

Abstract
Genetic polymorphisms in the malaria parasite Plasmodium falciparum mediate alterations in sensitivity to important antimalarial drugs. Surveillance for these polymorphisms is helpful in assessing the prevalence of drug resistance and designing strategies for malaria control. Multiple methods are available for the assessment of P. falciparum genetic polymorphisms, but they suffer from low throughput, technical limitations, and high cost. We have optimized and tested a multiplex ligase detection reaction-fluorescent microsphere (LDR-FM) assay for the identification of important P. falciparum genetic polymorphisms. For 84 clinical samples from Kampala, Uganda, a region where both transmission intensity and infection complexity are high, DNA was extracted from dried blood spots, genes of interest were amplified, amplicons were subjected to multiplex ligase detection reactions to add bead-specific oligonucleotides and biotin, fragments were hybridized to magnetic beads, and polymorphism prevalences were assessed fluorometrically in a multiplex format. A total of 19 alleles from the pfcrt, pfmdr1, pfmrp1, pfdhfr, and pfdhps genes were analyzed by LDR-FM and restriction fragment length polymorphism (RFLP) analyses. Considering samples with results from the two assays, concordance between the assays was good, with 78 to 100% of results identical at individual alleles, most nonconcordant results differing only between a mixed and pure genotype call, and full disagreement at individual alleles in only 0 to 3% of results. We estimate that the LDR-FM assay offers much higher throughput and lower cost than RFLP. Our results suggest that the LDR-FM system offers an accurate high-throughput means of classifying genetic polymorphisms in field samples of P. falciparum.
INTRODUCTION
Resistance of malaria parasites to commonly used antimalarial drugs is a large problem, in particular for Plasmodium falciparum, the most virulent human malaria parasite and the major malaria parasite infecting humans in Africa. Due to resistance to older drugs, in particular chloroquine and sulfadoxine-pyrimethamine (SP), the standard of care for the treatment of falciparum malaria is now artemisinin-based combination therapy (ACT) (1). ACTs each contain an artemisinin derivative and a longer-acting partner drug that clears parasites that survive 3-day exposure to artemisinins, thus improving therapeutic efficacy and limiting selection of artemisinin resistance. However, the long plasma exposure of partner drugs may facilitate selection for resistance to these agents when recurrent infections occur before clearance of the drugs. In addition, delayed clearance of parasites after treatment with artesunate suggests early signs of resistance to artemisinins in southeast Asia (2, 3). Thus, it is important that we maintain surveillance for resistance to artemisinins and partner drugs. Antifolates continue to be widely used for the treatment of malaria in some areas; artesunate plus SP is the standard treatment for falciparum malaria in India. In addition, antifolates have important roles in the prevention of malaria, with standard practices in areas of Africa where malaria is endemic including intermittent preventive therapy for pregnant women with SP (4), seasonal malaria chemoprevention for children with SP plus amodiaquine in areas with seasonal malaria transmission (5), and administration of trimethoprim-sulfamethoxazole, which protects against malaria, to HIV-infected children (6). Therefore, continued surveillance for resistance to antifolate antimalarials is also important.
Our understanding of mechanisms of resistance to antimalarials is incomplete. For chloroquine, mutations in the putative drug transporters pfcrt and pfmdr1 are the key mediators of resistance, with the pfcrt K76T polymorphism being of primary importance (7). Two related aminoquinolines are ACT partner drugs; resistance to amodiaquine is similarly mediated by pfcrt and pfmdr1 polymorphisms (8–10), but this does not appear to be the case with piperaquine (11). For artemisinins, and for the ACT partner drugs mefloquine and lumefantrine, parasite drug sensitivity is mediated in part by polymorphisms in pfmdr1, but interestingly, polymorphisms that decrease sensitivity to aminoquinolines lead to increased sensitivity to these drugs (9, 12, 13). Polymorphisms in one additional putative drug transporter, pfmrp1, may play a role in mediating drug sensitivity; the pfmrp1 I876V polymorphism is prevalent in Africa, and the wild-type sequence was selected by prior treatment with artemether-lumefantrine (14). Resistance to SP is well characterized, with a series of mutations in the target enzymes dihydrofolate reductase (DHFR) and dihydropteroate synthase (DHPS) leading to stepwise acquisition of resistance (15).
