison was made of the colonization of 3 cell lines (HT29, HT29-MTX, and HT29-MTX-E12) with the two organisms. All colonization studies were carried out on cells grown on Transwell filters for 21 days, because at this time the HT29-MTX-E12 cells had produced a mature adherent mucus layer and had formed tight junctions, while HT29-MTX cells were also fully differentiated and secreted mucins (21). Higher numbers of H. pylori and C. jejuni bacteria colonized the HT29-MTX-E12 cells than the HT29-MTX or HT29 cells (Fig. 4). Strikingly, H. pylori did not colonize the non-mucus-secreting HT29 cells. In addition, while the organism could colonize the mucin-secreting HT29-MTX cells (6.32 × 102 CFU/ml), the number of organisms recovered from the HT29-MTX-E12 cells, with an adherent mucus layer, was markedly higher (2.4 × 107 CFU/ml) (P < 0.01) (Fig. 4). The numbers of C. jejuni organisms recovered from the HT29 and HT29-MTX cells (9 × 106 CFU/ml and 1.5 × 107 CFU/ml, respectively) were also higher than the numbers of H. pylori organisms recovered from these cells. Levels of colonization of HT29 and HT29-MTX-E12 cells were comparable at 42°C and 37°C, an indication that the different body temperatures of humans and chickens do not influence mucus interaction (see Fig. S1 in the supplemental material).
Fig 4.

Infection of HT29, HT29-MTX, and HT29-MTX-E12 cells by mucosal pathogens. The HT29-MTX subclone E12, with an adherent mucus layer, mucus-secreting HT29-MTX cells, and non-mucus-secreting HT29 cells were grown for 21 days on Transwell filters and were subsequently infected with C. jejuni or H. pylori organisms for 24 h. C. jejuni infected all cell lines. The numbers of C. jejuni organisms colonizing HT29-MTX and HT29-MTX-E12 cells were statistically significantly higher than the number colonizing HT29 cells (P < 0.05). In contrast, H. pylori colonized HT29-MTX-E12 cells and, to a much lesser extent, HT29-MTX cells but not HT29 cells. The data presented are means ± standard deviations for 3 replicate experiments (n = 9).
Binding of C. jejuni and H. pylori to HT29-MTX-E12 mucus and mucin.
In light of the distinctly different mucin-binding signatures of H. pylori and C. jejuni, we explored further their interactions with purified mucus and mucin from HT29-MTX-E12 cells. An antibody was used to detect bacteria on either mucus or purified mucin from HT29-MTX-E12 cells immobilized on a PVDF membrane. C. jejuni was found to bind to both the mucus and mucin fractions. In contrast, H. pylori bound directly only to mucus; no appreciable binding to mucin was detected (Fig. 5A). Binding could be abolished by sodium metaperiodate treatment of the mucus or mucin (Fig. 5B), indicating that the organisms were adhering to glycan epitopes. Probing of HT29-MTX-E12 mucin printed on microarray slides also demonstrated markedly lower levels of binding by H. pylori than by C. jejuni (Fig. 5C).
Fig 5.

Interactions of C. jejuni and H. pylori with mucins from the mucus-secreting cell lines HT29-MTX-E12 and LS174T. (A) Mucus and purified mucin from HT29-MTX-E12 cells were immobilized on a PVDF membrane and were probed with C. jejuni strain 81-176 or H. pylori strain 26695. Both C. jejuni and H. pylori bound to immobilized mucus, but only C. jejuni bound directly to purified mucin. (B) Oxidation of the glycan structures by sodium metaperiodate reduced the binding of C. jejuni and H. pylori to mucus as well as the interaction of C. jejuni with purified E12 mucin. (C and D) Interrogation of mucin microarrays with C. jejuni and H. pylori showed differential binding of both pathogens to mucins from two colonic cell lines, HT29-MTX-E12 (C) and LS174T (D). Histograms show mean fluorescence intensities from duplicate subarrays on three replicate microarray slides; values for each subarray are medians for six feature replicates. Error bars are standard deviations of the means for 3 experiments.
