

Abstract
The genetic diversity of human immunodeficiency virus type 1 (HIV-1) has significant implications for diagnosis, vaccine development, and clinical management of patients. Although HIV-1 subtype B is predominant in the United States, factors such as global travel, immigration, and military deployment have the potential to increase the proportion of non-subtype B infections. Limited data are available on the prevalence and distribution of non-B HIV-1 strains in the United States. We sought to retrospectively examine the prevalence, geographic distribution, diversity, and temporal trends of HIV-1 non-B infections in samples obtained by ARUP Laboratories, a national reference laboratory, from all regions of the United States. HIV-1 pol sequences from 24,386 specimens collected from 46 states between 2004 and September 2011 for drug resistance genotyping were analyzed using the REGA HIV-1 Subtyping Tool, version 2.0. Sequences refractory to subtype determination or reported as non-subtype B by this tool were analyzed by PHYLIP version 3.5 and Simplot version 3.5.1. Non-subtype B strains accounted for 3.27% (798/24,386) of specimens. The 798 non-B specimens were received from 37 states and included 5 subtypes, 23 different circulating recombinant forms (CRFs), and 39 unique recombinant forms (URFs). The non-subtype B prevalence varied from 0% in 2004 (0/54) to 4.12% in 2011 (201/4,884). This large-scale analysis reveals that the diversity of HIV-1 in the United States is high, with multiple subtypes, CRFs, and URFs circulating. Moreover, the geographic distribution of non-B variants is widespread. Data from HIV-1 drug resistance testing have the potential to significantly enhance the surveillance of HIV-1 variants in the United States.
INTRODUCTION
One of the hallmarks of human immunodeficiency virus type 1 (HIV-1) is a high level of genetic diversity. HIV-1 is currently classified into four distinct lineages designated groups M (major), O (outlier), N (non-M, non-O), and P (1, 2). Group M strains, accounting for the vast majority of global HIV-1 infections, are further subdivided into subtypes (designated by letters) and sub-subtypes (denoted by numbers) as follows: A1 to A4, B, C, D, F1, F2, G, H, J, and K (2, 3). Genetic variation in env between subtypes and groups ranges from 15 to 25% and up to 50%, respectively (HIV Sequence Database [http://www.hiv.lanl.gov/]). Recombination further exacerbates the overall HIV-1 diversity (3). Full-genome sequence characterization of HIV-1 strains has led to the identification of circulating recombinant forms (CRFs) and unique recombinant forms (URFs) (4; HIV Sequence Database). Reflecting the dynamic nature of the HIV-1 pandemic, the number of recognized CRFs has been increasing steadily, with 51 described to date (HIV Sequence Database). Based on recent figures, CRFs account for 16% of worldwide HIV-1, with total recombinants (CRFs and URFs) estimated at >20% of infections (4). This trend of increasing diversity and genomic complexity can be anticipated to continue for the foreseeable future.
Analysis of HIV-1 strains has revealed an uneven global distribution of groups, subtypes, and CRFs (4). Subtype B is predominant in Australia, Europe, and the Americas. In contrast, the distribution of HIV-1 strains across Africa is quite variable, with detection of all subtypes and predominantly CRF02_AG in West Central Africa, subtypes A, C, and D in East Africa, and subtype C in Southern Africa and India. CRF01_AE predominates in South and Southeast Asia (4). HIV-1 genetic diversity has significant implications for diagnostics, blood screening, patient-monitoring assays, treatment, clinical progression, and vaccine development (5–11). Natural polymorphisms occurring within primer and/or probe binding sites can result in underquantitation or lack of detection in molecular assays designed for viral load measurement and blood screening (9, 12–16). Similarly, polymorphisms that modify or ablate key epitopes can compromise the performance of serological diagnostic and screening assays (7, 17). Although subtype B infections represent only 11% of HIV-1 infections worldwide (4), most serological and molecular assays and controls (including the WHO International Standard) have been developed and optimized utilizing subtype B strains.
Due to a variety of factors, including immigration, travel, military deployment, and commerce, the global distribution and regional prevalence of the various forms of HIV-1 are dynamic and unpredictable. Non-B subtypes are becoming increasingly common in countries where subtype B viruses predominate. For example, there has been a dramatic increase in HIV-1 diversity in France over the past 15 years, and based on national surveillance data, non-B strains account for 48% of newly diagnosed HIV-1 infections (18–21). Although non-B strains were primarily associated with African-born persons, a substantial proportion of French-born individuals (19%) harbored these divergent strains (20, 21). A national surveillance program in the United Kingdom yielded similar results, with 25% of HIV-1 infections due to non-B subtype or recombinant strains and a strong association with heterosexual transmission and birth in Africa (22). Moreover, analysis of nearly 2,800 prospective samples collected between 2002 and 2005 from patients in 20 European countries and Israel revealed that 33% were non-B infections (23).
The lack of a comprehensive national surveillance program in the United States has complicated the accurate determination of the prevalence of non-subtype B HIV-1 infections (24). Most studies have focused on specific populations, such as military personnel, immigrants, or blood donors (25–31). There is evidence of increasing non-B prevalence in the United States, and it may be higher than generally recognized (29, 32–34). Although data are limited, studies have shown that the prevalence of non-subtype B varies greatly depending on the population analyzed, ranging from 0.6% in a northern California clinic-based setting (35) to 95% in African-born immigrants in Minneapolis, MN (28, 36), and New York City (27). The largest published population-based study to date was performed by the Centers for Disease Control and Prevention. Analysis of 2,030 pol sequences from newly diagnosed HIV-1 infections collected in 2006 from 11 U.S. surveillance areas revealed that 3.8% were due to non-B strains (37). Notably, this survey did not include the five metropolitan areas in the United States with the highest numbers of African-born immigrants, nor did it include the four states (California, Florida, New York, and Texas) with the highest proportion (>5%) of foreign-born arrivals since 2005 (38); thus, it likely underestimates the prevalence and complexity of these divergent strains. Surveys among seropositive blood donors indicate that non-B prevalence has risen from 0% in the 1980s to 2 to 5% in post-2000 evaluations (30, 31, 39). This increase has occurred despite the implementation of deferral strategies that exclude donors at risk for infection with HIV-1 group O (West Central African countries) and at risk for malaria (regions of Central and South America, Africa, and Asia).
The goal of this study was to perform a large-scale retrospective analysis of HIV-1 diversity in the United States and to assess trends over time.
(Portions of this work were presented in poster form at the 16th Conference on Retroviruses and Opportunistic Infections, Montreal, Quebec, Canada, February 2009, and at the 28th Clinical Virology Symposium, Daytona Beach, FL, April 2012.)