Parasite genetic polymorphisms associated with drug resistance have been well studied with a number of assays, most notably restriction fragment length polymorphism (RFLP) analysis (16). RFLP analysis offers reliable assessment of known polymorphisms, but with rather low throughput. Other relatively low-throughput methodologies include direct DNA sequencing, mutation-specific PCR (17), dot blot probe hybridization (18), molecular beacons (19), and single-nucleotide primer extension (20). Systems examined to provide improved throughput have included polymorphism-specific microarrays (21), melting curve analysis (22, 23), quantitative PCR (24–26), and a ligase detection reaction-fluorescent microsphere (LDR-FM) assay (27, 28). Each of these assays has shown success, although each has challenges, including the cost of equipment and reagents and the uncertainty of success with field samples, which are typically stored on filter paper at room temperature and commonly consist of polyclonal infections. Our goal has been to develop a high-throughput system for the analysis of parasites from dried filter paper blood spots stored at room temperature, as these are typically available from research and surveillance programs across Africa. To this end, we have worked to optimize the LDR-FM assay (28), incorporating new-generation magnetic bead technology and use of field samples from Uganda extracted from filter paper. We present results demonstrating a robust assay that dramatically improves throughput over RFLP analysis and is appropriate for field conditions.
MATERIALS AND METHODS
Samples for analysis.
Control parasite DNA was from the Malaria Research and Reference Reagent Resource Center. Field samples were from a randomized trial comparing the antimalarial efficacy of 3 antimalarial treatments conducted in Kampala, Uganda, from the 2004-2008 period (29, 30). Data on parasite densities and multiplicities of infection from the same trial have been published previously (31). All clinical samples were collected as blood spots on filter paper, as previously described (29), and stored with desiccant at room temperature. DNA extraction and analyses were conducted in the 2012-2013 period, so samples were stored at room temperature for 4 to 8 years before study.
RFLP analyses.
DNA was extracted using Chelex 100 (Bio-Rad) (32), and analyses were conducted as previously described (16, 33, 34). Briefly, regions of interest were amplified, PCR products were treated with polymorphism-specific restriction endonucleases, and sizes of products were characterized by agarose gel electrophoresis to distinguish wild-type, mutant, and mixed alleles based on comparison with control reference strain DNA.
DNA extraction and allele amplification for LDR-FM.
DNA was extracted into 100 μl of water from blood spots dried on filter paper (Whatman 3MM) using Chelex 100 (Bio-Rad), as previously described (32). PCR amplification was performed in a 25-μl reaction mixture containing 160 nM upstream and downstream primers (Integrated DNA Technologies) (see Table S1 in the supplemental material), 160 μM deoxynucleoside triphosphates (dNTPs) (Invitrogen), 1 unit Taq DNA polymerase (New England BioLabs), and 4 μl of extracted DNA in 1× standard Taq buffer (New England BioLabs) with a Bio-Rad C1000 thermocycler. Thermocycling conditions were 95°C for 60 s; 11 cycles of 95°C for 15 s, 60°C for 30 s, and 68°C for 60 s; 39 cycles of 95°C for 15 s, 55°C for 30 s, and 68°C for 60 s; and 68°C for 5 min. To validate assays, PCR products from each primer set were resolved by agarose gel electrophoresis and examined to confirm the presence of expected amplicons.
Multiplex ligase detection reaction.
LDR mixtures contained allele-specific and common primers (Integrated DNA Technologies) (see Table S2 in the supplemental material). Each allele-specific primer contained a 5′ unique nucleotide sequence complementing a sequence attached to a MagPlex-Tag bead (Luminex) and a 3′ sequence corresponding to a particular P. falciparum polymorphism. Common primers were modified by 5′ phosphorylation and 3′ biotinylation. Two multiplex reactions were created by combining identical volumes of all PCR products for the studied transporter (pfcrt, pfmdr1, and pfmrp1) and antifolate (pfdhfr and pfdhps) alleles. LDRs (15 μl) contained 10 nM upstream and downstream primers, 2 units of Taq DNA ligase (New England BioLabs), and 1 μl of combined PCR products in 1× Taq ligase buffer (New England BioLabs). Thermocycling conditions were 95°C for 60 s followed by 32 cycles of 95°C for 15 s and 58°C for 2 min. Controls containing DNA from reference strains were included in all reactions.
