

Abstract
The p35 molecule is unique to interleukin-12 (IL-12), while p40 is shared by both IL-12 and IL-23. IL-12 promotes Th1 T cell responses, while IL-23 promotes Th17 T cell responses. The roles of IL-12p35- and IL-12p40-mediated responses in chlamydial infection were compared in mice following an intravaginal infection with Chlamydia muridarum. Mice deficient in either IL-12p35 or p40 both developed similar but prolonged infection time courses, confirming the roles of IL-12-mediated immune responses in clearing primary infection. However, all mice, regardless of genotype, cleared reinfection within 2 weeks, suggesting that an IL-12- or IL-23-independent adaptive immunity is protective against chlamydial infection. All infected mice developed severe oviduct hydrosalpinx despite the increased Th2 responses in IL-12p35- or IL-12p40-deficient mice, suggesting that Th2-dominant responses can contribute to Chlamydia-induced inflammatory pathology. Compared to IL-12p35 knockout mice, the IL-12p40-deficient mice exhibited more extensive spreading of chlamydial organisms into kidney tissues, leading to significantly increased incidence of pyelonephritis, which both confirms the role of IL-12 or IL-23-independent host responses in Chlamydia-induced pathologies and suggests that in the absence of IL-12/IFN-γ-mediated Th1 immunity, an IL-23-mediated response may play an important role in restricting chlamydial organisms from spreading into distal organs. These observations together provide important information for both understanding chlamydial pathogenesis and developing anti-Chlamydia vaccines.
INTRODUCTION
Chlamydia trachomatis infection in human urogenital tract epithelial tissues is a major cause of sexually transmitted bacterial diseases, potentially leading to severe complications, such as tubal factor infertility (1). However, the pathogenic mechanisms of C. trachomatis-induced diseases remain unknown, and there is still no licensed anti-C. trachomatis vaccine. Although Chlamydia muridarum organisms cause no known human diseases, they have been extensively used to study the mechanisms of C. trachomatis pathogenesis and immunity. By using the C. muridarum urogenital tract infection mouse model in combination with strategies such as in vivo depletion and gene knockout, CD4+ T cell-dependent and gamma interferon (IFN-γ)-mediated immunity has been identified as a major protective mechanism for mice to control chlamydial infection (2). However, the same Th1 response may also contribute to Chlamydia-induced inflammatory pathologies. In addition, the contribution of the IL-23/Th17 axis to Chlamydia-induced immunity and pathology remains unclear.
Interleukin-12 (IL-12), an essential inflammatory cytokine for the development of Th1 T cell immunity, consists of a p35 and p40 subunit (3). While p35 is unique to IL-12, p40 is shared with IL-23 (consisting of p40 and p19 subunits), another inflammatory cytokine required for the development of Th17 T cell immunity (4). Mice with a p35 knockout (KO) fail to develop IFN-γ-dominant Th1 immunity, while mice with a p40 KO produce neither IFN-γ nor IL-17 upon bacterial infection (5, 6). These KO mice have been extensively used to define the relative roles of the IL-12/IFN-γ and IL-23/IL-17 axes in controlling infections. IL-12 was found to play a dominant role in mouse resistance to toxoplasmosis, although in the absence of the IL-12, IL-23 could also play a limited role (7). The role of IL-23 in host defense is often overshadowed by IL-12. For example, only in the absence of IL-12 was an IL-23-dependent response associated with protective immunity to Salmonella enterica systemic infection (8). Both IL-12- and IL-23-mediated responses can also contribute to inflammatory pathologies. As a result, an anti-p40 neutralization antibody has been used to treat inflammatory diseases both in animal models (9) and clinic trials (10). Many studies have been carried out to investigate the roles of IL-12 and IL-23/IL-17 in chlamydial infection. IL-12 production by chlamydia-pulsed DCs was required for adoptively transferred DCs to confer protection against chlamydial infection in the recipient mice (11, 12); mice deficient in IL-12p35 or IL-12p40 were highly susceptible to pulmonary infection with C. muridarum (13, 14). Treatment of mice with anti-IL-17 neutralization antibodies led to increased susceptibility to airway infection with chlamydial organisms (15, 16). Using neutralization antibodies to IL-12 and IFN-γ, respectively, Perry et al. demonstrated an IL-12-dependent but IFN-γ-independent protection mechanism against urogenital infection with C. muridarum (17). This observation may suggest a role of the IL-23/IL-17 axis in anti-chlamydial urogenital infection, since the anti-IL-12 neutralization antibody might also block p40 in IL-23. However, a recent study showed that although IL-17 contributed to the generation of Th1 immunity and neutrophil recruitments but was not required for clearing chlamydial urogenital tract infection (18). Thus, the precise roles of IL-12/IFN-γ and IL-23/IL-17 axes in anti-chlamydial immunity and Chlamydia-induced pathology remain unclear.
In the present study, we compared the roles of IL-12p35- and IL-12p40-mediated responses in both protective immunity and pathology following an intravaginal infection with Chlamydia muridarum. Although mice deficient in IL-12p35 and those deficient in p40 both displayed a significant delay in resolving C. muridarum primary infection and developed severe oviduct pathology, IL-12p40 KO mice permitted more extensive chlamydial organism spreading into kidney tissues, leading to more severe renal pathology, which suggests that IL-23-mediated responses may play important roles in reducing chlamydial spreading into distal organs, while the IL-12/IFN-γ axis appeared to be critical in clearing primary infection. Furthermore, the IL-12p35 and IL-12p40 KO mouse models have also allowed us to uncover important roles of IL-12/IL-23-independent host responses in both anti-chlamydial immunity and chlamydial pathogenesis, since these KO mice were still able to both acquire a robust protective immunity against reinfection with C. muridarum and to develop severe pathologies in upper genital tracts and kidneys after primary infection.
MATERIALS AND METHODS
Chlamydial organisms and infection.
C. muridarum (Nigg strain) was propagated in HeLa cells (human cervical carcinoma epithelial cells, ATCC CCL2), purified, aliquoted, and stored as described previously (19). Female C57BL/6J mice, either wild type (stock number 000664) or with a deficiency in the IL-12p35 gene (B6.129S1-Il12atm1Jm/J, stock number 002692) or the IL-12p40 gene (B6.129S1-Il12btm1Jm/J, stock number 002693), were purchased at the age of 5 to 6 weeks from The Jackson Laboratory (Bar Harbor, ME). Each mouse was inoculated intravaginally with 1 × 104 inclusion-forming units (IFUs) of live C. muridarum organisms in 20 μl of SPG (sucrose-phosphate-glutamate buffer). Five days prior to infection, each mouse was injected subcutaneously with 2.5 mg Depo-provera (Pharmacia Upjohn, Kalamazoo, MI) to synchronize estrus cycles and increase mouse susceptibility to chlamydial infection. For some mice, a secondary infection was similarly carried out at day 114 after primary infection. Depo-provera was also applied 5 days prior to the secondary infection. For in vitro infection of HeLa cells, HeLa cells grown on coverslips in 24-well plates containing Dulbecco's modified Eagle medium (DMEM) (GIBCO BRL, Rockville, MD) with 10% fetal calf serum (FCS; GIBCO BRL) at 37°C in an incubator supplied with 5% CO2 were inoculated with C. muridarum organisms as described previously (20). The infected cultures were processed for immunofluorescence assay as described below.