MATERIALS AND METHODS
HIV-1 genotyping.
Samples received by ARUP Laboratories, a national reference laboratory based in Salt Lake City, UT, for HIV-1 antiretroviral resistance genotyping were analyzed. HIV-1 pol sequences were generated from plasma using the ViroSeq HIV-1 Genotyping System, version 2.0 (Celera, Alameda, CA), according to the manufacturer's instructions. ViroSeq generates a 1.8-kb PCR amplicon, comprising codons 1 to 99 of the protease gene and 1 to 335 of the reverse transcriptase gene, which is sequenced in seven overlapping reactions using redundant sequencing primers to produce a 1,302-nucleotide consensus sequence.
Study data and scope.
The data set contains 24,386 HIV-1 pol sequences obtained from specimens received between 2004 and September 2011, including patient age and gender and client geographic information. Prior to mid-2007, only New York State specimens were available. The sequences were deidentified by a third party using established institutional review board (IRB) protocols and procedures (IRB protocol 31050; University of Utah, Salt Lake City, UT) by removing all protected health information (PHI) from the database. Duplicate sequences (100% identical among the entire data set) (40) and sequences with evolutionary distances of 0 (among the non-subtype B samples, as assessed by DNADIST) were eliminated. Eighteen samples were removed because their sequences were 100% identical to another in the database. More-rigorous phylogenetic analysis of the non-B sequences resulted in the elimination of another 94 sequences. While these efforts minimize the possibility of duplicate patient sampling, the required deidentification process precludes absolute certainty that all duplicates have been eliminated.
HIV-1 subtype assignment and phylogenetic analysis.
All sequences were initially analyzed using the REGA HIV-1 Subtyping Tool, version 2.0 (http://dbpartners.stanford.edu/RegaSubtyping/). Sequences refractory to subtype determination (results of “Check the Bootscan” or “Sequence Error”) or reported as non-subtype B by this tool were analyzed using PHYLIP software (version 3.573; J. Felsenstein, University of Washington, Seattle, WA). Nucleotide sequences were aligned with reference sequences representing HIV-1 group M subtypes and CRFs (1 to 45), HIV-1 groups O and N, and simian immunodeficiency virus (SIV) (HIV Sequence Database) and gap stripped using BioEdit (version 7.0.4.1; Department of Microbiology, North Carolina State University, Raleigh, NC). Evolutionary distances were estimated with DNADIST (Kimura two-parameter method with a transition-transversion ratio of 2.0). Phylogenetic reconstructions were generated with NEIGHBOR using the neighbor-joining method. Branch reproducibility was evaluated using SEQBOOT on 100 replicates. Phylogenetic trees were displayed with TreeExplorer. Bootstrap values of >70 were considered acceptable for subtype assignment. In cases of subtype assignments that were discordant between the REGA Subtyping Tool and phylogenetic reconstructions, the PHYLIP/SimPlot designations were used.
Bootscanning analysis.
Nucleotide sequences were examined for intersubtype recombination using SimPlot software (version 3.5.1, http://sray.med.som.jhmi.edu/SCRoftware/SimPlot/; S. Ray, Johns Hopkins University, Baltimore, MD). Putative recombinant sequences were aligned with one reference sequence for each of the HIV-1 group M subtypes (A to D, F to H, J to K, and the major CRFs). SimPlot and BootScan were run with a sliding window of 300 or 400 nucleotides and 20-nucleotide steps.
Nucleotide sequence accession numbers.
The non-subtype B sequences have been deposited in GenBank with accession numbers JX459972 to JX460769.
RESULTS
Study population.
HIV-1 pol sequences generated from 24,386 patient specimens submitted by ARUP clients for resistance genotyping between 2004 and September 2011 were analyzed. Sequences were derived from samples collected from HIV-1-infected individuals residing in 46 states within the United States. Prior to mid-2007, only samples obtained from the state of New York were available. Subsequently, coverage was expanded to include clients in 46 states, which represented >90% of the total sequences analyzed (Table 1). Patient ages ranged from newborn to 85 years (average, 40.9; median, 42), and 70.1% were male, 29.7% female, and 0.2% unknown.
Table 1.
Percentages of HIV subtypes in samples collected in the United States between 2004 and 2011
| Subtype | % of samplesa from: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2004 (n = 54) | 2005 (n = 546) | 2006 (n = 742) | 2007 (n = 2,132) | 2008 (n = 4,643) | 2009 (n = 6,130) | 2010 (n = 5,255) | 2011 (n = 4,884) | All samples (n = 24,386) | |
| A | 0.00 | 0.18 | 0.00 | 0.47 | 0.56 | 0.83 | 0.51 | 0.68 | 0.61 |
| B | 100.00 | 98.90 | 98.25 | 97.47 | 96.81 | 96.93 | 96.42 | 95.88 | 96.73 |
| C | 0.00 | 0.00 | 0.67 | 0.61 | 1.31 | 0.91 | 1.27 | 1.45 | 1.12 |
| CRF02_AG | 0.00 | 0.55 | 0.27 | 0.47 | 0.45 | 0.54 | 0.80 | 0.66 | 0.59 |
| Other CRFs | 0.00 | 0.37 | 0.27 | 0.42 | 0.26 | 0.31 | 0.55 | 0.55 | 0.41 |
| URFs | 0.00 | 0.00 | 0.27 | 0.19 | 0.22 | 0.18 | 0.10 | 0.14 | 0.16 |
| D, F, G | 0.00 | 0.00 | 0.27 | 0.38 | 0.39 | 0.28 | 0.30 | 0.57 | 0.36 |
| Unclassified | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.04 | 0.06 | 0.02 |
The total does not always equal 100 due to rounding. For 2004 to mid-2007, only New York State samples were included. All states were included after mid-2007.
Prevalence of HIV-1 non-subtype B strains in our study population.
Of the 24,386 sequences analyzed, 23,588 (96.73%) were classified as subtype B and 798 (3.27%) as non-subtype B or recombinant forms. The prevalence of non-subtype B strains varied from 0% (0/54) in 2004 to 1.10% (6/546) in 2005, 1.75% (13/742) in 2006, 2.53% (54/2,132) in 2007, 3.19% (148/4,643) in 2008, 3.07% (188/6,130) in 2009, 3.58% (188/5,255) in 2010, and 4.12% (201/4,884) in 2011 (Table 1). Of the non-B strains, 353 (44.2%) were from male patients, 443 (55.5%) were from female patients, and 2 (0.3%) from patients where gender information was unavailable. Subtype C was the predominant non-B virus identified and comprised 1.12% of the total and 35.3% of non-B strains in 2011 (Table 1). Subtype A and CRF02_AG infections were the next most common non-B viruses.
