

Abstract
Krüppel-like factors 3 and 8 (KLF3 and KLF8) are highly related transcriptional regulators that bind to similar sequences of DNA. We have previously shown that in erythroid cells there is a regulatory hierarchy within the KLF family, whereby KLF1 drives the expression of both the Klf3 and Klf8 genes and KLF3 in turn represses Klf8 expression. While the erythroid roles of KLF1 and KLF3 have been explored, the contribution of KLF8 to this regulatory network has been unknown. To investigate this, we have generated a mouse model with disrupted KLF8 expression. While these mice are viable, albeit with a reduced life span, mice lacking both KLF3 and KLF8 die at around embryonic day 14.5 (E14.5), indicative of a genetic interaction between these two factors. In the fetal liver, Klf3 Klf8 double mutant embryos exhibit greater dysregulation of gene expression than either of the two single mutants. In particular, we observe derepression of embryonic, but not adult, globin expression. Taken together, these results suggest that KLF3 and KLF8 have overlapping roles in vivo and participate in the silencing of embryonic globin expression during development.
INTRODUCTION
Krüppel-like factors (KLFs) are DNA-binding transcriptional regulators that are involved in a wide range of biological processes (1, 2). The defining feature of KLFs is the presence of a conserved, tandem repeat of three Cys2-His2-type zinc fingers at their C termini, through which KLFs make sequence-specific contacts with GC-rich and CACCC-related elements of DNA (3–6). While as a whole this domain is highly conserved within the family, specific amino acid differences between the various KLFs result in different DNA-binding preferences (7). As such, individual KLFs have been shown to regulate overlapping, but also distinct, sets of target genes (8, 9). In addition, particular KLFs group together in their DNA-binding specificities. For instance, KLF1 and KLF3 recognize similar sequences and hence regulate overlapping sets of target genes in erythroid cells (8, 10, 11), while KLF4 and KLF5 together coregulate other genes in stem cells and other tissues (9, –16).
Outside of the DNA-binding domain, KLFs exhibit relatively little conservation and interact with a range of transcriptional coregulators and histone-modifying enzymes (2, 17). Some KLFs, such as KLF1, primarily function as activators of transcription (18), while other KLFs, for example KLF3, have typically been characterized as transcriptional repressors (19). It is also evident that some KLFs can act as either activators or repressors, depending on biological context and the gene regulatory region through which they are operating. This is indeed the case for KLF8 (20–24) and has also been reported in some instances for KLF1 (25–28) and KLF3 (29, 30).
KLF1, formerly known as erythroid Krüppel-like factor (EKLF), is primarily expressed in erythroid cells (18) and is a master regulator of multiple facets of erythropoietic differentiation (6, 28, 31–37). It is predominantly a transcriptional activator and binds to CACCC box motifs of the general consensus 5′-CCM CRC CCN-3′ (3, 6). Such motifs are present in the regulatory regions of many diverse genes but in particular have long been noted to be essential for the expression of various erythroid genes, such as those coding for the globins (38). A notable example is the CACCC box that resides within the promoter of the adult β-major globin (Hbb-b1) gene (3, 6, 18).
Hemoglobin is an oxygen-conveying metalloprotein that is expressed in erythrocytes and is composed of two α-like and two β-like globin peptide chains. Throughout ontogeny, there are different forms of both the α-like and β-like globins that are expressed to meet various developmental oxygen demands. This is known as globin switching, and it accompanies shifting sites of erythropoiesis during development. For instance, the embryonic α-like globin (Hba-x) and β-like globins (Hbb-y and Hbb-bh1) are expressed in primitive erythroid cells that are transiently produced in the yolk sac from around embryonic day 7.5 (E7.5) (39, 40). In contrast, the adult globin genes (Hba-a1 and Hbb-b1) are expressed in definitive cells produced in the fetal liver from around E9.5 up until birth, at which point erythropoiesis shifts to the bone marrow (39). In humans, the β-globin locus is subject to a similar yet distinct mechanism of switching: embryonic ε-globin is produced by primitive erythroid cells in the yolk sac; fetal γ-globin is expressed by definitive cells in the liver until birth, and transcription of the adult β- and δ-globin genes commences perinatally and continues throughout life.