Monitoring mouse shedding of live chlamydial organisms and titrating live organisms from kidney tissues.
To monitor live organism shedding, vaginal swabs were taken on different days after intravaginal infection (once every 3 to 4 days for the first 50 days and once per week thereafter). Each swab was suspended in 500 μl of SPG followed by sonication on ice, and the released organisms were titrated on HeLa cell monolayers in duplicate as described previously (9). The total number of IFUs per swab was calculated and converted into log10 values, and the log10 IFUs were used to calculate means and standard deviations for each group at each time point.
In some experiments, the whole kidney tissues were homogenized in 5 ml SPG using tissue grinders (Fisher Scientific, Pittsburgh, PA). Aliquots of the homogenates were used for titrating live chlamydial organisms or bacteria. For titrating chlamydial organisms, the homogenates were briefly sonicated. The released organisms were titrated on HeLa cell monolayers as described above. For titrating bacteria, the homogenates were serially diluted and plated on blood agar plates (BBL Trypticase soy agar with 10% sheep blood; BD, Franklin Lakes, NJ), and the plates were incubated for 2 days to allow as many bacteria as possible to grow for colony counting.
Evaluating mouse genital tract tissue and kidney pathologies and histological scoring.
Urogenital tract tissues and kidneys were harvested from mice sacrificed 114 (for those with primary infection only) or 143 (for those with a secondary infection) days (or as indicated for individual experiments) after the primary infection. Before the tissues were removed from mouse bodies, an in situ gross examination was performed for evidence of hydrosalpinx and enlarged, swollen, or necrotic kidneys or any other related abnormalities of both oviducts and kidneys. The severity of hydrosalpinx was scored based on the following criteria: no hydrosalpinx (0); hydrosalpinx detectable only under a stereoscope (1); hydrosalpinx clearly visible with the naked eye but smaller than (2), equal to (3), or larger than (4) the ovary on the same side. The excised tissues, after being photographed, were fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned longitudinally (5 μm/section). Efforts were made to include cervix, both uterine horns, and oviducts as well as lumenal structures of each tissue in each section. The sections were stained with hematoxylin and eosin (H&E) as described elsewhere (21). The H&E-stained sections were assessed by pathologists (I.-T.Y. and G.W.) who were blinded to mouse treatment and scored for severity of inflammation and pathologies based on the modified schemes established previously (21, 22). The uterine horns and oviducts were scored separately (only the oviduct scores were used in the present study). Scoring for dilatation of the uterine horns and oviducts was as follows: 0, no significant dilatation; 1, mild dilatation of a single cross section; 2, one to three dilated cross sections; 3, more than three dilated cross sections; and 4, confluent pronounced dilation. Scoring for inflammatory cell infiltrates was as follows (at the chronic stage of infection, the infiltrates mainly contain mononuclear cells): 0, no significant infiltration; 1, infiltration at a single focus; 2, infiltration at two to four foci; 3, infiltration at more than four foci; and 4, confluent infiltration. Scores were assigned to individual mice, and means ± standard errors were calculated for each group of animals.
For kidneys, gross pathology was determined based on size, color, shape, and surface smoothness. All kidneys identified as having gross pathology were marked in the corresponding relevant data figures. These kidneys exhibited typical gross pathology features of pyelonephritis as a result of urinary tract ascending infection, including being swollen and heavy, with a pale cortex and a dark hyperemic medulla. After being photographed, the kidneys were subjected to tissue sectioning as described above. The H&E-stained kidney sections were assessed by a pathologist (G.W.) who was blinded to mouse treatment and scored for severity of inflammation based on the modified schemes established previously (23). Briefly, the inflammation scoring criteria are as follows: normal renal histology, 0; mild inflammation (pyelitis) with infiltration of low to moderate numbers of neutrophils in the pelvic cavity and necrosis of individual epithelial cells but intact uroepithelium, 1; moderate pyelonephritis with diffuse damage to the uroepithelium and infiltration of moderate numbers of neutrophils in the uroepithelium and in the parenchyma adjacent to the pelvic cavity, 2; severe pyelitis with moderate inflammation in the parenchyma adjacent to the pelvis with moderate damage to the uroepithelium (which initially was disrupted because of necrosis and ulceration or later was thickened by regenerative hyperplasia), 3; acute pyelonephritis involving <50% of the kidney, 4; and severe acute pyelonephritis with extensive (>50%) damage, 5. Scores were assigned to individual mice for plotting and analyses. In addition, we also counted the polymorphic nuclear cells (PMNs) and mononuclear cells (MCs) in the inflammatory infiltrates. The total number of inflammatory cells counted from each section was 100. The ratio of PMNs/MCs was calculated for each section, and the average ratio from 3 sections cut from the same kidney (with an interval of 10 sections) was assigned to the corresponding mouse.
Immunofluorescence assay.
HeLa cells grown on coverslips with or without chlamydial infection were fixed and permeabilized for immunostaining as described previously (24). Hoechst (blue; Sigma) was used to visualize nuclear DNA. For titrating IFUs from mouse vaginal swab and kidney tissue homogenate samples, a mouse anti-chlamydial lipopolysaccharide antibody (clone MB5H9) (G. Zhong, unpublished observation) plus a goat anti-mouse IgG conjugated with Cy3 (red; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used to visualize chlamydial inclusions. For titrating and isotyping mouse antisera, C. muridarum-infected HeLa cells grown on coverslips were used as antigens. Binding of the serially diluted mouse antisera to antigens was detected with the fluorescence-conjugated goat antibodies against mouse μ (for IgM), γ (for total IgG), γ1 (for IgG1), γ2a (for IgG2a), γ2b (for IgG2b), and γ3 (for IgG3) chains (all from Jackson ImmunoResearch). The highest dilution of a given antiserum still showing a positive recognition of C. muridarum inclusions in HeLa cells was considered the titer of the antiserum, and the geometric titers were converted to log10 values for calculating means and standard deviations (19). For detecting C. muridarum inclusions in kidney sections, 3 sections were collected from each kidney tissue block with an interval of 10 cuts between sections. After deparaffinization and antigen retrieval treatments, the tissue section slides were blocked with 10% goat serum (catalog number 200-6210; Life Technologies, Grand Island, NY). A rabbit anti-antiserum against the mouse pneumonitis agent (G. Zhong, unpublished data) was applied to the slides followed by a Cy2-conjugated goat anti-rabbit IgG (green) (Jackson ImmunoResearch Laboratories) together with Hoechst dye (blue) (Sigma-Aldrich). All immunofluorescence-labeled samples were observed under an Olympus AX-70 fluorescence microscope equipped with multiple filter sets (Olympus, Melville, NY).