Non-B strain diversity.
Examination of the 798 sequences designated non-subtype B or recombinant strains revealed a high level of diversity (Table 2). A representative phylogenetic reconstruction with multiple subtypes and recombinant strains is shown in Figure 1. A subset of 510 (63.9%) were categorized within the recognized HIV-1 group M subtypes: 148 were subtype A (133 A1, 4 A2, 10 A3, and 1 A4), 273 subtype C, 42 subtype D, 12 subtype F (11 F1 and 1 F2), and 35 subtype G. Recombinant strains accounted for 282 (35.3%) of the non-B sequences. Of these, 243 (86.2%) were recognized CRFs: 143 were CRF02_AG, 52 CRF01_AE, 8 CRF06_cpx, 5 CRF14_BG, 3 CRF19_cpx, 3 CRF05_DF, 3 CRF08_BC, 3 CRF11_cpx, 3 CRF12_BF, 3 CRF39_BF, 2 CRF09_cpx, 2 CRF15_01B, 2 CRF20_BG, 2 CRF22_01A1, and 1 each of 9 other CRFs. Thirty-nine (13.8%) of the recombinants were classified as URFs (Table 2). Bootscan analysis revealed that although some shared similar subtype composition, each URF was a distinct recombinant with different recombination breakpoints (data not shown). The sequences generated from six samples were clearly divergent from subtype B reference strains but could not be reliably classified into any of the currently recognized forms and were designated U (unclassified).
Table 2.
Designations and numbers of all non-subtype B strains
| Designation | No. of specimens (n = 798) |
|---|---|
| Subtypes | |
| A1 | 133 |
| A2 | 4 |
| A3 | 10 |
| A4 | 1 |
| C | 273 |
| D | 42 |
| F1 | 11 |
| F2 | 1 |
| G | 35 |
| Ua | 6 |
| CRFs | |
| CRF01_AE | 52 |
| CRF02_AG | 143 |
| CRF05_DF | 3 |
| CRF06_cpx | 8 |
| CRF08_BC | 3 |
| CRF09_cpx | 2 |
| CRF10_CD | 1 |
| CRF11_cpx | 3 |
| CRF12_BF | 3 |
| CRF14_BG | 5 |
| CRF15_01B | 2 |
| CRF16_A2D | 1 |
| CRF17_BF | 1 |
| CRF18_cpx | 1 |
| CRF19_cpx | 3 |
| CRF20_BG | 2 |
| CRF21_A2D | 1 |
| CRF22_01A1 | 2 |
| CRF25_cpx | 1 |
| CRF28_BF | 1 |
| CRF29_BF | 1 |
| CRF35_AD | 1 |
| CRF39_BF | 3 |
| URFs | |
| URF_02_43 | 1 |
| URF_0206 | 1 |
| URF_02A3 | 3 |
| URF_02B | 1 |
| URF_02D | 1 |
| URF_08B | 1 |
| URF_A1A3 | 1 |
| URF_A1C | 5 |
| URF_A1D | 9 |
| URF_A1DK | 1 |
| URF_A1G | 1 |
| URF_A1H | 1 |
| URF_A1J | 1 |
| URF_A1JK | 1 |
| URF_A3G | 2 |
| URF_AB | 1 |
| URF_BC | 1 |
| URF_BF1 | 1 |
| URF_BG | 5 |
| URF_UC | 1 |
U, unclassified.
Fig 1.

A representative phylogenetic neighbor-joining tree for HIV-1 non-subtype B pol protease-reverse transcriptase sequences. HIV-1 subtypes are shown in pink, CRFs are shown in blue, and reference strains are shown in gray. Subtype and CRF designations for reference strains from the Los Alamos HIV Sequence Database are shown near each branch. Bootstrap values at key nodes are indicated.
Geographic distribution of non-B strains.
One unique attribute of this study was the scope of sampling, which involved submissions from clients in 46 states over multiple years (Tables 1 and 3). Figure 2 shows the unique non-subtype B and recombinant HIV-1 specimens identified in each state. The sampling depth across the states was highly variable. Texas accounted for 3,460 (14.19%) samples, and the next most sampled state was New York, with 3,066 (12.57%). Non-subtype B infections were detected in 37 states, from all regions across the United States. The percentage of non-B strains identified for each state ranged from 0 to 100%. However, the total number of samples from 17 states (for example, Hawaii, where the only sample received was CRF01_AE) was less than 100 for each. Notably, among the 7 states (California, Florida, Georgia, Indiana, Massachusetts, New York, and Texas) that each had >1,000 sequences analyzed, the prevalence of non-B strains ranged from 0.9% in Georgia to 8.9% in Massachusetts. More than half of the non-B samples were collected from four states: Massachusetts (8.9% of 2,028), Texas (3.2% of 3,460), Indiana (5.0% of 1,731), and New York (1.6% of 3,066). The diversity with respect to the various non-subtype B forms of HIV-1 was high in all 12 states with a sampling depth of at least 549 sequences (Fig. 2).
Table 3.