KLF1 plays a crucial role in the switching of β-like globins. It directly binds to the β-major globin promoter in vivo and is required for its transcriptional activity in definitive erythroid cells (6, 28, 32, 41, 42). Consequently, Klf1 null mice have depleted levels of β-major globin and die at around E15 of severe anemia (43, 44). Consistent with this, mutations within the orthologous KLF1-binding site in the human β-globin promoter are associated with β-thalassemia (45). In addition to being essential for the normal expression of adult β-globin, KLF1 also plays an indirect role in the silencing of fetal γ-globin in human erythroid cells. It achieves this by driving the expression of BCL11A, an established repressor of γ-globin (46–49). Lastly, in addition to its role in definitive erythroid cells, KLF1 has recently been shown to directly activate transcription of embryonic globins in primitive erythroid cells and accordingly, Hbb-y mRNA and Hbb-bh1 mRNA are reduced in Klf1−/− embryos (50–52).
In addition to globin genes, KLF1 drives the expression of many genes involved in erythropoietic pathways, such as heme biosynthesis, cell cycle control, and the establishment of membrane integrity (6, 28, 31–37). KLF1 also activates the transcription of two other family members in erythroid cells, Klf3 and Klf8 (10, 11). KLF3 (previously, basic Krüppel-like factor [BKLF]) is a potent transcriptional repressor that silences gene expression by recruiting the corepressor C-terminal binding protein (CtBP) (19, 53). KLF3 is expressed widely (29) but in particular is found at high levels in erythroid tissue owing to an erythroid cell-specific promoter that is directly activated by KLF1 (6, 10). KLF3 exhibits similar DNA-binding preferences to KLF1 in vitro (29). Consistent with this, KLF3 represses many genes that are activated by KLF1 in erythroid cells in vivo and is thought to fine-tune their expression during erythropoiesis (8). In the absence of KLF3, a set of genes is abnormally derepressed in mature erythroid cells. This is thought to explain the multiple erythroid defects of the Klf3 null mice, namely, reticulocytosis, increased nuclear inclusions (Howell-Jolly bodies) in peripheral blood, and mild, compensated anemia (8).
One such gene that is activated by KLF1 and repressed by KLF3 is Klf8 (11). KLF8 is highly related to KLF3, with the two proteins sharing 96% sequence similarity in their zinc finger domain (54). They recognize similar sequences of DNA that broadly fit the KLF DNA-binding consensus 5′-NCN CNC CCN-3′ (3, 20, 29). Moreover, both proteins are able to silence gene expression by recruiting CtBP corepressors via a conserved Pro-X-Asp-Leu-Ser-type motif (19, 20). In addition, KLF8 has been shown to activate transcription from some gene promoters (21–24). KLF8 is not expressed at readily detectable levels in most cell and tissue types studied to date (55), but numerous studies have reported its upregulation in various human cancers, including prostate (56), gastric (57, 58), hepatocellular (59, 60), glioma (61), breast (62, 63), renal (64), and ovarian (65). KLF8 has been shown to regulate oncogenesis by promoting cellular proliferation and tumor invasion and by inhibiting apoptosis (21, 65–68).
Despite a multitude of studies that have investigated the dysregulation of KLF8 expression in various cancers, little is known about its role in normal physiology. Here we report the generation of the first animal model with a gene trap (gt) insertion in the Klf8 locus and no detectable KLF8 protein. Mice with homozygous disruption of Klf8 (Klf8gt/gt) are viable but have a shortened life span. Crossing these mice with Klf3 null mice results in embryonic lethality, indicative of a genetic interaction between these two factors. This interaction is pronounced in erythroid tissue, in which we observe considerable derepression of KLF8 expression in the absence of KLF3. A cohort of genes is deregulated upon disruption of this network in fetal liver cells, and in particular, the embryonic globin genes are derepressed in the absence of both KLF3 and KLF8.
MATERIALS AND METHODS
Klf3−/− and Klf8gt/gt mice.
Murine embryonic stem (ES) cells (clone name AD0101; Sanger Institute Gene Trap Resource) containing a β-geo gene trap cassette in intron 2 of Klf8 were injected into C57BL/6 blastocysts by standard methods in order to generate chimeric mice. Germ line transmission of the Klf8 gene trap was confirmed, and mice were backcrossed for at least 10 generations to the FVB/NJ strain. Klf3−/− mice have previously been described and were also maintained on a pure FVB/NJ strain (69). Ethical approval for the use of these mice was obtained from the Animal Care and Ethics Committees at the University of Sydney (approval no. L02/6-2006/3/4344 and L02/7-2009/3/5079) and the University of New South Wales (approval no. 09/128A).