Enzyme-linked immunosorbent assay (ELISA).
Cytokines from supernatants of mouse spleen cells restimulated with chlamydial organisms were measured using commercially available ELISA kits. The kits for mouse IFN-γ (catalog no. DY485), IL-17 (catalog no. DY420), IL-4 (catalog no. DY404), and IL-5 (catalog no. DY405) were all obtained from R&D Systems, Inc. (Minneapolis, MN). The ELISA was performed following the instructions provided by the manufacturer or described elsewhere (25–27). A soluble colorimetric substrate [2-2′-azino-di-(3-ethylbenzthiazoline) sulfonic acid (ABTS)] was used to detect the secondary antibody conjugate binding. The absorbance taken at 405 nm using a microplate reader (Molecular Devices Corporation, Sunnyvale, CA) was used to calculate cytokine concentrations (pg per ml). To prepare mouse spleen cell in vitro restimulation cultures, splenocytes were harvested from mice infected with C. muridarum on day 114 (primary infection only) or 143 (primary and secondary infections) after the primary infection and cultured in 96-well plates at a density of 5 × 106 cells/well with or without stimulation with UV-inactivated C. muridarum elementary bodies at a final concentration of 1 × 106 IFUs/ml. After 3 days of culture, the culture supernatants were collected for cytokine measurements.
Statistical analyses.
Analysis of variance (ANOVA) was performed to analyze the cytokine concentrations from multiple groups and a two-tailed Student t test to compare each pair. The Kruskal-Wallis test was used to analyze the differences in IFUs recovered from mouse swabs and tissues and ratios of cell types and IgG isotype titers. The pathology score data were analyzed with the Wilcoxon rank sum test. Fisher's exact test was used to analyze category data, including the percentage of mice with oviduct hydrosalpinx or gross pathology-positive kidneys. Log transformation was used for analyzing the differences in live organism recovery (as IFUs) between different groups of mice. For calculation purpose, mice with zero IFUs were assigned a value of 1.
RESULTS
Mice deficient in IL-12p35 or p40 are highly susceptible to C. muridarum primary infection but are still able to develop robust protective immunity against reinfection.
We compared mice deficient in IL-12p35 (n = 13 mice), or p40 (n = 16) with wild-type mice (n = 15) for their susceptibility to C. muridarum infection in the urogenital tract (Fig. 1). All mice, regardless of genotype, shed similar levels of live organisms up to 3 weeks after infection. By day 21, the wild-type mice significantly reduced organism shedding, while both p35 and p40 KO mice still maintained significant levels of shedding. By day 27, only 3 out of 15 wild-type mice were positive for shedding of live organisms, while 10 out of 13 p35 KO and 14 out of 16 p40 KO mice were still positive. The p40 KO mice seemed to shed a higher level of live organisms than the p35 KO mice on day 27. However, the difference was not maintained. By day 31, the two IL-12 KO groups both released about 100 live organisms per mouse swab, while the wild-type mice completely cleared infection. The two KO groups continued to shed live organisms until day 87 after infection. Apparently, deficiency in IL-12p35 or p40 genes extended the primary infection time course by >50 days. On day 114, portion of the mice from each group (8 mice from the wild-type group and 7 from IL-12p35−/− or p40−/− group, respectively) were reinfected with the same amount of C. muridarum organisms. The mice without reinfection were sacrificed on day 114 for evaluating pathology and host immune responses. Upon reinfection, only up to 1,000 live organisms were recovered from any given mouse on day 3, and the reinfection was cleared within 2 weeks regardless of the mouse genotype, indicating that all mice developed protective immunity against C. muridarum reinfection, which suggests that IL-12p35 or p40-independent adaptive immunity can be protective against chlamydial infection.
Fig 1.

Effect of IL-12p35 or p40 deficiency on live organism shedding following chlamydial infection. (A) Wild-type mice or mice deficient in IL-12p35 (IL-12p35 KO) or IL-12p40 (IL-12p40 KO) were infected intravaginally with C. muridarum organisms, and vaginal swabs were taken on the days indicated on the x axis for measuring the number of live organisms (inclusion forming units [IFUs]). The number of IFUs from each swab was converted to a log10 value, and the log10 IFUs were used to calculate the mean and standard deviation for each mouse group at each time point. The wild-type group started with 15 mice, the IL-12p35KO group with 13, and the IL-12p40KO with 16. On day 114 after the primary infection, 7 (both KO groups) or 8 (wild-type group) mice from each group were reinfected with C. muridarum organisms. Mice that were negative for shedding 2 consecutive times were no longer swabbed. The number of mice that showed positive live organism shedding at each time point from each group were used to calculate the percentage of mice positive for shedding (B). All mice with primary infection only were sacrificed on day 114, while mice with both primary and secondary infection on day 143 after primary infection. The log10 IFUs and percent shedding-positive mice were compared between each two groups (**, P < 0.01). Note that the wild-type mice resolved their infections within 30 days after primary infection, while mice deficient in IL-12p35 or IL-12p40 maintained statistically significantly higher levels of live organism shedding starting on day 21, and the infection lasted up to 87 days. However, all mice cleared reinfection within 2 weeks.
Mice deficient in IL-12p35 or p40 are able to develop severe upper genital tract pathology.
All mice, regardless of genotype, developed severe upper genital tract pathology after primary infection or primary plus secondary infection (Fig. 2). Neither the incidence nor the severity of hydrosalpinx was significantly different among the mouse groups. The consistent severe upper genital tract pathology was further confirmed under microscopy, which revealed no significant differences in either the extent of inflammation infiltration (severity score) or lumenal dilation of oviduct among the three groups of mice. These observations suggest that IL-12p35 or p40-independent host responses can significantly contribute to Chlamydia-induced upper genital tract pathology.
Fig 2.