Statistics by statea
| State | No. of non-B samples | Total no. of samples | Proportion (%b) of: |
||
|---|---|---|---|---|---|
| Total samples from state that are non-B | All non-B samples | All samples | |||
| AL | 2 | 190 | 1.1 | 0.3 | 0.78 |
| AR | 8 | 539 | 1.5 | 1.0 | 2.21 |
| AZ | 15 | 261 | 5.7 | 1.9 | 1.07 |
| CA | 40 | 2,267 | 1.8 | 5.0 | 9.30 |
| CO | 19 | 378 | 5.0 | 2.4 | 1.55 |
| CT | 2 | 13 | 15.4 | 0.3 | 0.05 |
| FL | 13 | 1,293 | 1.0 | 1.6 | 5.30 |
| GA | 19 | 2,144 | 0.9 | 2.4 | 8.79 |
| HI | 1 | 1 | 100.0 | 0.1 | 0.00 |
| IA | 14 | 289 | 4.8 | 1.8 | 1.19 |
| ID | 0 | 8 | 0.03 | ||
| IL | 28 | 730 | 3.8 | 3.5 | 2.99 |
| IN | 87 | 1,731 | 5.0 | 10.9 | 7.10 |
| KS | 0 | 31 | 0.13 | ||
| KY | 9 | 239 | 3.8 | 1.1 | 0.98 |
| LA | 6 | 279 | 2.2 | 0.8 | 1.14 |
| MA | 181 | 2,028 | 8.9 | 22.7 | 8.32 |
| MD | 6 | 268 | 2.2 | 0.8 | 1.10 |
| ME | 0 | 90 | 0.37 | ||
| MI | 16 | 835 | 1.9 | 2.0 | 3.42 |
| MN | 2 | 7 | 28.6 | 0.3 | 0.03 |
| MO | 6 | 181 | 3.3 | 0.8 | 0.74 |
| MS | 0 | 2 | 0.01 | ||
| MT | 0 | 10 | 0.04 | ||
| NC | 3 | 397 | 0.8 | 0.4 | 1.63 |
| ND | 1 | 8 | 12.5 | 0.1 | 0.03 |
| NE | 17 | 152 | 11.2 | 2.1 | 0.62 |
| NJ | 23 | 597 | 3.9 | 2.9 | 2.45 |
| NM | 0 | 1 | 0.00 | ||
| NV | 0 | 8 | 0.03 | ||
| NY | 50 | 3,066 | 1.6 | 6.3 | 12.57 |
| OH | 23 | 590 | 3.9 | 2.9 | 2.42 |
| OK | 1 | 57 | 1.8 | 0.1 | 0.23 |
| OR | 1 | 55 | 1.8 | 0.1 | 0.23 |
| PA | 6 | 150 | 4.0 | 0.8 | 0.62 |
| RI | 1 | 2 | 50.0 | 0.1 | 0.01 |
| SC | 8 | 510 | 1.6 | 1.0 | 2.09 |
| SD | 12 | 78 | 15.4 | 1.5 | 0.32 |
| TN | 21 | 549 | 3.8 | 2.6 | 2.25 |
| TX | 111 | 3,460 | 3.2 | 13.9 | 14.19 |
| UT | 7 | 112 | 6.3 | 0.9 | 0.46 |
| VA | 9 | 214 | 4.2 | 1.1 | 0.88 |
| WA | 12 | 206 | 5.8 | 1.5 | 0.84 |
| WI | 18 | 346 | 5.2 | 2.3 | 1.42 |
| WV | 0 | 5 | 0.02 | ||
| WY | 0 | 9 | 0.04 | ||
| Total | 798 | 24,386 | N/A | 100.3 | 99.99 |
The 46 states from which samples were received are shown. For each state, the number of non-B samples received, the total number of samples received, and the proportions of non-B samples from that state, of all non-B samples, and of all samples are listed. NA, not applicable.
Totals do not equal 100 due to rounding.
Fig 2.
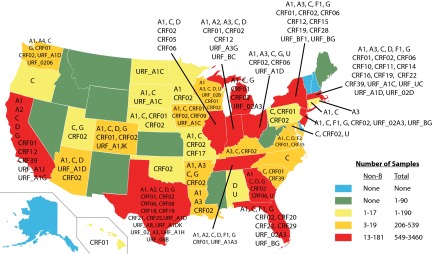
Geographic distribution of HIV-1 non-B subtype specimens. Samples were received from 46 states; non-B viruses were detected in 37 states. Non-B and recombinant forms identified in each state are listed. Colors indicate the total number of samples received and the number of non-subtype B strains identified per state. The subtype/CRF designations normally shown after the underscore in the formal CRF nomenclature have been deleted to enhance legibility. (Modified from a map by Theshibboleth from Wikimedia Commons under the terms of the GNU Free Documentation License.)
DISCUSSION
In North America and Europe, HIV-1 subtype B strains were responsible for the initial spread of HIV-1. The primary modes of exposure were men who have sex with men and intravenous drug use. Consequently, the HIV-1 pandemic in these regions was dominated by subtype B infections. Moreover, since the first HIV-1 strains to be fully characterized were from these regions, they formed the basis for the development of diagnostic, screening, and viral-load assays. With the recognition of the global nature of the HIV-1 pandemic and characterization of strains collected from other parts of the world, it became evident that HIV-1 exhibits a high degree of genetic diversity and that subtype B infections represent a relatively small proportion of infections worldwide (4; HIV Sequence Database). The first case of non-subtype B HIV-1 reported in the United States (Alabama) was in 1994 and involved a student from Zaire infected with a subtype D strain (41). Soon thereafter, the first documented cases of native U.S. residents becoming infected with non-B subtypes (A, D, and E [CRF01_AE]) were reported (42). These cases involved American servicemen who had acquired HIV-1 during overseas deployment. In a subsequent study involving active surveillance of military personnel, 7.4% of recent HIV-1 infections were due to subtype E (CRF01_AE) strains acquired abroad (25). Nine cases of non-subtype B infections among military health care beneficiaries were subsequently identified by astute physicians who suspected non-B infections based on low or undetectable viral load measurements and either declining CD4 cell counts or history of foreign travel (43). The first well-documented case of HIV-1 non-B (CRF01_AE) transmission to a native U.S. resident with no history of travel abroad was published in 1996 (44). Already by 2001, there was evidence of indigenous transmission of non-subtype B strains in rural Georgia (45). Several subsequent studies documented the presence of non-subtype B infections among immigrant residents in various U.S. settings, with a prevalence ranging from 6 to 95% (27–29, 34, 36, 46, 47). Since the majority of available data are from specific populations, accurate estimation of the overall prevalence and diversity of non-B infections in the United States is challenging.
In the present study, the diversity of HIV-1 in the United States was evaluated using 24,386 pol sequences generated by resistance genotyping of patients in 46 states. In addition, the availability of sequences from specimens collected during the time period from 2004 through September 2011 provided an opportunity to explore temporal trends. However, since the samples available from 2004 to mid-2007 were from New York State exclusively, the trends from 2008 to 2011 are the most informative. This analysis suggests that, although subtype B infections still predominate in the United States, the prevalence of non-subtype B strains is on the rise. The overall prevalence was 3.27%, and in 2011, non-B strains represented 4.12% of this study population (Table 1). Moreover, the diversity of the non-B variants is high, with an ever-expanding geographic distribution (Table 3 and Fig. 2). Non-B strains were detected in 37 of the 46 states from which samples were collected. The scope of genetic diversity observed is unprecedented for U.S. studies, with representatives of most HIV-1 group M subtypes (all sub-subtypes), 23 different CRFs, and 39 URFs. Of the 798 non-B strains identified, the majority were subtype C (34.2%), followed by subtype A (18.5%), CRF02_AG (17.9%), and CRF01_AE (6.5%). These four forms represented more than 77% of all non-B strains, which is consistent with several other U.S. studies (32, 34, 48). The documentation of 23 different CRFs in this study population is indicative of continued introductions of HIV-1 from many geographic sources. While several of the observed CRFs are consistent with links to West Central Africa, CRF14_BG was initially identified in injecting drug users in Spain and Portugal, and the subtype B/F-derived CRFs were first identified in Brazil (4; HIV Sequence Database). Of interest, although 70.1% of the total sequences were obtained from males, the majority (55.5%) of non-subtype B infections were in females. The bias toward females harboring non-B infections presumably reflects predominantly heterosexual transmission of these variants and is consistent with results from another recent U.S. study (34). However, due to the design of our study, information on the route of transmission and patients' country of origin is not available.