Hybridization and labeling of magnetic beads.
Products of LDRs were hybridized to MagPlex-Tag beads by adding 1,000 beads corresponding to each allele-specific primer in the LDR to 1.5× tetramethylammonium chloride (TMAC) buffer (3 M tetramethylammonium chloride, 50 mM Tris-HCl [pH 8], 3 mM EDTA, and 0.1% N-lauroylsarcosine sodium salt; all from Sigma-Aldrich) and then adding the LDR multiplex product (5 μl) to 60 μl of the TMAC bead mixture and running in a Bio-Rad C1000 thermocycler at 95°C for 90 s, followed by 37°C for 40 min. Each hybridized LDR product was then labeled by adding 6 μl of a 1:50 dilution of streptavidin-R-phycoerythrin (SA-PE) (1.7 μg/ml final concentration; Invitrogen) with 0.1% bovine serum albumin (BSA) (Enzo Life Sciences) in 1.5× TMAC buffer and incubating at 37°C for 20 min. To quantify the abundance of different alleles, labeled products were then run on a MAGPIX instrument with xPonenet 4.2 software (Luminex). Results were read as fluorescence intensity for each reaction in a 96-well format.
DNA sequencing.
For allele reads that were fully discrepant between LDR-FM and RFLP assessments, dideoxy sequencing was performed by standard methods at the University of California San Francisco (UCSF) Genomics Core Facility.
Statistical analysis.
The correlation of results between the assays was assessed using kappa statistics.
RESULTS
Analysis of P. falciparum reference strains.
We initially studied polymorphisms of interest in 7 P. falciparum reference strains. We assessed sequences at 5 single nucleotide polymorphisms (SNPs) in pfmdr1 that are common in different parts of the world, a 5 amino acid haplotype in pfcrt that distinguishes chloroquine-sensitive and -resistant parasites with different geographic backgrounds, an SNP in pfmrp1 that is common in Africa and has been selected by prior treatment, 4 known SNPs in the antifolate target gene pfdhfr, and 5 known SNPs in the antifolate target gene pfdhps (7, 14, 15). Sequence determinations at all studied polymorphisms were unequivocal, generally with background readings for unidentified SNPs 5- to 10-fold lower than the readings for correct identifications (Fig. 1). To assess the ability of the LDR-FM assay to identify mixed alleles in complex samples, we mixed different percentages of DNA from two reference strains and studied the identification of two alleles. Minority percentages greater than 10% were clearly identified (Fig. 2).
Fig 1.

LDR-FM readings for 19 SNPs in P. falciparum reference strains. Values shown are uncorrected mean fluorescence readings for 3 to 5 assays, each run in triplicate. For pfcrt, the haplotype represents amino acids 72 to 76 in the gene product. Readings representing the known sequences at each allele are in bold type.
Fig 2.
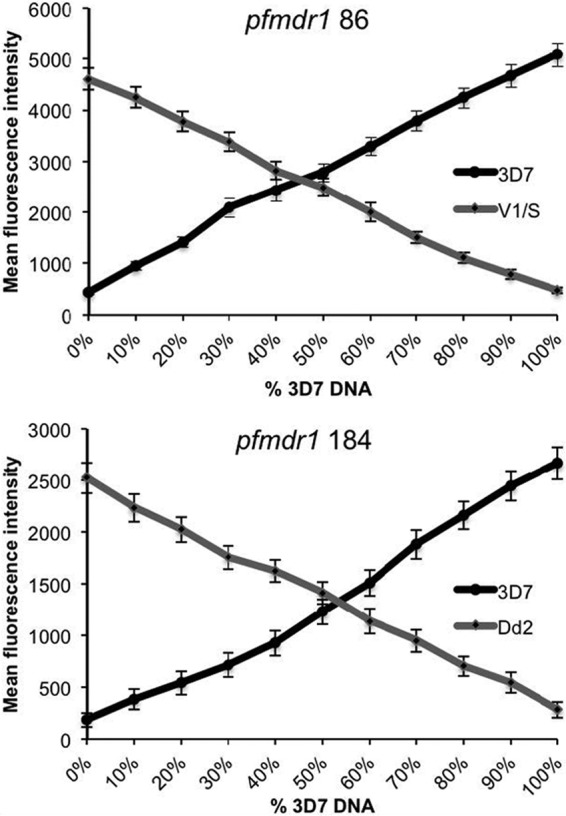
Mixing experiments. DNA from the indicated reference strains was mixed at the ratios shown, and the prevalences of the pfmdr1 86 and 184 alleles were assessed by LDR-FM. Mean fluorescence intensities, each based on triplicate readings, are shown. Error bars represent standard deviations from triplicate readings.