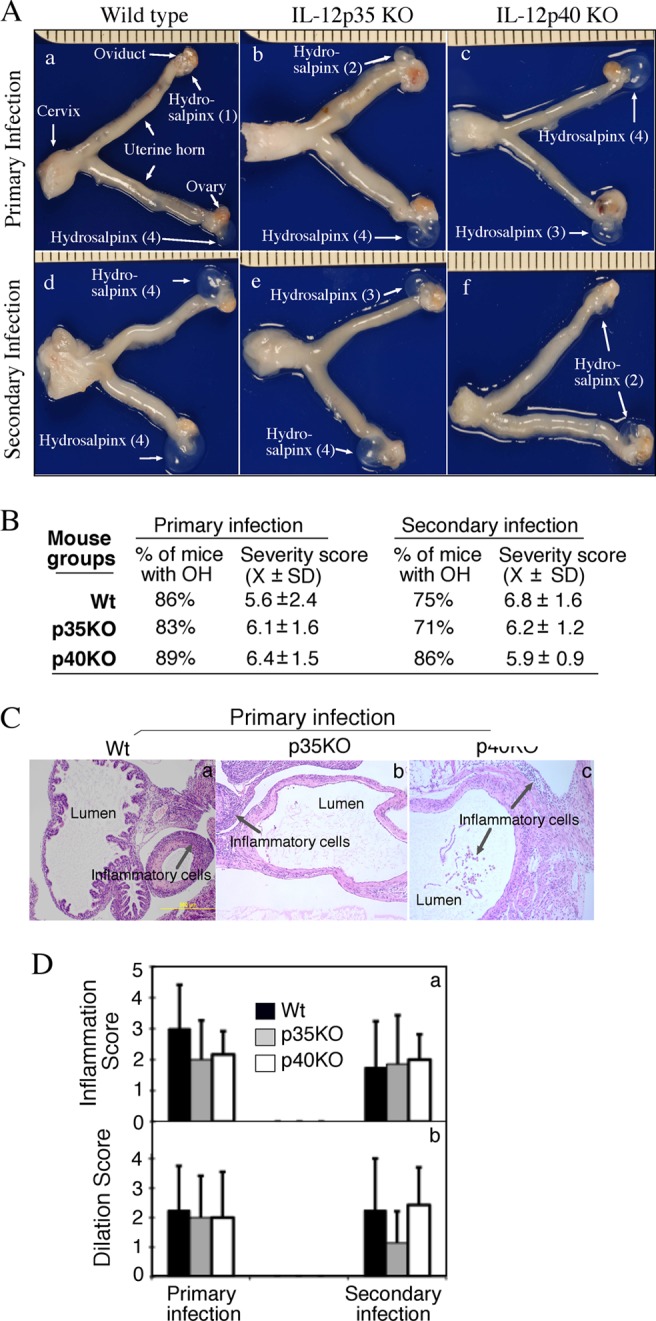
Effect of IL-12p35 or p40 deficiency on mouse hydrosalpinx development following chlamydial infection. (A) When urogenital tract tissues from wild-type (panel a, n = 7 with primary infection; panel d, n = 8 with secondary infection), IL-12p35 KO (b, n = 6; e, n = 7), and IL-12p40 KO (c, n = 9; f, n = 7) mice were examined for gross appearance, obvious hydrosalpinx (OH) was noted; representative images from all groups are shown. The hydrosalpinx severity was semiquantitatively graded from 0 to 4, and scores are given in parentheses. The total score from both oviducts was assigned as the score for a given mouse. Both the incidence (percentage of mice with either unilateral or bilateral OH) and severity (mean ± standard deviation) of hydrosalpinx (but the medians were used for statistical analyses) are listed (B). There were no significant differences in either the number of mice positive for OH or the severity of hydrosalpinx between different groups. After images were taken and gross pathology was evaluated, the oviduct tissues were further examined microscopically (C). Both inflammatory infiltration (gray arrows) and dilated lumen were noted and further semiquantitated according to the criteria listed in the Materials and Methods section; data are expressed as medians plus standard deviations (D). As with gross pathology, there was no significant difference in oviduct histopathology between different groups of mice.
Mice deficient in IL-12p40 are able to develop more severe renal pathology.
When mouse kidneys were examined for gross appearance, enlarged and edematous kidneys were noted (Fig. 3). These kidneys exhibited gross pathology consistent with pyelonephritis. The kidneys were swollen and heavy, with a pale cortex and a dark hyperemic medulla. The abnormal kidneys were found in most mice deficient in IL-12p40, while no obvious abnormality was noted in any of the kidneys from wild-type (WT) mice. The abnormal kidneys were found in only some IL-12p35−/− mice (Fig. 3A). The number of mice with abnormal kidneys in the p40 KO group was significantly higher than that in the wild-type group after both primary and secondary infections and than that in the p35 KO group after primary infection (Fig. 3B). Despite the lack of statistical significance, 86% of IL-12p40 KO mice had kidneys with pathology, while only 43% of IL-12p35 KO mice did after the secondary infection. The lack of statistical difference might be due to the limited number of animals in each group. Neither the age-matched IL-12p35 KO nor p40 KO mice developed any gross kidney pathology in the absence of chlamydial infection, suggesting that the observed kidney pathology is dependent on chlamydial infection. The kidney gross pathology was further confirmed using microscopy (Fig. 4). Microscopic examination of sections from enlarged kidneys revealed very thin renal cortex and extremely enlarged cavities of renal pelvis and calices with phlogistic exudation (Fig. 4A). When PMNs and MCs were counted separately, we found that ratios of PMNs/MCs were within the range of 0.41 (KO groups) to 0.80 (WT group) among all mice (there were not enough inflammatory foci in kidneys of wild-type mice after secondary infection, and thus, no ratio was calculated), and there were no statistically significant differences between any two groups (data not shown), probably due to large variations between different mice within a given group and small sample sizes. Nevertheless, the low ratios of PMNs to MCs may reflect the chronic nature of the kidney inflammation (kidneys harvested >100 days after chlamydial infection). The overall renal inflammation was semiquantitatively scored (Fig. 4B). Both the knockout mouse groups displayed significantly higher renal inflammation scores than the wild-type mice. Furthermore, there was a significant difference in renal inflammation scores between p40 and p35 KO groups after primary but not secondary infection. Thus, both gross appearance inspection and microscopic observation demonstrated that p40 KO mice developed more severe renal pathology than p35 KO mice.
Fig 3.