The observation that H. pylori bound poorly to HT29-MTX-E12 mucin was surprising given that the adherent mucus layer of this cell line contains the gastric mucin MUC5AC (21). H. pylori does not colonize the colon or adhere in vivo to colonic cells. Therefore, in order to determine whether the lack of binding of H. pylori to HT29-MTX-E12 mucin was a result of the colonic origin of the mucin, we compared the binding of H. pylori and C. jejuni to mucin purified from another colorectal adenocarcinoma cell line, LS174T. In contrast to the findings with HT29-MTX-E12 mucin, H. pylori interacted strongly with the LS174T mucin. In fact, C. jejuni also bound better to LS174T mucin than to E12 mucin (Fig. 5C and D).
O-glycan profile of purified secreted mucin from HT29-MTX-E12 cells.
Bacterial binding to mucins is mediated by interactions between bacterial adhesins and O-glycans on the mucins. The failure of H. pylori to interact with HT29-MTX-E12 mucin suggested that the relevant fucosylated O-glycan structures were not present on mucin originating from these cells. In order to investigate this finding further, we determined the O-glycan profile of purified secreted mucin from HT29-MTX-E12 cells. The structure of O-glycans detected in HT29-MTX-E12 mucins (Table 2) was not dissimilar to those reported for mucins secreted from related cell lines, such as HT29 (42) and HT29-MTX (43). They comprised mainly core 1- and core 2-related structures, with a high proportion of truncated structures (lower GU values) (Fig. 6). Analysis of O-glycans by WAX chromatography showed that a substantial proportion of the O-glycans present in mucins secreted from HT29-MTX-E12 cells were acidic. The same analysis after sialidase digestion showed no remnant acidic structures, confirming that all the charged species observed are sialylated glycans (Fig. 7). A noticeable paucity of fucosylated structures was detected, suggesting that a lack of relevant receptors could explain the weak interaction of H. pylori with E12 purified mucin.
Table 2.
Structures of O-glycans released from E-12 mucins, described by HILIC-HPLC and exoglycosidase digestion arrays

Glucose unit (GU) values are averages of triplicates, with a standard deviation of 0.06. Asterisks indicate proposed isomeric structures.
Structures are represented using the Oxford system for monosaccharides and linkages (Fig. 6, key) (59). ND, not determined.
The enzymes used were bovine testis β-galactosidase (BTG), Arthrobacter ureafaciens sialidase (ABS), Streptococcus pneumoniae sialidase (NAN1), Streptococcus pneumoniae β-N-acetylglucosaminidase (GUH), Streptococcus pneumoniae β-galactosidase (SPG), bovine kidney α-fucosidase (BKF), and almond meal α-fucosidase (AMF).
Fig 6.
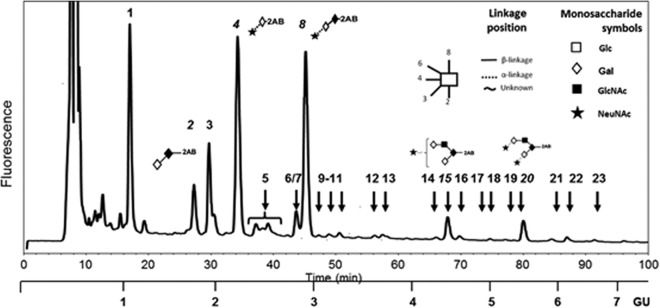
HILIC profile of fluorescently labeled O-glycans released from HT29-MTX-E12 mucins. O-glycans present in this sample were structurally characterized by HILIC-HPLC and exoglycosidase digestion patterns (see Materials and Methods). O-glycans are represented using the Oxford system for monosaccharides and linkages (59). A complete description of all structures can be found in Table 2; structures represented in this figure correspond to major peaks in the sample, for which numbers are italicized. Peak 4 is not a natural O-glycan but a peeling product obtained due to the chemical degradation of sialylated structures, most probably from structure 8 (sialyl-T antigen). The scale of glucose units (GU) is based on the elution of the 2-AB-labeled glucose ladder.
Fig 7.