Histological examination of mice.
Necropsies were conducted on four wild-type and four litter-matched Klf8gt male mice, ages 12 to 15 weeks (Australian Phenomics Network, The University of Melbourne, Melbourne, Australia, and IMVS, Adelaide, Australia). A wide range of organs were sectioned and subjected to standard histological examination, including heart, liver, spleen, pancreas, kidney, brain, thyroid, trachea, lungs, thymus, skin, testes, epididymis, seminal vesicles, prostate gland, penis, preputial gland, bladder, gallbladder, stomach, duodenum, jejunum, ileum, colon, mesenteric lymph node, adrenal glands, tail, eyes, Harderian glands, spinal cord, salivary glands, and regional lymph nodes. Paraffin embedding of five Klf3−/− Klf8gt/gt and three Klf3+/+ Klf8gt/gt E13.5 embryos and subsequent staining with hematoxylin and eosin were performed by the Histology and Microscopy Unit, University of New South Wales.
Real-time qRT-PCR.
Total RNA from murine tissues was extracted, DNase treated, and analyzed by real-time quantitative reverse transcription (qRT)-PCR as described previously (8, 10, 70). Adult tissues were from 3- to 4-month-old, sex-matched littermates. The sequences of primers used are as follows and as listed previously (10): Klf8, 5′-TGGATGTCCGAATTAAATCAGAAA-3′ and 5′-GAAGGATCTCTGGTCGGAACAG-3′ or 5′-CCAAAAGCTCTCACCTGAAAGC-3′ and 5′-AGCGAGCAAATTTCCAGGAA-3′; εy-globin (Hbb-y), 5′-GGCCTGTGGAGTAAGGTCAA-3′ and 5′-GCAGAGGACAAGTTCCCAAA-3′; βh1-globin (Hbb-bh1), 5′-CTCAAGGAGACCTTTGCTCA-3′ and 5′-AATCACCAGCTTCTGCCAGGC-3′; β-major globin (Hbb-b1), 5′-CACTGTGACAAGCTGCATGT-3′ and 5′-TAGTGGTACTTGTGAGCCAG-3′; ζ-globin (Hba-x), 5′-ATGCGGTTAAGAGCATCGAC-3′ and 5′-GGGACAGGAGCTTGAAGTTG-3′; and α-globin (Hba-a1), 5′-GTCACGGCAAGAAGGTCGC-3′ and 5′-GGGGTGAAATCGGCAGGGT-3′.
Protein overexpression in COS cells.
COS cells were cultured and transfected as previously described (10) using 1 to 2 μg pMT3-mKlf8 (11), pMT2-Klf3 (29), or empty pMT3 vector.
Western blotting.
Western blotting of nuclear extracts and the anti-KLF3 and anti-KLF8 sera have been described previously (11, 29, 71). To detect KLF8, 0.1% (vol/vol) anti-KLF8 in Tris-buffered saline–Tween 20 (TBST) (50 mM Tris [pH 7.4], 150 mM NaCl, 0.05% Tween 20) with 5% (wt/vol) skim milk powder was allowed to hybridize with the blot for 12 to 18 h at room temperature. KLF3 was detected using 0.03% anti-KLF3 in TBST for 1 to 1.5 h. Secondary labeling was achieved using 1:15,000 horseradish peroxidase (HRP)-linked anti-rabbit antibody (Amersham Pharmacia Biotech) in TBST for 1 h at room temperature. Signals were visualized using Immobilon Western chemiluminescent HRP substrate (Millipore). Blots were stripped in a mixture of 62.5 mM Tris (pH 6.8), 2% SDS, and 0.7% β-mercaptoethanol at 70°C for 30 min and were subsequently washed six times with TBST over 30 min. Blots were then reblocked and probed using anti-β-actin (1:10,000) (Sigma) and horseradish peroxidase-linked anti-mouse antibody (1:15,000) (Amersham Pharmacia Biotech) in TBST, each for 1 h at room temperature.
Determination of the precise genomic location of the Klf8 gene trap.