Analysis of Ugandan samples.
Clinical samples were stored on filter paper for a number of years before analysis, contained highly variable quantities of parasite DNA, and frequently consisted of polyclonal infections. We analyzed 84 samples by RFLP analysis, using established methods, and LDR-FM analysis, using conditions optimized for this study. LDR-FM methods used previously published oligonucleotide primers and reaction conditions (27, 28) as a starting point, but extensive modification of methods was required to obtain optimal results with the field samples. Representative uncorrected fluorescence data for 10 samples showed generally good discrimination of wild-type, mutant, and mixed samples (Fig. 3).
Fig 3.

Representative LDR-FM results for 10 Ugandan samples. Results shown are for the same 10 clinical specimens for all studied alleles. Values shown are uncorrected mean fluorescence readings. For pfcrt, the haplotype represents amino acids 72 to 76 in the gene product. Readings representing the sequence call at each allele after correction by subtraction of background are in bold type.
Based on results with reference strains, we developed an algorithm for the classification of results for each SNP of interest, including the consideration of a mixed sequence at each allele. MFI readings varied considerably for each SNP (Fig. 1 and 2). Therefore, a separate background threshold and correction factor were devised for each SNP to best eliminate spurious readings due to background intensity but not eliminate valid readings (Table 1). Specifically, through iterative analysis of reference samples we established a minimum threshold of 400 MFI, and then, for each SNP, we used means of triplicate readings to establish correction factors to allow correct reads of reference samples with known sequences at each allele of interest.
Table 1.
Mean fluorescence readings, thresholds, and correction factors for each SNPa
| Gene | Allele | MFI | Threshold | Correction factor |
|---|---|---|---|---|
| pfdhfr | 51N | 2,017 | 538 | 1.5 |
| 51I | 1,603 | 623 | 1.5 | |
| 59R | 3,113 | 625 | 1.5 | |
| 59C | 2,021 | 605 | 1.5 | |
| 108S | 1,759 | 920 | 1.6 | |
| 108T | NA | 400 | 1.8 | |
| 108N | 3,974 | 831 | 1.6 | |
| 164I | 915 | 400 | 1.5 | |
| 164L | 981 | 405 | 2 | |
| pfdhps | 436-437 SA | 1,385 | 400 | 1.5 |
| 436-437 SG | 4,063 | 1,537 | 1.5 | |
| 436-437 AG | 1,069 | 400 | 1.5 | |
| 436-437 FG | 4,017 | 2,115 | 1.5 | |
| 540K | 2,099 | 412 | 1.5 | |
| 540E | 1,180 | 400 | 1.5 | |
| 581A | 3,254 | 582 | 1.9 | |
| 581G | 2,554 | 1,410 | 1.5 | |
| 613A | 1,398 | 475 | 1.5 | |
| 613S | 4,181 | 474 | 1.5 | |
| pfmdr1 | 86N | 3,033 | 443 | 1.5 |
| 86Y | 1,999 | 400 | 1.5 | |
| 184Y | 2,887 | 400 | 1.75 | |
| 184F | 1,892 | 571 | 1.75 | |
| 1034S | 3,912 | 400 | 1.5 | |
| 1034C | 5,016 | 1,716 | 2.75 | |
| 1042N | 3,134 | 400 | 1.5 | |
| 1042D | 4,789 | 932 | 2.25 | |
| 1246D | 2,117 | 400 | 1.5 | |
| 1246Y | 1,644 | 400 | 1.5 | |
| pfcrt | CVMNK | 1,523 | 839 | 2 |
| CVIET | 2,044 | 464 | 1.6 | |
| *SVMNT | 1,723 | 400 | 1.5 | |
| pfmrp1 | 876I | 1,858 | 667 | 2 |
| 876V | 4,802 | 1,624 | 4.25 |
For each allele, the mean fluorescence intensity (MFI) was based on 9 readings from clinical samples (3 readings for pfmrp1), except for pfcrt SVMNT and pfdhps 436F/437G, 581G, and 613S, for which the values were obtained from reference strains. For a positive call at a particular allele, the MFI must be greater than the assigned threshold and also greater than the mean value for negative controls on the same plate multiplied by the correction factor.