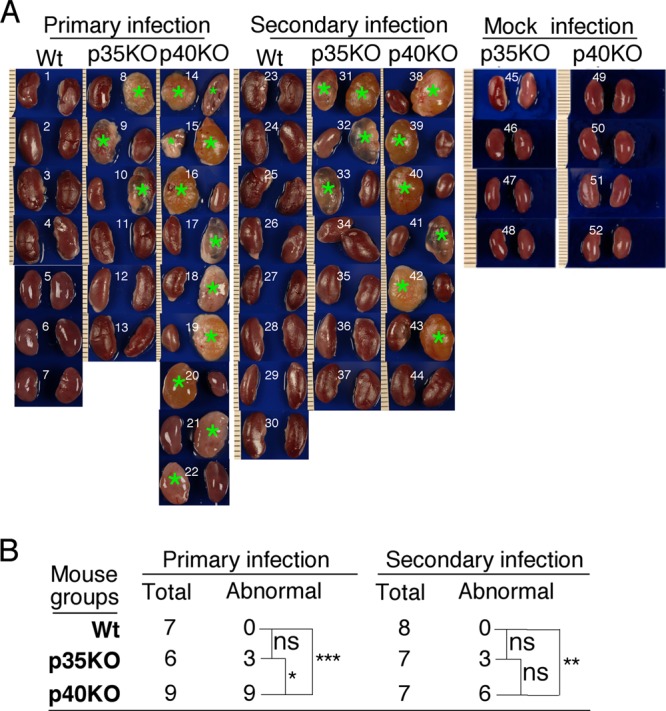
Effect of IL-12p35 or p40 deficiency on renal gross pathology following chlamydial infection. (A) Kidneys from wild-type (images 1 to 7 are from mice with primary infection and 23 to 30 are from those with secondary infection), IL-12p35 KO (images 8 to 13 and 31 to 37), and IL-12p40 KO (images 14 to 22 and 38 to 44) mice were examined for gross appearance, and enlarged, edematous, or necrotic kidneys were noted. Most of these kidneys exhibited typical gross pathology of pyelonephritis. All kidneys identified as abnormal are marked with green stars. Abnormal kidneys were found in most mice deficient in IL-12p40, while no obvious kidney abnormality was noted in any wild-type (WT) mice. The abnormal kidneys were also found in some IL-12p35−/− mice. The numbers of mice with abnormal kidneys from different groups are summarized in panel B (*, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant). The number of mice with abnormal kidneys was the highest in the IL-12p40 KO group. The age-matched KO mice with mock infection (images 45 to 48 for IL-12p35 KO and images 49 to 52 for IL-12p40 KO) were sacrificed on day 114 after primary mock infection for kidney gross pathology evaluation. Vaginal swabs were also taken from these mock-infected mice. No abnormal kidneys were found in these mice.
Fig 4.

Effect of IL-12p35 or p40 deficiency on renal inflammatory pathology following chlamydial infection. (A) Kidneys harvested from wild-type (representative images in panels a and d), IL-12p35 KO (panels b and e), and IL-12p40 KO (c and f) mice as described in the legend to Fig. 3 were further examined for inflammatory pathologies under a microscope. The inflammation was semiquantitatively scored based on the criteria listed in Materials and Methods, and the scores are given in each image as examples. Inflammatory cells or infiltrates are marked with white arrows. The inset in the lower left corner of each panel shows how the polymorphic nuclear cells (PMNs; green arrows) and mononuclear cells (MCs; red arrows) in the corresponding inflammatory infiltration loci were differentiated under a 40× objective lens. Renal tubules (white star) and glomeruli (white arrowheads) are marked. (B) Inflammatory scores for mouse groups, including wild type (n = 7 for primary and n = 8 for secondary infection groups), IL-12p35 KO (n = 6 for primary and n = 7 for secondary infection), and IL-12p40 KO (n = 9 for primary and n = 7 for secondary infection). Each symbol indicates the score from two kidneys of one mouse. Note that the most severe inflammation was in kidney sections from IL-12p40 KO mice (*, P < 0.05; **, P < 0.01; NS, not significant).
Mice deficient in IL-12p40 permit more extensive chlamydial organism spreading into kidneys.
To test whether the severe renal pathology correlates with more extensive spreading of chlamydial organisms in IL-12p40 KO mice, we compared the levels of chlamydial organisms in the kidneys of the three mouse groups (Fig. 5). An immunofluorescence assay revealed that C. muridarum inclusions were detected only in kidneys with gross pathology from either IL-12p35 or p40 KO groups. The chlamydial inclusions were mainly observed in the wall of the renal calices and in the phlogistic exudation but not in the blood vessels, the lymphatic vessels, or the glomeruli, suggesting that the chlamydial organisms might come from the transitional epithelium of the urinary urethral, bladder, and ureter, along which chlamydial organisms could ascend. However, these observations alone cannot exclude the contribution of chlamydial spreading via other routes to renal pathology. Since no chlamydial inclusions were detected in wild-type mouse kidneys (Fig. 5A, panels a and d), we can conclude that IL-12p35 or p40-mediated responses play significant roles in limiting chlamydial spreading. Interestingly, more chlamydial inclusions were detected in abnormal kidneys from IL-12p40 KO (Fig. 5A, panels c and f) than p35 KO (panels b and e) mice, suggesting that the p40-mediated host responses play a more important role in reducing chlamydial spreading into the renal calyces.
Fig 5.

Effect of IL-12p35 or p40 deficiency on chlamydial organism spreading into kidneys. (A) Kidney sections from wild-type (panels a and d), IL-12p35 KO (b and e), and IL-12p40 KO (c and f) mice were subjected to immunolabeling with anti-chlamydial antibodies (green, indicated with white arrows) and a DNA dye (blue). The intact chlamydial inclusions were counted from each section, and 3 sections were used from each kidney. To ensure that the inclusions counted from the 3 sections were not the same inclusions, each of the 3 sections was selected by skipping 5 sections in between. The total number of chlamydial inclusions counted from the 6 sections (from both kidneys) of the same mouse was used to calculate the average number of inclusions per section that was assigned to each mouse. The number of inclusions per section for all mice was summarized (B) and expressed as the mean and standard deviation. Note that chlamydial inclusions were detected only in enlarged and edematous kidneys from the IL-12 gene KO mice, not in kidneys from the wild-type mice. Kruskal-Wallis analysis revealed that the IL-12p40 KO kidney sections contained significantly more chlamydial inclusions than IL-12p35 KO kidney sections. No chlamydial inclusions were detected in wild-type mouse kidney sections using this method. (C) To test whether infectious live organisms could be recovered from the kidney tissues, a parallel experiment with 3 to 5 mice in each group listed on the x axis was carried out. Both kidneys were harvested from each mouse 100 days after C. muridarum infection for making homogenates. The live chlamydial organisms from each renal homogenate were titrated, and the numbers of live organisms per kidney were calculated for each mouse (values are means and standard deviations). Note that the IL-12p40 KO mice had the highest number of live chlamydial organisms in their kidneys. (D) Aliquots of the same kidney homogenates were used for titrating live bacteria on sheep blood agar plates. The number of bacterial colonies counted from each plate was used to calculate the total number of bacteria recovered from each kidney, and the data are expressed as total number of bacteria per mouse. Similar numbers of bacteria were recovered from IL-12p35 KO and IL-12p40 KO mice.
The number of chlamydial inclusions was counted from all 6 renal sections of a mouse (3 sections from each kidney) and was used to calculate the average number of inclusions per section for each mouse. As shown in Fig. 5B, kidneys from the p40 KO mice had significantly more chlamydial inclusions than those from the p35 KO mice, while no inclusions were detected in kidneys from wild-type mice. Clearly, the p40 KO mice underwent more chlamydial spreading.