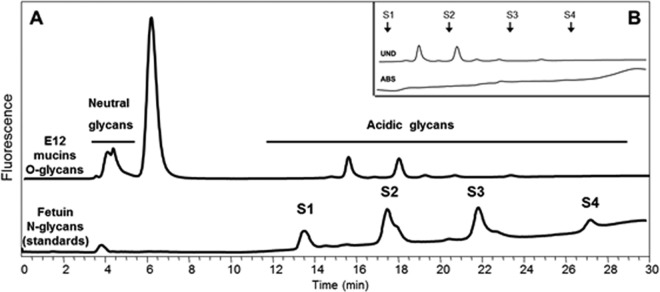
Weak anionic-exchange chromatography of O-glycans released from HT29-MTX-E12 mucins. Separation by charge results in different fractions of glycans, from neutral (retention time, 4.5 min) to charged (retention time, 11 to 28 min) species. The peak at 5.5 min corresponds to excess 2-aminobenzamide. The retention times of the different O-glycans are compared to those of the fetuin N-glycans, used as standards, where S1 corresponds to monosialylated N-glycans, and S2 to S4 correspond to di-, tri-, and tetrasialylated N-glycans, respectively. (B) Prevalence of sialylated structures in the acidic fraction of O-glycans from HT29-MTX-E12 mucins. This panel shows a zoom-in of the chromatogram in panel A (area between 11 and 30 min) before and after A. ureafaciens sialidase (ABS) digestion.
Presence of Lewisb and sialyl Lewisx antigens in HT29-MTX-E12 cells.
Despite the lack of detection of fucosylated structures in mucin purified from HT29-MTX-E12 cells, staining of the cells with an antibody against Lewisb clearly demonstrated the presence of Lewisb blood group antigen in the adherent mucus layer of the cells. Sialyl-Lewisx staining of the mucus layer was weaker, but cellular staining for this antigen showed the presence of this structure on HT29-MTX-E12 cells (Fig. 8).
Fig 8.

Expression of Lewisb and sialyl-Lewisx by HT29-MTX-E12 cells. Immunofluorescence with specific antibodies was used to assess the expression of the Lewisb or sialyl-Lewisx antigen by HT29-MTX-E12 cells grown for 21 days on Transwell filters. DAPI staining was used to visualize cell nuclei. Arrowheads indicate staining of the extracellular mucus layer, and arrows indicate cellular staining.
Interactions of H. pylori and C. jejuni with non-mucin-containing fractions of E12 mucus.
To determine the nonmucin component(s) of HT29-MTX-E12 mucus that contained Lewisb blood group antigens, mucus fractions were obtained from a CsCl gradient and were probed with an antibody against Lewisb. The first two fractions collected from the top of the gradient (densities, 1.27 to 1.28 g/ml) reacted with the anti-Lewisb antibody (Fig. 9A). Cellular glycolipids are known to act as receptors for the binding of both H. pylori and C. jejuni (14, 44) and have been found in CsCl density gradient fractions in the range in which we found Lewisb structures (45). Therefore, we asked whether HT29-MTX-E12 mucus contained glycolipids and, if so, whether these could provide a source of receptors for bacterial binding. Orcinol staining demonstrated the presence of glycolipids in the organic extract of the mucus separated on TLC plates (Fig. 9B). Probing of these glycolipids with antibodies against both Lewisb and sialyl Lewisx revealed that both glycans were expressed on the glycolipids (Fig. 9C). Furthermore, overlay assays with H. pylori and C. jejuni revealed that the organisms could bind to glycolipids isolated from HT29-MTX-E12 mucus (Fig. 9D). These results highlight the importance of non-mucin-based glycan interactions in mediating bacterial colonization of mucus.
Fig 9.

Interactions of H. pylori and C. jejuni with the non-mucin-containing fractions of HT29-MTX-E12 cell mucus. (A) Mucus from HT29-MTX-E12 cells was separated on a CsCl gradient and was then divided into 1-ml fractions. A sample of each non-mucin-containing fraction was blotted onto a PVDF membrane, which was probed with an antibody against Lewisb. Shown are 3 fractions taken from the top of the density gradient. (B) TLC separation and orcinol staining of glycolipids extracted from secreted HT29-MTX-E12 mucus. (C) TLC-separated glycolipids were probed with antibodies against Lewisb (Le B) and sialyl-Lewisx (Sialyl Le X). (D) TLC plates containing glycolipids were probed for the binding of either C. jejuni strain 81-176 or H. pylori strain 26695 or J99. When identical TLC plates that were not overlaid with microorganisms were probed with either anti-H. pylori or anti-C. jejuni and secondary antibodies, no signals were detected.