Confirmation that the gene trap lies within intron 2 of Klf8 was achieved by RT-PCR analysis of AD0101 ES cells using a forward primer specific for Klf8 exon 2 (5′-TGGATGTCCGAATTAAATCAGAAA-3′) and reverse primers specific for the β-geo gene trap (5′-AGTATCGGCCTCAGGAAGATCG-3′ and 5′-ATTCAGGCTGCGCAACTGTTGGG-3′). The precise site of integration was determined by conducting genomic PCR using these reverse primers together with 23 forward primers evenly spaced across the 11.1 kb of intron 2. A single forward primer (5′-GGAACCTGTGACTGATTTGACTAGGC-3′) yielded a strong PCR band with both of the reverse primers. Sequencing of the PCR products revealed that the site of integration lies 434 bp upstream of exon 3.
Genotyping.
Genomic DNA was extracted from tail snips or embryonic tissue using DirectPCR lysis buffer (Viagen Biotech) as per the manufacturer's instructions. Klf8 gene trap mice were genotyped by multiplex PCR using the primer pairs 5′-GGAACCTGTGACTGATTTGACTAGGC-3′ and 5′-GCATTGTGCTAAGTCCACTGACAGC-3′, which flank the site of gene trap insertion and generate a 210-bp product, indicative of an intact, wild-type allele, and 5′-CAGTATCTGCAACCTCAAGCTAGCTTGG-3′ and 5′-ATTCAGGCTGCGCAACTGTTGGG-3′, which recognize the gene trap itself and produce a 348-bp product. PCRs were conducted using REDTaq DNA polymerase (Roche Molecular Biochemicals) as recommended by the supplier and in the presence of 10% dimethyl sulfoxide (DMSO). The following PCR parameters were used: 1 cycle of 94°C for 2 min, 31 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and 1 cycle of 72°C for 5 min. Klf3 knockout line mice were genotyped as previously described (69). Klf3 Klf8 double mutant mice were genotyped by multiplex PCR using the Klf8 primers listed above together with primers specific for wild-type Klf3 (5′-CATCCTTCCGTCATCGTGCAG-3′ and 5′-TTTCAAGTGCGAGCTCTTAGTGTAGACC-3′, 135-bp product) and for the Klf3 Neo cassette (5′-TCCATGTCTGTCTCCCCCTA-3′ and 5′-ATTAAGGGCCAGCTCATTCC-3′, 250-bp product). PCRs were prepared using MangoTaq DNA polymerase (Bioline) as per the manufacturer's instructions and using the following thermocycler parameters: 1 cycle of 94°C for 2 min, 29 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and 1 cycle of 72°C for 5 min.
Full blood count analysis.
Full blood counts were performed for 21 Klf8 mutant mice (Klf8gt males or Klf8gt/gt females) and 18 wild-type mice at 11 to 12 weeks of age as previously described (8). For phenylhydrazine treatment of mice, 7 wild-type and 9 Klf8gt males (18 to 22 weeks of age) were treated by sequential intraperitoneal injections of 1-acetyl-2-phenylhydrazine (0.5% [wt/vol] in Hanks' balanced salt solution) at a dose of 0.04 mg per g body mass at time 0 and 16 h. Peripheral blood was collected by cardiac puncture at 120 h and was subjected to full blood count analysis as described above.
Sorting of Ter119+ erythroid cells.
Ter119+ cells were sorted from E13.5, E14.5, and E16.5 fetal livers using mass spectrometry (MS) columns and anti-Ter119 Microbeads (Miltenyi Biotech) as per the supplier's protocol. Following elution, purified cells were centrifuged for 10 min at 300 g (4°C), the supernatant was removed, and pellets were harvested for RNA as described above.
Cytospins of peripheral blood and fetal livers.
Peripheral blood and livers from individual E14.5 embryos were disaggregated in 1 ml filtered fluorescence-activated cell sorter (FACS) buffer (10 mM EDTA, 5% fetal calf serum, 0.05% NaN3 in phosphate-buffered saline [PBS]), and cells were counted using a Countess automated cell counter (Life Technologies). Cells were diluted to 4 × 105/ml in FACS buffer, and 100 to 150 μl was centrifuged for 5 min at 300 rpm in Shandon Cytofunnels (Thermoscientific). Slides were subsequently stained using Diff-Quik (Lab Aids, Pty., Ltd.), and the cells were counted.
ChIP analysis of MEL cells.