Results with the LDR-FM methodology were compared to those with RFLP analysis. It is important to note that both systems have potential for errors in misclassification, in particular due to challenges in distinguishing pure and mixed genotypes at each allele of interest. Concordance between results from the two assays was generally good, although results varied between the studied alleles (Fig. 4). Most discrepancies between results were due to a mixed reading with one assay, compared to a pure mutant or wild-type reading with the other assay. Fully discrepant readings were seen in up to 3% of readings at 6 of the studied alleles. For the 9 discrepant alleles, dideoxy sequencing results agreed with the LDR-FM read in 8/9 and the RFLP read in 1/9 (1 of 2 pfdhfr 164 reads).
Fig 4.

Agreement between RFLP and LDR-FM assays. Results are shown for samples with readings from both assays at the indicated alleles. For the 84 samples studied, the RFLP assay was unsuccessful for 0 to 14 (mean, 6.2) samples, and the LDR-FM assay was unsuccessful for 0 to 11 (mean, 3.1) samples for the indicated alleles. Kappa statistics were 0.9 to 1 for all comparisons except for pfmdr1 1246 (0.72) and pfdhps 540 (0.55), with most discrepancies explained by differences in the percentages of mixed alleles. RFLP assays were not done for the other alleles studied by LDR-FM.
DISCUSSION
We have optimized a new LDR-FM methodology for the high-throughput detection of P. falciparum genetic polymorphisms associated with varied drug sensitivity in samples from a field trial in Uganda. The methodology involves amplification of alleles of interest from filter paper blood samples, multiplex ligase detection reactions to allow binding of DNA to magnetic beads and biotinylation, and then discrimination of allele sequences based on the fluorescence of magnetic beads recognizing different alleles. We built on a previously published system (27, 28) to establish good performance of multiplex assays using a new magnetic bead LDR-FM methodology for the characterization of 19 alleles in 5 relevant genes in reference P. falciparum DNA. More importantly, LDR-FM provided characterization of parasites from complex Ugandan blood samples that had been stored for 4 to 8 years on filter paper at room temperature, suggesting that the technique offers an accurate and efficient means of high-throughput assessment of field samples of P. falciparum.
A number of different methodologies for the analysis of drug-resistance-mediating SNPs in P. falciparum have been reported in recent years. Direct DNA sequencing of samples has become simpler and cheaper, but it remains a rather low-throughput technology, and it is not well suited for characterizing mixed genotypes, which are common in Africa. The most widely used technique for SNP characterization has been RFLP analysis, which utilizes PCR and electrophoresis technology that is routinely available at many developing world laboratories. However, RFLP is fairly expensive and labor-intensive, and its throughput is quite low. Thus, a number of groups have attempted to develop higher-throughput methods for the analysis of P. falciparum genotypes. A microarray-based method proved robust, sensitive, and accurate, but this methodology requires access to microarray slides and a fluorescence scanner, which may not be available in many laboratories (21). Melting curve analysis (23) and quantitative PCR (25, 26) offer other high-throughput systems for detection of P. falciparum SNPs for laboratories with appropriate infrastructure, and these systems are particularly suited to identify small minority populations in mixed samples. However, for microarray, melting curve, and quantitative PCR analyses, results have not been reported for filter paper field samples from populations with polymorphisms at a number of alleles of importance in Africa, e.g., pfmdr1 1246Y and pfdhps 540E. Direct comparisons of the accuracy, throughput, and cost of high-throughput systems for SNP detection have not been reported. In many cases the key consideration in choice of analysis, especially in field laboratories, will be availability of necessary equipment.