DISCUSSION
The gastrointestinal tracts of animals and humans are rich sources of glycans (46, 47) due to the presence of highly glycosylated mucin molecules in the mucus layer overlying the epithelial cells. These glycans are often exploited by pathogens to facilitate colonization and disease (46, 48, 49). The expression and glycosylation of mucins differ depending on a number of factors, including the species, the location within the body, inflammation, and the presence of microbes (48, 50, 51). To further the study of bacterial interactions with complex glycoproteins, a mucin microarray was developed containing a wide range of natural mucins, including those from a number of gastrointestinal sites in several animal species (22). A major strength of this technique is that the binding chemistry of the array slides allows for optimal presentation of the glycans, thereby maximizing the access of the bacteria to potential glycan receptors. This enables a quantitative analysis of the interaction of bacteria with secreted mucins in a high-throughput format. Indeed, this improved access to potential binding sites may explain why we observed a degree of binding of H. pylori to mucin from the HT29-MTX-E12 cells on the array that was not detectable when mucin immobilized on a PVDF membrane was used.
These natural mucin arrays have been shown previously to be an effective tool for the profiling of mucin glycoepitopes (22). Our analysis indicated that C. jejuni and H. pylori each interact directly with a specific subset of mucins. Each pathogen showed a marked preference for mucins from specific sites in certain animal species. While the interaction of H. pylori with gastric mucins has been extensively studied, little is known about the mechanisms used by C. jejuni to interact with mucins from either the human or the chicken gut. C. jejuni is most commonly identified as a commensal of chickens, which are the primary source of human infection, although it has been found in other species, including cattle, swine, and sheep (52, 53). However, despite the importance of C. jejuni colonization of chickens in the transmission of the organism to humans, there are no studies examining directly the interaction of C. jejuni with chicken mucins. C. jejuni showed a marked tropism for mucin obtained from chicken intestine. In addition to this species preference, C. jejuni also showed a preference for mucin from specific sites in the chicken gut (in descending order of preference: the large intestine, proximal small intestine, and cecum). While a previous study demonstrated an interaction between C. jejuni lipopolysaccharide (LPS) and rabbit intestinal mucus (54), this is the first study to report this tropism of C. jejuni for chicken mucin. In light of the current interest in bacteria and how they interact with mucosal surfaces (3–5), our results highlight the importance of selecting an appropriate species for the collection of mucus for such studies. The finding that the binding of C. jejuni to chicken mucin is influenced by the anatomical location from which the mucus was sourced echoes the results of our previous studies showing a gradient of inhibition of C. jejuni invasiveness by chicken mucin, with large intestinal mucus and mucin having the most profound effect (16, 17). Coupled with recent data suggesting differences in the presence or accessibility of glycans and/or differing proportions of sulfated glycans in different parts of the chicken intestine (22), these data point to the fundamental role of mucin glycosylation in dictating C. jejuni tropism and lifestyle (commensal versus pathogenic).
The strong interaction of C. jejuni with chicken mucin suggests that this is an important colonization factor for the organism. Therefore, alteration of the glycosylation profile of the chicken intestinal tract may be a viable means of decreasing the Campylobacter burden in chickens in order to reduce the risk of transmission to humans. In support of this, previous studies have shown that constituents of poultry feed alter mucin carbohydrates of the chick intestinal tract and that these alterations result in reduced numbers of C. jejuni bacteria colonizing the chicks (55).