To test whether the chlamydial inclusions detected in renal tissues are live and infectious, in a parallel experiment, we compared the live organisms harvested from each kidney 100 days after primary infection (Fig. 5C). The two KO mouse groups had significantly higher numbers of live chlamydial organisms than the wild-type group. More importantly, the p40 KO mice had significantly higher numbers than the p35 KOs. These observations established a strong correlation between chlamydial spreading into the renal calyces and inflammatory pathologies in the kidneys. Aliquots of frozen homogenates from the same mice were thawed and used for titrating potential live bacteria using sheep blood agar plates. Surprisingly, significant numbers of live bacteria were detected in mice deficient in either p35 or p40 but not wild-type mice. However, there was no significant difference in the numbers of live bacteria recovered from p35 and p40 KO mice, which was in contrast to the differential severity of renal pathology between these two types of KO mice.
Both IL-12p35 KO and p40 KO mice develop Th2 dominant responses with a significantly impaired Th17 response in p40 KO mice.
All mice regardless of genotypes developed robust anti-C. muridarum organism IgG antibody responses during the course of both primary and secondary infections (Fig. 6), suggesting that either IL-12p35 or p40 deficiency did not affect mouse overall antibody responses to chlamydial infection. Isotyping analyses of the chlamydial antigen-specific IgG antibodies revealed that the titers of IgG2a and -2b were higher than those of IgG1 and -3 in wild-type mice, while the titers of IgG2b and -1 seemed to be higher than those of IgG2a and -3 in the IL-12 gene KO mice (Fig. 6A). Comparison of the ratios of IgG2a to IgG1 revealed that wild-type mice had a significantly higher ratio than either IL-12p35 or p40 KO mice (Fig. 6B), indicating that the wild-type mice maintained a Th1 cytokine environment after chlamydial infection. This is consistent with the well-established concept that intracellular infection favors the development of a Th1-dominant response (28). The lower ratio of IgG2a to IgG1 in IL-12p35 KO and p40 KO mice indicated that these mice developed Th2-biased responses after chlamydial infection. This is consistent with the well-known function of IL-12 in promoting Th1 responses (29). We further compared T cell cytokine production between the three groups of mice using an in vitro spleen cell restimulation assay (Fig. 7). Splenocytes from C. muridarum-infected wild-type mice produced high levels of IFN-γ upon restimulation with chlamydial antigens, while cells from mice deficient in either IL-12p35 or p40 failed to do so (Fig. 7A). Splenocytes from both wild-type and IL-12p35 KO mice produced significantly large amounts of IL-17, while splenocytes from IL-12p40 KO mice failed to do so. These observations are consistent with the well-established concepts that p35 is a critical component of IL-12 (that promotes Th1 responses), while p40 is shared by both IL-12 and IL-23 (which promotes Th17 responses [30]).
Fig 6.
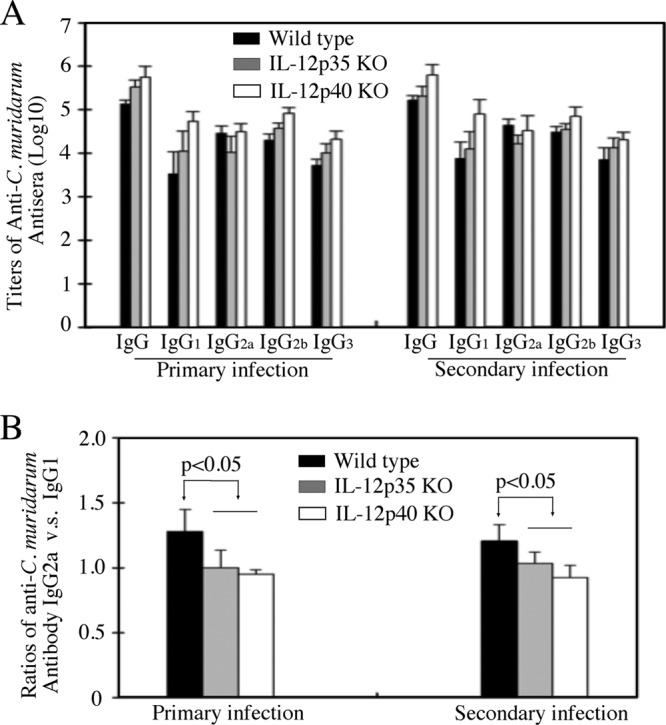
Effect of IL-12p35 or p40 deficiency on mouse antibody responses after chlamydial infection. (A) The 3 groups of mice infected with chlamydial organisms as described in the legend to Fig. 1 were bled on day 114 (after primary infection; n = 4 for wild-type and 6 for IL-12p35 and p40 KO groups) or day 143 (after secondary infection; n = 8 for wild-type and 7 for IL-12p35 and p40 KO groups) for titrating C. muridarum-specific IgG antibodies, including both total IgG and IgG isotypes. The highest dilution at which a given mouse serum still positively stained the C. muridarum inclusions was determined as the titer of that serum. The serum dilutions were converted into log10 titers for calculating means and standard deviations. (B) The ratios of IgG2a titer to IgG1 titer were calculated for each group of mice. Note that the wild-type mice displayed the highest IgG2/IgG1 ratio during both primary and secondary infections (ratios are expressed as means and standard deviations).
Fig 7.
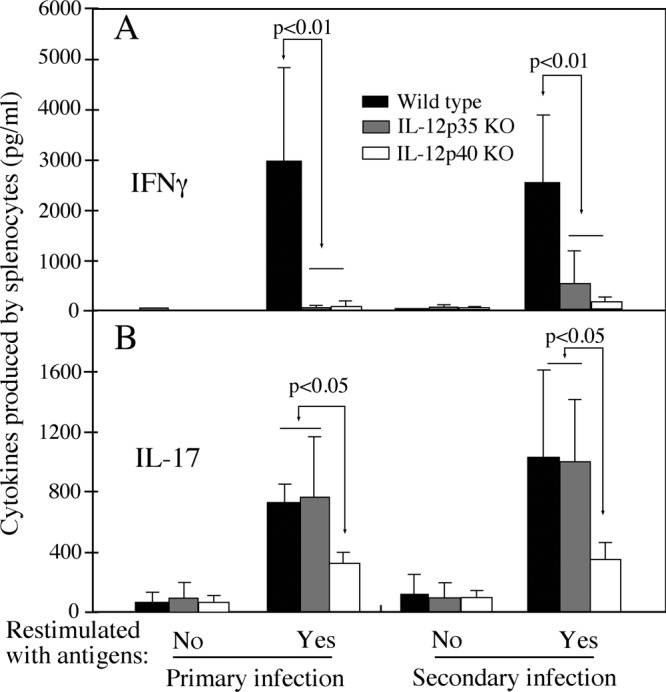
Effect of IL-12p35 or p40 deficiency on mouse T cell responses following chlamydial infection. Splenocytes were harvested from IL-12p35 KO, IL-12p40 KO, and wild-type mice on day 114 (after primary infection) or day 143 (after secondary infection). These mice were infected with C. muridarum as described in the legend to Fig. 1, and numbers of mice in each group are the same as in Fig. 6. The splenocytes were restimulated in vitro with UV-inactivated C. muridarum organisms for 72 h. IFN-γ (A) and IL-17 (B) in culture supernatants were measured using ELISA, and the results are expressed in picograms per ml (data are means and standard deviations). Note that levels of IFN-γ produced by splenocytes from IL-12p35 or IL-12p40 KO mice and IL-17 from IL-12p40 KO mice were significantly lower than those from wild-type mice.