To our knowledge, this is the largest and most comprehensive survey of HIV-1 non-subtype B strains in the United States to date. However, this study has several limitations and likely underestimates their prevalence and overall diversity in the United States. First, although >24,000 sequences were analyzed, this represents only ∼2% of the estimated total HIV-1 infections in the United States. Second, sampling was uneven across the country and across the states, as it was dependent on the ARUP client base. Seventeen states each had fewer than 100 sequences represented (385 sequences, 1.58% of the total), and nine of these states had no non-B sequences, as shown in Figure 2 and Table 3. The potential for large regional differences in the proportion of non-subtype B even within a state has been demonstrated (34). Third, the determination of subtype is based on analysis of resistance genotyping sequence information, which is ∼15% of the complete HIV-1 genome. To ensure the identification of all recombinants and to confirm the subtype/recombinant classification, complete-genome sequencing would be required. Fourth, the sequences were generated by utilizing a resistance genotyping assay validated for subtype B; any divergent HIV strains not successfully genotyped are excluded from our analysis. Fifth, some bias is likely introduced due to capturing data only from patients accessing treatment. It is possible that immigrant or lower-socioeconomic-status groups may be underrepresented. Due to the study design, the treatment status (naive versus experienced) of the patients is unknown. Finally, the sampling early in the study was from New York state exclusively, and so diversity in the 2004 to 2007 time frame is likely underestimated. However, the vast majority (>90%) of sequences analyzed were collected from a client base in 46 states.
Since the sequence data were deidentified, it is theoretically possible that some resampling occurred. Analysis of sequence similarity was performed to reduce this possibility. However, such a filter does not discern temporal changes in sequence, such as viral sequence drift, evolution of resistance-associated substitutions, or variability associated with the genotyping assay. Too-stringent criteria would result in the inadvertent elimination of transmission cluster sequences. Thus, the determination of an ideal percentage for identity cutoff is challenging. More-rigorous phylogenetic analysis of the non-subtype B subset of sequences resulted in the elimination of ∼10% of candidate sequences due to resampling. This is in line with previous resampling estimates based on an analysis of a similar database with patient identifiers (40). A similar level of resampling would be expected for the subtype B sequences. Thus, although the non-B prevalence is slightly underestimated, resampling likely has a minimal impact on the overall study conclusions.
Although the HIV-1 pol gene sequence is relatively conserved and sequences can be influenced by antiretroviral drug pressure, it provides ample resolution for distinguishing between subtype B and non-B strains (26). The utility of resistance genotyping sequences for HIV-1 characterization has been established by numerous studies (23, 26, 34–37, 46–49). Current U.S. guidelines recommend resistance genotype testing at the initiation of antiretroviral therapy and for subsequent patient monitoring (50). The availability of HIV-1 nucleotide sequence data from resistance testing provides an opportunity to determine the genomic form of HIV-1 responsible for infection; unfortunately, this is an opportunity not generally capitalized on. Online software such as the REGA Subtyping Tool simplifies mining of subtype information from resistance genotyping sequences. Although the performance of the REGA Subtyping Tool is not perfect (51), it successfully categorized >90% of our sequences. Of the untypeable sequences, ∼90% were subtype B based on subsequent PHYLIP analysis. Most non-B sequences were categorized correctly, although more-rigorous phylogenetic analysis resulted in the reclassification of 22. Thus, this automated and widely accessible online tool provides a useful means for subtype/recombinant form assignment.
An important issue to be explored is the extent of indigenous transmission of non-subtype B strains in the United States. Although studies in the 1990s provided evidence that this was already occurring (44, 45), the degree to which it contributes to ongoing transmission in U.S.-born patients is unknown. Unfortunately, because of the study design, demographic data related to country of birth, ethnicity, risk factors, and travel history are not available, so this issue cannot be addressed here. Increasing movement of non-B viruses from predominantly immigrant populations to native-born individuals has been documented in France, the United Kingdom, and Canada (20, 21, 52, 53). A recent study examining HIV-1 in Maryland revealed that non-B viruses were responsible for 6.5% of infections in U.S.-born individuals (34). Interestingly, a recent study encompassing 15 states and one county that focused on persons whose birthplace was known revealed that, of the non-subtype B variants identified, 28% were in U.S.-born residents (49). This crossover trend warrants future monitoring. Since the commercial immunoassays used to diagnose HIV-1 infection are not designed to discriminate between subtypes and recombinant forms, the spread of these viruses could go largely undetected.
Among the practical considerations associated with increasing HIV-1 diversity is the potential impact on diagnostic, screening, and patient-monitoring assays. Natural genetic polymorphisms have the potential to modify or ablate epitopes utilized for the detection of p24 antigen and HIV-1-specific antibodies, thereby compromising assay performance characteristics (54, 55). It should be recognized that the performance of fourth-generation HIV-1 antigen-antibody combination assays used widely throughout the world varies substantially with respect to the sensitivity of detection of antibodies and antigens of non-subtype B and recombinant strains (7, 17). HIV-1 genetic heterogeneity occurring within primer and/or probe binding sites can also influence the performance of commercial patient-monitoring assays (5, 6, 9, 12–14, 43, 56). Reflecting the degree of challenge that HIV-1 diversity presents, even simultaneous targeting of two genomic regions for viral load determination by one commercial manufacturer does not completely prevent underquantitation in some cases (16, 57). Given the ongoing diversification and continual redistribution of HIV-1, it has become increasingly important to utilize assays whose performance is transparent to group/subtype and recombinant form diversity.
This study shows that the level of HIV-1 strain diversity in the United States is high, with multiple non-B subtypes and many of the recognized CRFs, as well as many URFs. Moreover, the geographic distribution of these HIV variants is widespread. For the years where the coverage was most comprehensive (2008 to 2011), there was a general trend of increasing prevalence of non-B strains in our study population. Factors such as increases in global travel, shifting immigration policies, and indigenous transmission are likely to contribute to an increasing prevalence of non-B variants in the United States. Given the potential implications for diagnostics, patient monitoring, transmissibility, disease pathogenesis, response to treatment, and vaccine development (3, 7–10), continual and more-comprehensive surveillance of the prevalence and distribution of HIV-1 variants in the United States would be prudent.