Natural infection with H. pylori occurs only in humans and nonhuman primates. Perhaps, therefore, it is not surprising that the interactions between H. pylori and mucins from different animals were not as strong as that between C. jejuni and chicken mucin. It is of interest that H. pylori bound best to mucin isolated from porcine stomach mucus, since the gnotobiotic piglet was one of the first animal species to be experimentally infected with H. pylori (56). The BabA adhesins of H. pylori strains J99 and G27 were demonstrated to interact with porcine stomach mucin. However, strain 26695, though able to interact with porcine stomach mucin, did not appear to have a functional BabA adhesin, as assessed both by the mucin overlay blotting experiment and by testing of direct binding to an NGC array. This finding is in agreement with other studies (12). While the predicted protein sequences of the BabA adhesins are ∼91% identical (using BLAST algorithms from GenBank and the Institute for Genomic Research, now the J. Craig Venter Institute) for all three strains, the gene locations differ (11). In strains G27 and J99, babA is located at what is termed the babA locus, while in 26695, it is at a locus normally associated with a BabA paralogue, BabB (11). Despite the lack of a functional BabA protein, 26695 did not show a reduction in binding to mucins. This is in contrast to strain G27ΔbabA, which had significantly lower levels of binding to mucins than the wild-type strain G27. The BabA mutant was not complemented, and therefore, the possibility that spontaneous mutation or phase variation played a role in the reduction in binding cannot be ruled out. Nevertheless, these findings suggest that while BabA is an important H. pylori adhesin, other adhesin-glycan interactions must also occur (as in the case of 26695) to fulfill a similar function. The binding of H. pylori to sialyl-Lewisx is mediated by the OMP SabA (10). However, strain 26695 showed low binding to sialyl-Lewisx on the NGC arrays. A previous study showed that strain 26695 contains predominately out-of-frame sabA alleles that do not express functional SabA protein (57). What adhesins mediate the binding of strain 26695 to mucin has yet to be elucidated.
We have shown previously that HT29-MTX-E12 cells can be infected by both H. pylori and C. jejuni (20, 21). In this study, C. jejuni infection was enhanced in, but not dependent on, the presence of mucus, whereas H. pylori required the presence of the adherent mucus layer for effective infection. This suggests that either the mucus layer on HT29-MTX-E12 cells offered additional H. pylori receptors not present on the other cell lines or the mucus induced an alteration in adhesins produced by the organism that enabled interaction with the cells. Gastric mucins have been shown to promote the proliferation of H. pylori and to alter gene expression by the organism (58). Recently, we have shown that the interaction of H. pylori with the trefoil peptide TFF1, present in the adherent mucus layer of HT29-MTX E12 cells, promotes infection (21). This finding highlights the role of nonmucin components of mucus in mediating the colonization of mucosal surfaces and warrants their further investigation.
While C. jejuni bound strongly to purified E12 mucin, we detected markedly reduced interaction of H. pylori with E12 mucins. Structural analysis showed a paucity of fucosylated structures on mucin from HT29-MTX-E12 cells, despite the strong immunoreactivity of the adherent mucus layer with an antibody to Lewisb blood group antigen. CsCl gradient fractionation suggested that the Lewisb immunoreactive fractions colocated with glycolipids, a finding corroborated by the detection of both Lewisb and sialyl-Lewisx on purified glycolipids (Fig. 9). Moreover, both H. pylori and C. jejuni could interact with these glycolipid fractions. These findings emphasize the importance of considering mucus-bound glycolipids as a reservoir of microbial receptors that can be exploited by mucosal pathogens.
In summary, this study highlights the role mucus can play in promoting bacterial infection. It demonstrates that two closely related organisms interact very differently with mucins from various sources, most likely reflecting the site-specific nature of glycosylation. Further analysis of the glycans present in mucus may lead to the identification of unique ligands for these interactions and, subsequently, to the development of novel therapeutic or prophylactic compounds to reduce the burden of infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from Science Foundation Ireland (08/SRC/B1393).
We thank Marian Kane for help with this work and also Steffen Backert for donating H. pylori isolates G27 and G27ΔbabA.
Footnotes
Published ahead of print 28 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00415-13.