Murine erythroleukemia (MEL) cells were cultured and induced to differentiate for 72 h in 1.8% DMSO as described previously (70). Chromatin immunoprecipitation (ChIP) assays were conducted in triplicate as described previously (72, 73), using rabbit polyclonal anti-KLF3 serum (29) or Pierce anti-KLF3 antibody (PA5-18030) and preimmune serum or IgG (Santa Cruz sc-2028) as negative controls. The primer sequences are found elsewhere (11, 50, 74) and as follows: Hba-a1 promoter, 5′-GTTTGAGGGACTTGCTTCTCTGA-3′ and 5′-GCCCGGACACACTTCTTACC-3′; Hba-x promoter, 5′-AGCCCATTGGCACTGAGACT-3′ and 5′-CAATCCCTCTTCTGACCTGCTTA-3′; and Hbb-y promoter, 5′-CATGACCTGGCTCCACCCATGAG-3′ and 5′-CTGCTGCTAGAAGTGGTGGCCTT-3′.
Microarrays.
Total RNA was extracted from Ter119+ fetal liver cells purified from four wild-type, four Klf8gt/gt, three Klf3−/−, and four Klf3−/− Klf8gt/gt embryos (E13.5), litter matched where possible. RNA was prepared and analyzed by Affymetrix GeneChIP 1.0 ST arrays as previously described (8). Genes showing a greater than 2-fold change in expression and passing a false discovery rate (FDR) threshold of 0.3 to correct for multiple testing were considered to be significantly differentially expressed.
Microarray data accession numbers.
Microarray data have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/projects/geo) under accession no. GSE43524.
RESULTS
Generation of a Klf8 gene trap mutant mouse model.
While a KLF3 knockout mouse line has been previously characterized and shown to have mild erythropoietic, adipogenic, and B cell defects (8, 69, 75, 76), a mutant mouse model for KLF8 has not been described. Therefore, to investigate the biological role of KLF8, we generated mice from ES cells (Sanger Institute Gene Trap Resource) containing a gene trap (gt) cassette within intron 2 of the Klf8 gene (Fig. 1A and B). Sequencing of genomic PCRs using a series of primers spread across Klf8 intron 2 revealed that the precise site of gene trap integration is 434 bp upstream of exon 3 (Fig. 1C) (data not shown). The gene trap contains a potent splice acceptor site to disrupt expression of KLF8 and instead generate a β-geo fusion protein. Such splicing was confirmed by RT-PCR analysis using a forward primer specific for Klf8 exon 2 and reverse primers specific for the gene trap (data not shown).
Fig 1.

The Klf8 gene trap disrupts normal KLF8 expression and results in increased mortality. (A and B) Schematic of the wild-type (WT) murine Klf8 locus (A) and the location of the β-geo gene trap (B). Klf8 exons are shown as lightly shaded boxes. The gene trap contains a splice acceptor (SA) site, a β-galactosidase–Neomycin resistance fusion gene (β-geo), and a poly(A) signal. The β-geo fusion protein contains a short (24-amino-acid) N-terminal portion encoded by exon 2 of Klf8. (C) Multiplex genotyping PCRs confirming the integration site of the gene trap in the Klf8 locus. Primer pairs were used that flank the site of gene trap insertion (210-bp product for a wild-type, intact allele) and that recognize the gene trap (348-bp product). (D) Western blot of KLF8 expression (top panel) in the fetal brain (E14.5) for the genotypes indicated. KLF8 expressed in COS cells has been included as a positive control, while β-actin levels are shown as loading controls (bottom panel). (E) Western blots of KLF8 (top panel) and β-actin (bottom panel) in E10.5 placenta. In panels D and E, the band corresponding to KLF8 is marked with an asterisk. (F) Percentage of animals surviving for the genotypes indicated over a 6-month period. n = 40 wild-type males, n = 45 Klf8gt males, n = 52 wild-type females, n = 87 Klf8gt/+ females, and n = 16 Klf8gt/gt females. * and ***, P < 0.05 and P < 0.001, respectively, compared to wild-type females; **, P < 0.01 compared to wild-type males (log rank tests).
While KLF8 protein is normally expressed at very low levels in noncancerous tissues (55), we have been able to detect endogenous KLF8 in wild-type fetal brain and placental tissue (Fig. 1D and E). Mice that are homozygous for the gene trap have no detectable KLF8 protein in these tissues (Fig. 1D and E). Similarly, qRT-PCR analysis of a range of erythroid tissues (E11.5 yolk sac, E14.5 fetal liver, and adult peripheral blood) revealed that the gene trap reduced Klf8 mRNA below the level of detection (data not shown).