DISCUSSION
Although the role of IL-12 in chlamydial infection is well established (11, 17, 31), the relative contributions of IL-12p35 and IL-12p40 to Chlamydia-induced protective immunity and pathologies remain unclear. Here, we used a mouse intravaginal infection model to evaluate the roles of IL-12p35 and IL-12p40 in C. muridarum infection and C. muridarum-induced pathologies in both upper genital tracts and kidneys. We found that although mice deficient in either IL-12p35 or p40 developed prolonged infection courses after chlamydial primary infection, the same mice rapidly cleared reinfection, which suggests that adaptive immunity that develops independently of IL-12 or IL-23 is protective against chlamydial infection. Mice with or without deficiency in IL-12p35 or IL-12p40 developed severe hydrosalpinx, suggesting that Th2-dominant responses can contribute to Chlamydia-induced inflammatory pathology. Mice deficient in IL-12p40 but not IL-12p35 underwent more extensive spreading of chlamydial organisms into kidney tissues, leading to significantly increased incidence of pyelonephritis, suggesting that, in the absence of IL-12/IFN-γ-mediated Th1 immunity, an IL-23-dependent response may be able to restrict chlamydial organisms from spreading into distal organs. These findings have not only demonstrated the relative contributions of IL-12p35 or IL-12p40-mediated responses to but also uncovered the roles of IL-12- or IL-23-independent responses in chlamydial infection and pathogenesis. These findings together provide important information for both understanding chlamydial pathogenesis and developing vaccines.
The role of Th1-dominant responses in protective immunity against C. trachomatis primary infection has been well established (31). However, a Th1-dominant immunity may not be required for resistance to reinfection (32). Our study has confirmed both. In addition, we have also demonstrated that the IL-23/TH17 axis may not be required for host resistance to either primary infection or reinfection with C. muridarum organisms in the genital tract, since mice deficient in IL-12p35 and those deficient in p40 developed similar prolonged infection time courses after primary infection and developed similar levels of resistance to reinfection. The precise immune mechanisms involved in the protective immunity of the IL-12p40 KO mice against chlamydial reinfection are worth further investigation. Interestingly, the IL-12p35 KO and IL-12p40 KO mice all developed severe pathology in the upper genital tract, suggesting that chlamydial primary infection-induced immune responses that are independent of p35 and p40 can contribute to Chlamydia-induced pathology. The question is whether the same immune mechanisms are responsible for both protecting against reinfection in the lower genital tract and exacerbating pathology in the upper genital tract during primary infection. Obviously, these IL-12 gene KO mice can provide a good model for investigating the non-Th1- and non-Th17-dominant immune mechanisms involved in Chlamydia-induced protective immunity or pathology.
Although both the IL-12p35 KO and IL-12p40 KO mice developed similar prolonged infection time courses in the genital tract, careful examination of kidney tissues revealed a significant difference in incidence of pyelonephritis between these two KO strains. The IL-12p40 KO mice developed a significantly higher rate of pyelonephritis compared to the IL-12p35 KO mice, which correlated well with a significantly increased spreading of chlamydial organisms into the kidney tissues of IL-12p40 KO mice. These observations suggest that an IL-23-mediated response is important in controlling chlamydial organism spreading and pathology in distal organs in the absence of IL-12/IFN-γ, which may provide an explanation for an earlier finding that protective immunity against chlamydial spreading was abolished by an anti-IL-12p40 neutralization antibody but not by an anti-IFN-γ neutralization antibody (17). A p40-mediated response has also been shown to play a critical role in controlling Trypanosoma cruzi dissemination in the spinal cord (33). Furthermore, in the absence of IL-12, an IL-23-dependent response was associated with protective immunity to Salmonella enterica systemic infection (8). Similarly, we found that in the absence of p40, chlamydial organisms inoculated into the vagina spread to upper urinary tract organs. This finding is consistent with a previous observation that Chlamydia trachomatis DNA was detected in the upper urinary tract of patients with pyelonephritis (34). However, neither the spreading of C. muridarum organisms into mouse kidneys nor the detection of C. trachomatis DNA in human kidneys could prove that infection with C. trachomatis contribute to pyelonephritis in humans. Nevertheless, it will be interesting to know whether pyelonephritis patients with positive C. trachomatis DNA detection in the upper urinary tract have any deficiency in IL-23-involved pathways. It is worth noting that significant live bacterial infection was also detected in the kidneys of the KO mice, suggesting a potential role of bacterial superinfection in renal pathology. However, p35 and p40 KO mice displayed similar levels of renal bacterial infection, which was in contrast with the observation that more severe pathology was detected in the kidneys of p40 KO mice. Thus, we can conclude that the bacterial superinfection in the kidney may play a limited role in renal pathology of Chlamydia-infected mice.
We have demonstrated that IL-23-mediated responses play an essential role in preventing chlamydial spreading. The next question is what pathways IL-23 may use to offer the protection. There are two obvious pathways, IL-17 and IL-22. IL-17 is known to play important roles in controlling both extracellular and intracellular bacterial infection (35, 36). It has been shown that IL-17 is critical in controlling pulmonary infection with chlamydial organisms (15, 16). However, the role of IL-17 in urogenital tract infection with chlamydial organisms remains unclear. A recent study reported that neutralization of IL-17 in IFN-γ KO mice only decreased neutrophil influx to urogenital tract without increasing bacterial burden in the genital tract (18). It is worth noting that this observation can't necessarily exclude the potential roles of IL-17 in reducing chlamydial genital infection. The present study also revealed that IL-12p35 KO and IL-12p40 KO mice displayed similar levels of bacterial burden and pathology in the urogenital tract. The lack of differences in genital tract tissue pathology in these KO mice may be due to the fact that the levels of pathology and organism burden were saturated in the genital tracts of all mice regardless of genotypes. Importantly, although chlamydial organisms can spread to and induce pathology in kidneys in IFN-γ KO (32) and IL-12p35 KO (present study) mice, both the spreading and pathology were significantly enhanced in kidneys of IL-12p40 KO mice, suggesting a critical role of IL-23-mediated responses in controlling chlamydial spreading into distal organs. It will be interesting to test whether treatment with an anti-IL-17 neutralization antibody can further increase bacterial burden in the kidneys of IFN-γ KO mice. Alternatively, IL-23 may be able to use an IL-17-independent pathway for reducing chlamydial spreading. IL-23 can both induce the production of IL-22 in innate lymphoid cells and promote Th22 T cell development (37). IL-22 has been shown to play important roles in host defense against various infections (38, 39). This alternative hypothesis can be tested by evaluating the effect of an anti-IL-22 neutralization antibody treatment on chlamydial spreading in either IL-12p35 KO or IFN-γ KO mice.