The LDR-FM methodology requires specialized equipment, but this device can provide a range of assays, including measurements of serum cytokine levels and antibody reactivity, suggesting that it may be of value for field-based laboratories. In our laboratory, reagent costs for LDR-FM assays to evaluate the 19 SNPs considered in this study are estimated at ∼$3.50/sample, assuming 96-well reactions and 12 control wells per plate. About 60% of this cost is for magnetic beads and ∼17% for PCR and LDR enzymes and reagents, and the remainder is for disposable supplies. Reagent costs for RFLP assays for the same SNPs (omitting pfmrp1 876) in our laboratory are estimated at $6.90/sample, with substantial costs for the multiple PCR reagents and restriction endonucleases required to assess 18 SNPs. Importantly, the LDR-FM methodology offers substantial improvement in throughput compared to RFLP. We estimate routine performance by one investigator of 8 to 10 LDR-FM assays, each containing 84 sample wells plus controls, per week, and thus evaluation of 672 to 840 samples per week. In our experience some reactions will need repeating due to inadequate fluorescence readings and other factors. Some reactions benefited from a nested PCR step, with improved fluorescent signals, but the nested reaction was not consistently helpful and was not included in the data presented in this report. For RFLP analysis, considering the time for multiple nested PCRs and restriction endonuclease digestions, we estimate full evaluation of ∼100 samples per week. As with the LDR-FM assay, some RFLP assays will need to be repeated to optimize outputs. Overall, we estimate that, compared to RFLP analysis, the LDR-FM assay will require half the cost per SNP, with throughput improved 5- to 10-fold. However, LDR-FM methodology may be less advantageous in settings where only a small number of parasite polymorphisms are of interest, and direct sequencing, RFLP analyses, or other systems may be more practical in these settings.
The LDR-FM methodology that we have optimized seems well suited for the high-throughput analysis of P. falciparum genetic polymorphisms. Our results were robust despite the use of samples that were stored as dried blood spots on filter paper for a number of years before DNA extraction and analysis. The system offers accuracy similar to that of other reported systems, with substantial cost and throughput advantages over RFLP analysis. Thus, we suggest that the LDR-FM system may be appropriate for laboratories in developing- or developed-world settings wishing to improve throughput of assessment of known P. falciparum resistance-mediating genetic polymorphisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Peter Zimmerman, Rina Wong, and Melinda Zikursh for generous and valuable advice regarding the LDR-FM assay, which was initially developed in their laboratory, Jenny Legac for technical support, and the children, parents, and guardians who participated in the clinical trial that provided samples for this study.
This work was supported by an International Center of Excellence in Malaria Research grant from the National Institutes of Health (AI089674).
Footnotes
Published ahead of print 29 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00904-13.
REFERENCES
- 1. Nosten F, White NJ. 2007. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 77(6 Suppl):181–192 [PubMed] [Google Scholar]
- 2. Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359:2619–2620 [DOI] [PubMed] [Google Scholar]
- 4. ter Kuile FO, van Eijk AM, Filler SJ. 2007. Effect of sulfadoxine-pyrimethamine resistance on the efficacy of intermittent preventive therapy for malaria control during pregnancy: a systematic review. JAMA 297:2603–2616 [DOI] [PubMed] [Google Scholar]