REFERENCES
- 1. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. 2008. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phillipson M, Johansson ME, Henriksnäs J, Petersson J, Gendler SJ, Sandler S, Persson AE, Hansson GC, Holm L. 2008. The gastric mucus layers: constituents and regulation of accumulation. Am. J. Physiol. Gastrointest. Liver Physiol. 295:G806–G812 [DOI] [PubMed] [Google Scholar]
- 3. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. 2009. Bacterial community variation in human body habitats across space and time. Science 326:1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. U. S. A. 108:6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R, Gordon JI. 2011. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332:970–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corfield AP, Carroll D, Myerscough N, Probert CS. 2001. Mucins in the gastrointestinal tract in health and disease. Front. Biosci. 6:D1321–D1357 [DOI] [PubMed] [Google Scholar]
- 7. Yamaoka Y. 2010. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 7:629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomsen LL, Gavin JB, Tasman-Jones C. 1990. Relation of Helicobacter pylori to the human gastric mucosa in chronic gastritis of the antrum. Gut 31:1230–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van de Bovenkamp JH, Mahdavi J, Korteland-Van Male AM, Buller HA, Einerhand AW, Boren T, Dekker J. 2003. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter 8:521–532 [DOI] [PubMed] [Google Scholar]
- 10. Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, Takahashi N, Yoshida M, Azuma T, Hattori M, Uchiyama I, Kobayashi I. 2011. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 11:104. 10.1186/1471-2180-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Odenbreit S, Swoboda K, Barwig I, Ruhl S, Boren T, Koletzko S, Haas R. 2009. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect. Immun. 77:3782–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cooke CL, An HJ, Kim J, Canfield DR, Torres J, Lebrilla CB, Solnick JV. 2009. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterology 137:1061–1071.e8 [DOI] [PubMed] [Google Scholar]
- 14. Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, Benghezal M, Marshall BJ, Peek RM, Jr, Boren T, Solnick JV. 2010. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect. Immun. 78:1593–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tu QV, McGuckin MA, Mendz GL. 2008. Campylobacter jejuni response to human mucin MUC2: modulation of colonization and pathogenicity determinants. J. Med. Microbiol. 57:795–802 [DOI] [PubMed] [Google Scholar]
- 16. Alemka A, Whelan S, Gough R, Clyne M, Gallagher ME, Carrington SD, Bourke B. 2010. Purified chicken intestinal mucin attenuates Campylobacter jejuni pathogenicity in vitro. J. Med. Microbiol. 59:898–903 [DOI] [PubMed] [Google Scholar]
- 17. Byrne CM, Clyne M, Bourke B. 2007. Campylobacter jejuni adhere to and invade chicken intestinal epithelial cells in vitro. Microbiology 153:561–569 [DOI] [PubMed] [Google Scholar]
- 18. Ruiz-Palacios GM, Cervantes LE, Ramos P, Chavez-Munguia B, Newburg DS. 2003. Campylobacter jejuni binds intestinal H(O) antigen (Fucα1, 2Galβ1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J. Biol. Chem. 278:14112–14120 [DOI] [PubMed] [Google Scholar]
- 19. Day CJ, Tiralongo J, Hartnell RD, Logue CA, Wilson JC, von Itzstein M, Korolik V. 2009. Differential carbohydrate recognition by Campylobacter jejuni strain 11168: influences of temperature and growth conditions. PLoS One 4:e4927. 10.1371/journal.pone.0004927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alemka A, Clyne M, Shanahan F, Tompkins T, Corcionivoschi N, Bourke B. 2010. Probiotic colonization of the adherent mucus layer of HT29MTXE12 cells attenuates Campylobacter jejuni virulence properties. Infect. Immun. 78:2812–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dolan B, Naughton J, Tegtmeyer N, May FE, Clyne M. 2012. The interaction of Helicobacter pylori with the adherent mucus gel layer secreted by polarized HT29-MTX-E12 cells. PLoS One 7:e47300. 10.1371/journal.pone.0047300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kilcoyne M, Gerlach JQ, Gough R, Gallagher ME, Kane M, Carrington SD, Joshi L. 2012. Construction of a natural mucin microarray and interrogation for biologically relevant glyco-epitopes. Anal. Chem. 84:3330–3338 [DOI] [PubMed] [Google Scholar]