ACKNOWLEDGMENTS
This work was supported in part by grants from Natural Science Foundation of China (L.C.) and the National Institutes of Health (G.Z.).
Footnotes
Published ahead of print 10 June 2013
REFERENCES
- 1. Sherman KJ, Daling JR, Stergachis A, Weiss NS, Foy HM, Wang SP, Grayston JT. 1990. Sexually transmitted diseases and tubal pregnancy. Sex Transm Dis. 17:115–121 [DOI] [PubMed] [Google Scholar]
- 2. Morrison RP, Feilzer K, Tumas DB. 1995. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect. Immun. 63:4661–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watford WT, Moriguchi M, Morinobu A, O'Shea JJ. 2003. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 14:361–368 [DOI] [PubMed] [Google Scholar]
- 4. Hunter CA. 2005. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 5:521–531 [DOI] [PubMed] [Google Scholar]
- 5. Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. 2005. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202:761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175:788–795 [DOI] [PubMed] [Google Scholar]
- 7. Lieberman LA, Cardillo F, Owyang AM, Rennick DM, Cua DJ, Kastelein RA, Hunter CA. 2004. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893 [DOI] [PubMed] [Google Scholar]
- 8. Schulz SM, Kohler G, Schutze N, Knauer J, Straubinger RK, Chackerian AA, Witte E, Wolk K, Sabat R, Iwakura Y, Holscher C, Muller U, Kastelein RA, Alber G. 2008. Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J. Immunol. 181:7891–7901 [DOI] [PubMed] [Google Scholar]
- 9. Vom Berg J, Prokop S, Miller KR, Obst J, Kalin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. 2012. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat. Med. 18:1812–1819 [DOI] [PubMed] [Google Scholar]
- 10. Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley LT, Lebwohl M. 2007. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N. Engl. J. Med. 356:580–592 [DOI] [PubMed] [Google Scholar]
- 11. Lu H, Zhong G. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect. Immun. 67:1763–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaw J, Grund V, Durling L, Crane D, Caldwell HD. 2002. Dendritic cells pulsed with a recombinant chlamydial major outer membrane protein antigen elicit a CD4(+) type 2 rather than type 1 immune response that is not protective. Infect. Immun. 70:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jupelli M, Selby DM, Guentzel MN, Chambers JP, Forsthuber TG, Zhong G, Murthy AK, Arulanandam BP. 2010. The contribution of interleukin-12/interferon-gamma axis in protection against neonatal pulmonary Chlamydia muridarum challenge. J. Interferon Cytokine Res. 30:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu H, Shen C, Brunham RC. 2000. Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J. Immunol. 165:1463–1469 [DOI] [PubMed] [Google Scholar]
- 15. Bai H, Cheng J, Gao X, Joyee AG, Fan Y, Wang S, Jiao L, Yao Z, Yang X. 2009. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J. Immunol. 183:5886–5895 [DOI] [PubMed] [Google Scholar]
- 16. Zhang X, Gao L, Lei L, Zhong Y, Dube P, Berton MT, Arulanandam B, Zhang J, Zhong G. 2009. A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum. J. Immunol. 183:1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perry LL, Feilzer K, Caldwell HD. 1997. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J. Immunol. 158:3344–3352 [PubMed] [Google Scholar]
- 18. Scurlock AM, Frazer LC, Andrews CW, Jr, O'Connell CM, Foote IP, Bailey SL, Chandra-Kuntal K, Kolls JK, Darville T. 2011. Interleukin-17 contributes to generation of Th1 immunity and neutrophil recruitment during Chlamydia muridarum genital tract infection but is not required for macrophage influx or normal resolution of infection. Infect. Immun. 79:1349–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. 2008. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect. Immun. 76:515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen C, Chen D, Sharma J, Cheng W, Zhong Y, Liu K, Jensen J, Shain R, Arulanandam B, Zhong G. 2006. The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect. Immun. 74:4826–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm. Dis. 32:49–56 [DOI] [PubMed] [Google Scholar]
- 22. Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. 2010. Mice deficient in MyD88 develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J. Immunol. 184:2602–2610 [DOI] [PubMed] [Google Scholar]
- 23. Svensson M, Yadav M, Holmqvist B, Lutay N, Svanborg C, Godaly G. 2011. Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCxcr2 heterozygous mice. Kidney Int. 80:1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sharma J, Bosnic AM, Piper JM, Zhong G. 2004. Human antibody responses to a Chlamydia-secreted protease factor. Infect. Immun. 72:7164–7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong G, Toth I, Reid R, Brunham RC. 1993. Immunogenicity evaluation of a lipidic amino acid-based synthetic peptide vaccine for Chlamydia trachomatis. J. Immunol. 151:3728–3736 [PubMed] [Google Scholar]
- 27. Zhong GM, Brunham RC. 1990. Immunoaccessible peptide sequences of the major outer membrane protein from Chlamydia trachomatis serovar C. Infect. Immun. 58:3438–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cooper AM, Khader SA. 2008. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 226:191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. 2009. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug Targets 8:40–52 [DOI] [PubMed] [Google Scholar]
- 30. Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. 2004. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol. Rev. 202:96–105 [DOI] [PubMed] [Google Scholar]
- 31. Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 70:2741–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. 1997. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect. Immun. 65:2145–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bombeiro AL, Goncalves LA, Penha-Goncalves C, Marinho CR, D'Imperio Lima MR, Chadi G, Alvarez JM. 2012. IL-12p40 deficiency leads to uncontrolled Trypanosoma cruzi dissemination in the spinal cord resulting in neuronal death and motor dysfunction. PLoS One 7:e49022. 10.1371/journal.pone.0049022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dimitrakov J, Ganev V, Zlatanov T, Detchev I, Horvat A, Kirov S, Vatchkova I, Dimitrakov D. 1998. PCR studies on the presence of Chlamydia trachomatis in the upper urinary tract of patients with obstructive pyelonephritis. Folia Med. (Plovdiv) 40:24–28 [PubMed] [Google Scholar]
- 35. Khader SA, Gopal R. 2010. IL-17 in protective immunity to intracellular pathogens. Virulence 1:423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wozniak TM, Saunders BM, Ryan AA, Britton WJ. 2010. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect. Immun. 78:4187–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, Brown GD, Steele C. 2012. The beta-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J. Immunol. 189:3653–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, Ouyang W. 2011. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12:941–948 [DOI] [PubMed] [Google Scholar]