ACKNOWLEDGMENTS
We thank Heather Beagley, Dave Davis, and Yelena Demura for performing the database queries, Denise Jones and Haley Elmer for removing PHI from the data, Mark Ebbert for performing the sequence similarity analysis, and Priscilla Swanson for critical review of the manuscript.
No funding is applicable to this study. M.T.P. is a consultant for Roche Diagnostics. V.H. and J.H. are employees and shareholders of Abbott Laboratories. D.R.H. is a consultant for Thermo Fisher, Roche Diagnostics, and Primera Diagnostics.
M.T.P., V.H., J.H., and D.R.H. designed the study. M.T.P. and V.H. analyzed the data. J.H., M.T.P., V.H. and D.R.H. drafted the manuscript. All authors reviewed and approved the final manuscript.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Plantier JC, Leoz M, Dickerson JE, de Oliveira F, Cordonnier F, Lemée V, Damond F, Robertson DL, Simon F. 2009. A new human immunodeficiency virus derived from gorillas. Nat. Med. 15:871–872 [DOI] [PubMed] [Google Scholar]
- 2. Kuiken C, Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B. (ed). 2011. HIV Sequence Compendium 2011. Los Alamos National Laboratory, Los Alamos, NM [Google Scholar]
- 3. McCutchan FE. 2006. Global epidemiology of HIV. J. Med. Virol. 78:S7–S12 [DOI] [PubMed] [Google Scholar]
- 4. Hemelaar J, Gouws E, Ghys PD, Osmanov S. WHO-UNAIDS Network for HIV Isolation and Characterisation 2011. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 25:679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swanson P, de Mendoza C, Joshi J, Goldan A, Hodinka RL, Soriano V, Devare SG, Hackett J., Jr 2005. Impact of human immunodeficiency virus type 1 (HIV-1) genetic diversity on performance of four commercial viral load assays: LCx HIV RNA quantitative, AMPLICOR HIV-1 MONITOR version 1.5, VERSANT HIV-1 RNA 3.0, and NucliSens HIV-1 QT. J. Clin. Microbiol. 43:3860–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gueudin M, Plantier JC, Lemée V, Schmitt MP, Chartier L, Bourlet T, Ruffault A, Damond F, Vray M, Simon F. 2007. Evaluation of the Roche Cobas TaqMan and Abbott RealTime extraction-quantification systems for HIV-1 subtypes. J. Acquir. Immune Defic. Syndr. 44:500–505 [DOI] [PubMed] [Google Scholar]
- 7. Ly TD, Ebel A, Faucher V, Fihman V, Laperche S. 2007. Could the new HIV combined p24 antigen and antibody assays replace p24 antigen specific assays? J. Virol. Methods 143:86–94 [DOI] [PubMed] [Google Scholar]
- 8. Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. 2008. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 358:1590–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Church D, Gregson D, Lloyd T, Klein M, Beckthold B, Laupland K, Gill MJ. 2011. Comparison of the RealTime HIV-1, COBAS TaqMan 48 v1.0, Easy Q. v1.2, and Versant v3.0 assays for determination of HIV-1 viral loads in a cohort of Canadian patients with diverse HIV subtype infections. J. Clin. Microbiol. 49:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wainberg MA, Brenner BG. 2012. The impact of HIV genetic polymorphisms and subtype differences on the occurrence of resistance to antiretroviral drugs. Mol. Biol. Int. 2012:256982. 10.1155/2012/256982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saison J, Tardy JC, Scholtes C, Icard V, Trabaud MA, Perpoint T, Chidiac C, Ecohard R, André P, Ferry T. Lyon HIV Cohort Study 2013. Low-level viremia is associated with non-B subtypes in patients infected with HIV with virological success following HAART introduction. J. Med. Virol. 85:953–958 [DOI] [PubMed] [Google Scholar]
- 12. Pyne MT, Brown KL, Hillyard DR. 2010. Evaluation of the Roche Cobas AmpliPrep/Cobas TaqMan HIV-1 test and identification of rare polymorphisms potentially affecting assay performance. J. Clin. Microbiol. 48:2852–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bolivar H, Geffin R, Manzi G, Fischl MA, Holzmayer V, Mak W-B, Abravaya K, Hackett J., Jr 2009. The challenge of HIV diversity: discordant CD4+ T-cell count and viral load in an untreated patient infected with a subtype F strain. J. Acquir. Immune Defic. Syndr. 52:659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jennings C, Harty B, Granger S, Wager C, Crump JA, Fiscus SA, Bremer JW. 2012. Cross-platform analysis of HIV-1 RNA data generated by a multicenter assay validation study with wide geographic representation. J. Clin. Microbiol. 50:2737–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chudy M, Weber-Schehl M, Pichl L, Jork C, Kress J, Heiden M, Funk MB, Nübling CM. 2012. Blood screening nucleic acid amplification test for human immunodeficiency virus type 1 may require two different amplification targets. Transfusion 52:431–439 [DOI] [PubMed] [Google Scholar]
- 16. Karasi JC, Dziezuk F, Quennery L, Förster S, Reischl U, Colucci G, Schoener D, Sequin-Devaux C, Schmit JC. 2011. High correlation between Roche COBAS Ampliprep/COBAS TaqMan HIV-1, v2.0 and the Abbott m2000 RealTime HIV-1 assays for quantification of viral load in HIV-1 B and non-B subtypes. J. Clin. Virol. 52:181–186 [DOI] [PubMed] [Google Scholar]
- 17. Ly TD, Plantier JC, Lebaillais L, Gonzalo S, Lemée V, Laperche S. 2012. The variable sensitivity of HIV Ag/Ab combination assays in the detection of p24 Ag according to genotype could compromise the diagnosis of early HIV infection. J. Clin. Virol. 55:121–127 [DOI] [PubMed] [Google Scholar]