- 23. Hu L, Kopecko DJ. 1999. Campylobacter jejuni 81-176 associates with microtubules and dynein during invasion of human intestinal cells. Infect. Immun. 67:4171–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Korlath JA, Osterholm MT, Judy LA, Forfang JC, Robinson RA. 1985. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J. Infect. Dis. 152:592–596 [DOI] [PubMed] [Google Scholar]
- 25. Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668 [DOI] [PubMed] [Google Scholar]
- 26. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 27. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180 [DOI] [PubMed] [Google Scholar]
- 28. Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. 2009. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191:447–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tom BH, Rutzky LP, Jakstys MM, Oyasu R, Kaye CI, Kahan BD. 1976. Human colonic adenocarcinoma cells. I. Establishment and description of a new line. In Vitro 12:180–191 [DOI] [PubMed] [Google Scholar]
- 30. von Kleist S, Chany E, Burtin P, King M, Fogh J. 1975. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J. Natl. Cancer Inst. 55:555–560 [DOI] [PubMed] [Google Scholar]
- 31. Gouyer V, Leteurtre E, Zanetta JP, Lesuffleur T, Delannoy P, Huet G. 2001. Inhibition of the glycosylation and alteration in the intracellular trafficking of mucins and other glycoproteins by GalNAcα-O-bn in mucosal cell lines: an effect mediated through the intracellular synthesis of complex GalNAcα-O-bn oligosaccharides. Front. Biosci. 6:D1235–D1244 [DOI] [PubMed] [Google Scholar]
- 32. Behrens I, Stenberg P, Artursson P, Kissel T. 2001. Transport of lipophilic drug molecules in a new mucus-secreting cell culture model based on HT29-MTX cells. Pharm. Res. 18:1138–1145 [DOI] [PubMed] [Google Scholar]
- 33. Kilcoyne M, Gerlach JQ, Kane M, Joshi L. 2012. Surface chemistry and linker effects on lectin–carbohydrate recognition for glycan microarrays. Anal. Methods 4:2721–2728 [Google Scholar]
- 34. Reeves EP, Ali T, Leonard P, Hearty S, O'Kennedy R, May FE, Westley BR, Josenhans C, Rust M, Suerbaum S, Smith A, Drumm B, Clyne M. 2008. Helicobacter pylori lipopolysaccharide interacts with TFF1 in a pH-dependent manner. Gastroenterology 135:2043–2054.e2 [DOI] [PubMed] [Google Scholar]
- 35. van Amsterdam K, van der Ende A. 2004. Nutrients released by gastric epithelial cells enhance Helicobacter pylori growth. Helicobacter 9:614–621 [DOI] [PubMed] [Google Scholar]
- 36. Cottet S, Corthesy-Theulaz I, Spertini F, Corthesy B. 2002. Microaerophilic conditions permit to mimic in vitro events occurring during in vivo Helicobacter pylori infection and to identify Rho/Ras-associated proteins in cellular signaling. J. Biol. Chem. 277:33978–33986 [DOI] [PubMed] [Google Scholar]
- 37. Mooney A, Byrne C, Clyne M, Johnson-Henry K, Sherman P, Bourke B. 2003. Invasion of human epithelial cells by Campylobacter upsaliensis. Cell. Microbiol. 5:835–847 [DOI] [PubMed] [Google Scholar]
- 38. Royle L, Campbell MP, Radcliffe CM, White DM, Harvey DJ, Abrahams JL, Kim YG, Henry GW, Shadick NA, Weinblatt ME, Lee DM, Rudd PM, Dwek RA. 2008. HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal. Biochem. 376:1–12 [DOI] [PubMed] [Google Scholar]
- 39. Merry AH, Neville DC, Royle L, Matthews B, Harvey DJ, Dwek RA, Rudd PM. 2002. Recovery of intact 2-aminobenzamide-labeled O-glycans released from glycoproteins by hydrazinolysis. Anal. Biochem. 304:91–99 [DOI] [PubMed] [Google Scholar]
- 40. Keely S, Rawlinson LA, Haddleton DM, Brayden DJ. 2008. A tertiary amino-containing polymethacrylate polymer protects mucus-covered intestinal epithelial monolayers against pathogenic challenge. Pharm. Res. 25:1193–1201 [DOI] [PubMed] [Google Scholar]