- 5. Konate AT, Yaro JB, Ouedraogo AZ, Diarra A, Gansane A, Soulama I, Kangoye DT, Kabore Y, Ouedraogo E, Ouedraogo A, Tiono AB, Ouedraogo IN, Chandramohan D, Cousens S, Milligan PJ, Sirima SB, Greenwood B, Diallo DA. 2011. Intermittent preventive treatment of malaria provides substantial protection against malaria in children already protected by an insecticide-treated bednet in Burkina Faso: a randomised, double-blind, placebo-controlled trial. PLoS Med. 8:e1000408. 10.1371/journal.pmed.1000408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gasasira AF, Kamya MR, Ochong EO, Vora N, Achan J, Charlebois E, Ruel T, Kateera F, Meya DN, Havlir D, Rosenthal PJ, Dorsey G. 2010. Effect of trimethoprim-sulphamethoxazole on the risk of malaria in HIV-infected Ugandan children living in an area of widespread antifolate resistance. Malar. J. 9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valderramos SG, Fidock DA. 2006. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol. Sci. 27:594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dokomajilar C, Lankoande ZM, Dorsey G, Zongo I, Ouedraogo JB, Rosenthal PJ. 2006. Roles of specific Plasmodium falciparum mutations in resistance to amodiaquine and sulfadoxine-pyrimethamine in Burkina Faso. Am. J. Trop. Med. Hyg. 75:162–165 [PubMed] [Google Scholar]
- 9. Humphreys GS, Merinopoulos I, Ahmed J, Whitty CJ, Mutabingwa TK, Sutherland CJ, Hallett RL. 2007. Amodiaquine and artemether-lumefantrine select distinct alleles of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for uncomplicated malaria. Antimicrob. Agents Chemother. 51:991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nawaz F, Nsobya SL, Kiggundu M, Joloba M, Rosenthal PJ. 2009. Selection of parasites with diminished drug susceptibility by amodiaquine-containing antimalarial regimens in Uganda. J. Infect. Dis. 200:1650–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Some AF, Sere YY, Dokomajilar C, Zongo I, Rouamba N, Greenhouse B, Ouedraogo JB, Rosenthal PJ. 2010. Selection of known Plasmodium falciparum resistance-mediating polymorphisms by artemether-lumefantrine and amodiaquine-sulfadoxine-pyrimethamine but not dihydroartemisinin-piperaquine in Burkina Faso. Antimicrob. Agents Chemother. 54:1949–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dokomajilar C, Nsobya SL, Greenhouse B, Rosenthal PJ, Dorsey G. 2006. Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob. Agents Chemother. 50:1893–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mwai L, Kiara SM, Abdirahman A, Pole L, Rippert A, Diriye A, Bull P, Marsh K, Borrmann S, Nzila A. 2009. In vitro activity of piperaquine, lumefantrine and dihydroartemisinin in Kenyan Plasmodium falciparum isolates and polymorphisms in pfcrt and pfmdr1. Antimicrob. Agents Chemother. 53:5069–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dahlstrom S, Ferreira PE, Veiga MI, Sedighi N, Wiklund L, Martensson A, Farnert A, Sisowath C, Osorio L, Darban H, Andersson B, Kaneko A, Conseil G, Bjorkman A, Gil JP. 2009. Plasmodium falciparum multidrug resistance protein 1 and artemisinin-based combination therapy in Africa. J. Infect. Dis. 200:1456–1464 [DOI] [PubMed] [Google Scholar]
- 15. Gregson A, Plowe CV. 2005. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol. Rev. 57:117–145 [DOI] [PubMed] [Google Scholar]
- 16. Duraisingh MT, Curtis J, Warhurst DC. 1998. Plasmodium falciparum: detection of polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes by PCR and restriction digestion. Exp. Parasitol. 89:1–8 [DOI] [PubMed] [Google Scholar]
- 17. Dorsey G, Kamya MR, Singh A, Rosenthal PJ. 2001. Polymorphisms in the Plasmodium falciparum pfcrt and pfmdr-1 genes and clinical response to chloroquine in Kampala, Uganda. J. Infect. Dis. 183:1417–1420 [DOI] [PubMed] [Google Scholar]
- 18. Ranford-Cartwright LC, Johnston KL, Abdel-Muhsin AM, Khan BK, Babiker HA. 2002. Critical comparison of molecular genotyping methods for detection of drug-resistant Plasmodium falciparum. Trans. R. Soc. Trop. Med. Hyg. 96:568–572 [DOI] [PubMed] [Google Scholar]
- 19. Durand R, Eslahpazire J, Jafari S, Delabre JF, Marmorat-Khuong A, di Piazza JP, Le Bras J. 2000. Use of molecular beacons to detect an antifolate resistance-associated mutation in Plasmodium falciparum. Antimicrob. Agents Chemother. 44:3461–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair S, Brockman A, Paiphun L, Nosten F, Anderson TJ. 2002. Rapid genotyping of loci involved in antifolate drug resistance in Plasmodium falciparum by primer extension. Int. J. Parasitol. 32:852–858 [DOI] [PubMed] [Google Scholar]