- 18. Barin F, Courouce A-M, Pillonel J, Buzelay L. 1997. Increasing diversity of HIV-1M serotypes in French blood donors over a 10-year period (1985-1995). AIDS 11:1503–1508 [DOI] [PubMed] [Google Scholar]
- 19. Couturier E, Damond F, Roques P, Fleury H, Barin F, Brunet JB, Brun-Vézinet F, Simon F. 2000. HIV-1 diversity in France, 1996–1998. AIDS 14:289–296 [DOI] [PubMed] [Google Scholar]
- 20. Lot F, Samaille C, Cazein F, Barin F, Pinget R, Pillonel J, Desenclos JC. 2004. Preliminary results from the new HIV surveillance system in France. Euro Surveill. 9:34–37 [PubMed] [Google Scholar]
- 21. Semaille C, Barin F, Cazein F, Pillonel J, Lot F, Brand D, Plantier JC, Bernillon P, Le Vu S, Pinget R, Desenclos JC. 2007. Monitoring the dynamics of the HIV epidemic using assays for recent infection and serotyping among new HIV diagnoses: experience after 2 years in France. J. Infect. Dis. 196:377–383 [DOI] [PubMed] [Google Scholar]
- 22. Parry JV, Murphy G, Barlow KL, Lewis K, Rogers PA, Belda FJ, Nicoll A, McGarrigle C, Cliffe S, Mortimer PP, Clewley JP. 2001. National surveillance of HIV-1 subtypes for England and Wales: design, methods, and initial findings. J. Acquir. Immune Defic. Syndr. 26:381–388 [DOI] [PubMed] [Google Scholar]
- 23. Vercauteren J, Wensing AMJ, van de Vijver DAMC, Alvert J, Balotta C, Hamouda O, Kücherer C, Struck D, Schmit JC, Asjö B, Bruckova M, Camacho RJ, Clotet B, Coughlan S, Grossman Z, Horban A, Korn K, Kostrikis L, Nielsen C, Paraskevis D, Poljak M, Puchhammer-Stöckl E, Riva C, Ruiz L, Salminen M, Schuurman R, Sonnerborg A, Stanekova D, Stanojevic M, Vandamme AM, Boucher CA. 2009. Transmission of drug-resistant HIV-1 is stabilizing in Europe. J. Infect. Dis. 200:1503–1508 [DOI] [PubMed] [Google Scholar]
- 24. Bennett D. 2005. HIV-1 genetic diversity surveillance in the United States. J. Infect. Dis. 192:4–9 [DOI] [PubMed] [Google Scholar]
- 25. Brodine SK, Shaffer RA, Starkey MJ, Tasker SA, Gilcrest JL, Louder MK, Barile A, VanCott TC, Vahey MT, McCutchan FE, Birx DL, Richman DD, Mascola JR. 1999. Drug resistance patterns, genetic subtypes, clinical features, and risk factors in military personnel with HIV-1 seroconversion. Ann. Intern. Med. 131:502–506 [DOI] [PubMed] [Google Scholar]
- 26. Wegner SA, Larder B, Hertogs K, Carr J, McCutchan F. 2000. Use of a drug resistance sequence database to determine the prevalence of non-subtype B HIV-1 infections in US and European clinical cohorts, abstr 177. Antiviral Ther. 5(Suppl 3):136–145 [Google Scholar]
- 27. Achkar JM, Burda ST, Konings FAJ, Urbanski MM, Silliams CA, Seifen D, Kahirimbanyi MN, Vogler M, Parta M, Lupatkin HC, Zolla-Pazner S, Nyambi PN. 2004. Infection with HIV type 1 group M non-B subtypes in individuals living in New York City. J. Acquir. Immune Defic. Syndr. 36:835–844 [DOI] [PubMed] [Google Scholar]
- 28. Sides TL, Akinsete O, Henry K, Wotton JT, Carr PW, Bartkus J. 2005. HIV-1 subtype diversity in Minnesota. J. Infect. Dis. 192:37–45 [DOI] [PubMed] [Google Scholar]
- 29. Lin H-H, Gaschen BK, Collie M, El-Fishaway M, Chen Z, Korber BT, Beatrice ST, Zhang L. 2006. Genetic characterization of diverse HIV-1 strains in an immigrant population living in New York City. J. Acquir. Immune Defic. Syndr. 41:399–404 [DOI] [PubMed] [Google Scholar]
- 30. Brennan CA, Stramer SL, Holzmayer V, Yamaguchi J, Foster GA, Notari Iv EP, Schochetman G, Devare SG. 2009. Identification of human immunodeficiency virus type 1 non-B subtypes and antiretroviral drug-resistant strains in United States blood donors. Transfusion 49:125–133 [DOI] [PubMed] [Google Scholar]
- 31. Delwart E, Slikas E, Stramer SL, Kamel H, Kessler D, Krysztof D, Tobler LH, Carrick DM, Steele W, Todd D, Wright DJ, Kleinman SH, Busch MP, NHLBI-REDS-II Study Group 2012. Genetic diversity of recently acquired and prevalent HIV, hepatitis B virus, and hepatitis C virus infections in US blood donors. J. Infect. Dis. 205:875–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wheeler W, Mahle K, Bodnar U, Pienaizek D, Kline R, Hall I, McKenna M, US Variant, Atypical and Resistant HIV Surveillance (VARHS) Group 2007. Antiretroviral drug-resistance mutations and subtypes in drug-naïve persons newly diagnosed with HIV-1 infection, US, March 2003 to October 2006. CDC, Atlanta, GA: http://www.retroconference.org/2007/PDFs/648.pdf [Google Scholar]
- 33. Brennan CA. 2007. Review of status of HIV strain diversity in the United States. J. Med. Virol. 79:S27–S31 [Google Scholar]
- 34. Carr JK, Osinusi A, Flynn CP, Gilliam BL, Maheshwari V, Zhao RY. 2010. Two independent epidemics of HIV in Maryland. J. Acquir. Immune Defic. Syndr. 54:297–303 [DOI] [PubMed] [Google Scholar]
- 35. Gonzales MJ, Machekano RN, Shafer RW. 2001. Human immunodeficiency virus type 1 reverse-transcriptase and protease subtypes: classification, amino acid mutation patterns, and prevalence in a northern California clinic-based population. J. Infect. Dis. 184:998–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cartwright CP. 2006. The changing epidemiology of HIV/AIDS at a Minnesota hospital: impact of demographic change and viral diversity. J. Med. Virol. 78:S19–S21 [DOI] [PubMed] [Google Scholar]
- 37. Wheeler WH, Ziebell RA, Zabina H, Pieniazek D, Prejean J, Bodnar UR, Mahle KC, Heneine W, Johnson JA, Hall HI, Variant Atypical and Resistant HIV Surveillance Group 2010. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, U.S.-2006. AIDS 24:1203–1212 [DOI] [PubMed] [Google Scholar]