- 41. Muindi K, Cernadas M, Watts GF, Royle L, Neville DC, Dwek RA, Besra GS, Rudd PM, Butters TD, Brenner MB. 2010. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d. Proc. Natl. Acad. Sci. U. S. A. 107:3052–3057 (Corrected.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huet G, Gouyer V, Delacour D, Richet C, Zanetta JP, Delannoy P, Degand P. 2003. Involvement of glycosylation in the intracellular trafficking of glycoproteins in polarized epithelial cells. Biochimie 85:323–330 [DOI] [PubMed] [Google Scholar]
- 43. Capon C, Laboisse CL, Wieruszeski JM, Maoret JJ, Augeron C, Fournet B. 1992. Oligosaccharide structures of mucins secreted by the human colonic cancer cell line CL.16E. J. Biol. Chem. 267:19248–19257 [PubMed] [Google Scholar]
- 44. Lingwood CA. 1999. Glycolipid receptors for verotoxin and Helicobacter pylori: role in pathology. Biochim. Biophys. Acta 1455:375–386 [DOI] [PubMed] [Google Scholar]
- 45. Cheung MD, Albers JJ. 1982. Distribution of high density lipoprotein particles with different apoprotein composition: particles with A-I and A-II and particles with A-I but no A-II. J. Lipid Res. 23:747–753 [PubMed] [Google Scholar]
- 46. Hooper LV, Gordon JI. 2001. Commensal host-bacterial relationships in the gut. Science 292:1115–1118 [DOI] [PubMed] [Google Scholar]
- 47. Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. 2005. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307:1955–1959 [DOI] [PubMed] [Google Scholar]
- 48. Corfield AP, Wiggins R, Edwards C, Myerscough N, Warren BF, Soothill P, Millar MR, Horner P. 2003. A sweet coating—how bacteria deal with sugars. Adv. Exp. Med. Biol. 535:3–15 [PubMed] [Google Scholar]
- 49. Moran AP, Gupta A, Joshi L. 2011. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut 60:1412–1425 [DOI] [PubMed] [Google Scholar]
- 50. Lebeer S, Vanderleyden J, De Keersmaecker SC. 2010. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat. Rev. Microbiol. 8:171–184 [DOI] [PubMed] [Google Scholar]
- 51. Singh PK, Hollingsworth MA. 2006. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 16:467–476 [DOI] [PubMed] [Google Scholar]
- 52. Fricker CR, Park RW. 1989. A two-year study of the distribution of ‘thermophilic’ campylobacters in human, environmental and food samples from the Reading area with particular reference to toxin production and heat-stable serotype. J. Appl. Bacteriol. 66:477–490 [DOI] [PubMed] [Google Scholar]
- 53. Zanetti F, Varoli O, Stampi S, De Luca G. 1996. Prevalence of thermophilic Campylobacter and Arcobacter butzleri in food of animal origin. Int. J. Food Microbiol. 33:315–321 [DOI] [PubMed] [Google Scholar]
- 54. McSweegan E, Walker RI. 1986. Identification and characterization of two Campylobacter jejuni adhesins for cellular and mucous substrates. Infect. Immun. 53:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fernandez F, Sharma R, Hinton M, Bedford MR. 2000. Diet influences the colonisation of Campylobacter jejuni and distribution of mucin carbohydrates in the chick intestinal tract. Cell. Mol. Life Sci. 57:1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eaton KA, Morgan DR, Krakowka S. 1992. Motility as a factor in the colonisation of gnotobiotic piglets by Helicobacter pylori. J. Med. Microbiol. 37:123–127 [DOI] [PubMed] [Google Scholar]
- 57. Goodwin AC, Weinberger DM, Ford CB, Nelson JC, Snider JD, Hall JD, Paules CI, Peek RM, Jr, Forsyth MH. 2008. Expression of the Helicobacter pylori adhesin SabA is controlled via phase variation and the ArsRS signal transduction system. Microbiology 154:2231–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skoog EC, Sjoling A, Navabi N, Holgersson J, Lundin SB, Linden SK. 2012. Human gastric mucins differently regulate Helicobacter pylori proliferation, gene expression and interactions with host cells. PLoS One 7:e36378. 10.1371/journal.pone.0036378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harvey DJ, Merry AH, Royle L, Campbell MP, Dwek RA, Rudd PM. 2009. Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics 9:3796–3801 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.