- 21. Crameri A, Marfurt J, Mugittu K, Maire N, Regos A, Coppee JY, Sismeiro O, Burki R, Huber E, Laubscher D, Puijalon O, Genton B, Felger I, Beck HP. 2007. Rapid microarray-based method for monitoring of all currently known single-nucleotide polymorphisms associated with parasite resistance to antimalaria drugs. J. Clin. Microbiol. 45:3685–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Decuypere S, Elinck E, Van Overmeir C, Talisuna AO, D'Alessandro U, Dujardin JC. 2003. Pathogen genotyping in polyclonal infections: application of a fluorogenic polymerase-chain-reaction assay in malaria. J. Infect. Dis. 188:1245–1249 [DOI] [PubMed] [Google Scholar]
- 23. Daniels R, Ndiaye D, Wall M, McKinney J, Sene PD, Sabeti PC, Volkman SK, Mboup S, Wirth DF. 2012. Rapid, field-deployable method for genotyping and discovery of single-nucleotide polymorphisms associated with drug resistance in Plasmodium falciparum. Antimicrob. Agents Chemother. 56:2976–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Monbrison F, Raynaud D, Latour-Fondanaiche C, Staal A, Favre S, Kaiser K, Peyron F, Picot S. 2003. Real-time PCR for chloroquine sensitivity assay and for pfmdr1-pfcrt single nucleotide polymorphisms in Plasmodium falciparum. J. Microbiol. Methods 54:391–401 [DOI] [PubMed] [Google Scholar]
- 25. Purfield A, Nelson A, Laoboonchai A, Congpuong K, McDaniel P, Miller RS, Welch K, Wongsrichanalai C, Meshnick SR. 2004. A new method for detection of pfmdr1 mutations in Plasmodium falciparum DNA using real-time PCR. Malar. J. 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kamau E, Alemayehu S, Feghali KC, Tolbert LS, Ogutu B, Ockenhouse CF. 2012. Development of a TaqMan Allelic Discrimination assay for detection of single nucleotides polymorphisms associated with anti-malarial drug resistance. Malar. J. 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carnevale EP, Kouri D, DaRe JT, McNamara DT, Mueller I, Zimmerman PA. 2007. A multiplex ligase detection reaction-fluorescent microsphere assay for simultaneous detection of single nucleotide polymorphisms associated with Plasmodium falciparum drug resistance. J. Clin. Microbiol. 45:752–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong RP, Karunajeewa H, Mueller I, Siba P, Zimmerman PA, Davis TM. 2011. Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection reaction fluorescent microsphere assay. Antimicrob. Agents Chemother. 55:798–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dorsey G, Staedke S, Clark TD, Njama-Meya D, Nzarubara B, Maiteki-Sebuguzi C, Dokomajilar C, Kamya MR, Rosenthal PJ. 2007. Combination therapy for uncomplicated falciparum malaria in Ugandan children: a randomized trial. JAMA 297:2210–2219 [DOI] [PubMed] [Google Scholar]
- 30. Clark TD, Njama-Meya D, Nzarubara B, Maiteki-Sebuguzi C, Greenhouse B, Staedke SG, Kamya MR, Dorsey G, Rosenthal PJ. 2010. Incidence of malaria and efficacy of combination antimalarial therapies over 4 years in an urban cohort of Ugandan children. PLoS One 5:e11759. 10.1371/journal.pone.0011759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nsobya SL, Kiggundu M, Joloba M, Dorsey G, Rosenthal PJ. 2008. Complexity of Plasmodium falciparum clinical samples from Uganda during short-term culture. J. Infect. Dis. 198:1554–1557 [DOI] [PubMed] [Google Scholar]
- 32. Plowe CV, Djimde A, Bouare M, Doumbo O, Wellems TE. 1995. Pyrimethamine and proguanil resistance-conferring mutations in Plasmodium falciparum dihydrofolate reductase: polymerase chain reaction methods for surveillance in Africa. Am. J. Trop. Med. Hyg. 52:565–568 [DOI] [PubMed] [Google Scholar]
- 33. Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourte Y, Dicko A, Su XZ, Nomura T, Fidock DA, Wellems TE, Plowe CV, Coulibaly D. 2001. A molecular marker for chloroquine-resistant falciparum malaria. N. Engl. J. Med. 344:257–263 [DOI] [PubMed] [Google Scholar]
- 34. Duraisingh MT, Jones P, Sambou I, von Seidlein L, Pinder M, Warhurst DC. 2000. The tyrosine-86 allele of the pfmdr1 gene of Plasmodium falciparum is associated with increased sensitivity to the anti-malarials mefloquine and artemisinin. Mol. Biochem. Parasitol. 108:13–23 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.