- 38. United States Census Bureau 2011. The newly arrived foreign-born population of the United States: 2010. United States Census Bureau, U.S. Department of Commerce, Suitland, MD: http://www.census.gov/prod/2011pubs/acsbr10-16.pdf [Google Scholar]
- 39. de Oliveira CF, Diaz RS, Machado DM, Sullivan MT, Finlayson T, Gwinn M, Lackritz EM, Williams AE, Kessler D, Operskalski EA, Mosley JW, Busch MP. 2000. Surveillance of HIV-1 genetic subtypes and diversity in the US blood supply. Transfusion 40:1399–1406 [DOI] [PubMed] [Google Scholar]
- 40. Ebbert MTW, Mallory MA, Wilson AR, Dooley SK, Hillyard DR. 2013. Application of a new informatics tool for contamination screening in the HIV sequencing laboratory. J. Clin. Virol. 57:249–253 [DOI] [PubMed] [Google Scholar]
- 41. Gao F, Yue L, Hill SC, Robertson DL, Graves AH, Saag MS, Shaw GM, Sharp PM, Hahn BH. 1994. HIV-1 sequence subtype D in the United States. AIDS Res. Hum. Retroviruses 10:625–627 [DOI] [PubMed] [Google Scholar]
- 42. Brodine SK, Mascola JR, Weiss PJ, Ito SI, Porter KR, Artenstein AW, Garland FC, McCutchan FE, Burke DS. 1995. Detection of diverse HIV-1 genetic subtypes in the U. S. A. Lancet 346:1198–1199 [DOI] [PubMed] [Google Scholar]
- 43. Walter EA, Gilliam B, Delmar JA, Spooner K, Morris JT, Aronson N, Wegner SA, Michael NL, Jagodzinski LL. 2000. Clinical implications of identifying non-B subtypes of human immunodeficiency virus type 1 infection. Clin. Infect. Dis. 31:798–802 [DOI] [PubMed] [Google Scholar]
- 44. Artenstein AW, Ohl CA, VanCott TC, Hegerich PA, Mascola JR. 1996. Transmission of HIV-1 subtype E in the United States. JAMA 276:99–100 [PubMed] [Google Scholar]
- 45. Womack C, Roth W, Newman C, Rissing JP, Lovell R, Haburchak D, Essex M, Bond VC. 2001. Identification of non-B human immunodeficiency virus type 1 subtypes in rural Georgia. J. Infect. Dis. 183:138–142 [DOI] [PubMed] [Google Scholar]
- 46. Hanna GJ, Balaguera HU, Freedberg KA, Werner BG, Seger Craven KA, Craven DE, D'Aguila RT. 2003. Drug-selected resistance mutations and non-B subtypes in antiviral-naive adults with established human immunodeficiency virus infection. J. Infect. Dis. 188:986–991 [DOI] [PubMed] [Google Scholar]
- 47. Parker MM, Gordon D, Reilly A, Horowitz HW, Waters M, Bennett R, Hallack R, Smith J, Lamson D, Aydemir A, Dvali N, Agins BD, Drusano GL, Taylor J, Resistance Study Group 2007. Prevalence of drug resistant and nonsubtype B strains in antiretroviral-naïve, HIV-infected individuals in New York State. AIDS Patient Care STDS 21:644–652 [DOI] [PubMed] [Google Scholar]
- 48. Karchava M, Pulver W, Smith L, Philpott S, Sullivan TJ, Wethers J, Parker MM. 2006. Prevalence of drug-resistance mutations and non-subtype B strains among HIV-infected infants from New York State. J. Acquir. Immune Defic. Syndr. 42:614–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pieniazek D, Ziebell R, Ocfemia CB, Saduvala N, Kline R, Prejean J, Kim D, Hall HI, US Variant, Atypical and Resistant HIV Surveillance (VARHS) Group 2012. Phylogenetic surveillance of HIV-1 non-B subtypes among US-born and foreign-born individuals living in the United States revealed similar viral genetic diversity but different race and male transmission patterns. CDC, Atlanta, GA: http://www.retroconference.org/2012b/PDFs/516.pdf [Google Scholar]
- 50. Panel on Antiretroviral Guidelines for Adults and Adolescents 2013. Guidelines for the use of antiretroviral agents in HIV-1 infected adults and adolescents. National Institutes of Health, Rockville, MD: http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf [Google Scholar]
- 51. Yebra G, de Mulder M, Martín L, Pérez-Cachafeiro S, Rodríguez C, Labarga P, García F, Tural C, Jaén A, Navarro G, Holguín A, Cohort of Spanish AIDS Research Network (CoRIS) 2011. Sensitivity of seven HIV subtyping tools differs among subtypes/recombinants in the Spanish cohort of naïve HIV-infected patients (CoRIS). Antiviral Res. 89:19–25 [DOI] [PubMed] [Google Scholar]
- 52. Aggarwal I, Smith M, Tatt ID, Murad S, Osner N, Geretti AM, Easterbrook PJ. 2006. Evidence of onward transmission of HIV-1 non-B subtype strains in the United Kingdom. J. Acquir. Immune Defic. Syndr. 41:201–209 [DOI] [PubMed] [Google Scholar]
- 53. Luft LM, Beckthold B, Gill MJ. 2011. Increasing HIV subtype diversity in Canadian-born patients living in Southern Alberta, Canada. J. Acquir. Immune Defic. Syndr. 57:e27–e29 [DOI] [PubMed] [Google Scholar]
- 54. Gaudy C, Moreau A, Brunet S, Descamps JM, Deleplangue P, Brand D, Barin F. 2004. Subtype B human immunodeficiency virus (HIV) type 1 mutant that escapes detection in a fourth-generation immunoassay for HIV infection. J. Clin. Microbiol. 42:2847–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henquell C, Jacomet C, Antoniotti O, Chaib A, Regagnon C, Brunet S, Peigue-Lafeuille H, Barin F. 2008. Difficulties in diagnosing group O human immunodeficiency virus type 1 acute primary infection. J. Clin. Microbiol. 46:2453–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Rensburg EJ, Tait K, Watt A, Schall R. 2011. Comparative evaluation of the Roche Cobas AmpliPrep/Cobas TaqMan HIV-1 version 2 test using the TaqMan 48 analyzer and the Abbott RealTime HIV-1 assay. J. Clin. Microbiol. 49:377–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Troppan KT, Stelzl E, Violan D, Winkler M, Kessler HH. 2009. Evaluation of the new Versant HIV-1 RNA 1.0 assay (kPCR) for quantitative detection of human immunodeficiency virus type 1 RNA. J. Clin. Virol. 46:69–74 [DOI] [PubMed] [Google Scholar]