The Klf8 gene lies on the X chromosome, and hence males are hemizygous for the gene trap (Klf8gt), while females can either be heterozygous (Klf8gt/+) or homozygous (Klf8gt/gt). For simplicity, hemizygous males and homozygous females will be collectively referred to as Klf8gt/gt throughout. Klf8gt/gt males and females are both viable and fertile. They have a significantly shortened life span, with approximately 30% dying by the age of 6 months (P < 0.01 and P < 0.001 for males and females, respectively), compared to wild-type males and females, who exhibited only 5% and 4% lethality, respectively, over the same period (Fig. 1f). Heterozygous females exhibited a partially penetrant phenotype, with 16% of animals dying within 6 months (P < 0.05 compared to wild-type females). This might be explained by decreased KLF8 expression in these animals, the extent of which may vary between individuals due to nonrandom lyonization (X inactivation). Necropsies and extensive histological examination of Klf8gt/gt mice have not yet revealed any phenotypic abnormalities that might explain the shortened life span of these animals (see Materials and Methods). Similarly, full blood counts indicated that erythroid parameters and platelet numbers are normal in Klf8gt/gt mice (see Fig. S1 in the supplemental material). Moreover, phenylhydrazine treatment did not reveal any significant differences in blood count parameters between Klf8gt/gt and wild-type mice (data not shown), suggesting that stress erythropoiesis is not appreciably modulated by KLF8.
Mice lacking both KLF3 and KLF8 are embryonic lethal.
KLF3 and KLF8 are highly related proteins that recognize similar sequences of DNA, repress transcription via CtBP corepressors, and are directly upregulated by KLF1 in erythroid cells (11, 19, 20, 29). Given these similarities, we reasoned that they may have overlapping physiological roles in vivo and may be able to compensate functionally for each other's absence. This hypothesis is made all the more salient by our previous observation that KLF8 expression is elevated in Klf3 null mice (8, 11).
Therefore, to explore the possibility of functional redundancy between these two factors and to elucidate their biological roles, we crossed the Klf3 knockout (Klf3−/−) mouse line with the newly generated Klf8gt/gt line. We have found that Klf3−/− mice are essentially infertile, but using a series of different parental crosses involving Klf3 heterozygous animals, we analyzed the frequency of the nine possible resulting genotypes (see Fig. S2 in the supplemental material) at weaning age (Table 1). The observed numbers do not adhere to Mendelian expectance, with no mice observed that were deficient in both KLF3 and KLF8 (Klf3−/− Klf8gt/gt) and an underrepresentation of female mice containing only a single allele of Klf8, that is, Klf3−/− Klf8gt/+ (observed at 10% of the expected number). Klf3 knockout mice (Klf3−/− Klf8+/+) were also observed at lower than expected numbers (43% of expected), in line with a reduced viability of Klf3-deficient animals that we have previously reported (69). Investigations in utero revealed that Klf3−/− Klf8gt/gt embryos are viable and found at expected Mendelian numbers at E12.5 (Table 2) but start to die thereafter, with n = 10/15 embryos surviving at E13.5 and n = 1/5 surviving at E14.5. Lethality at this developmental stage is often caused by cardiovascular defects and/or disrupted liver hematopoiesis (77); however, histological examination of sections of E13.5 Klf3−/− Klf8gt/gt embryos provided no conclusive explanations as to their cause of death. Taken together, the observation that the Klf8gt/gt and Klf3−/− single-gene mutant mice are viable while the Klf3−/− Klf8gt/gt double mutants exhibit an embryonic lethal phenotype indicates a genetic interaction between KLF3 and KLF8 and suggests that these factors have at least partial functional redundancy in vivo.
Table 1.
Observed and expected numbers of mice from crosses between the Klf3−/− and Klf8gt/gt linesa
| Klf8 genotype | No. of mice (expected, observed) with Klf3 genotype: |
||
|---|---|---|---|
| +/+ | +/− | −/− | |
| +/+ | 81, 109 | 161, 226 | 81, 35 |
| gt/+ | 59, 90 | 118, 154 | 59, 6 |
| gt/gt | 82, 92 | 165, 175 | 82, 0 |
A total of 887 mice were genotyped at 3 to 4 weeks of age. Klf8gt males and Klf8gt/gt females have been collectively grouped as Klf8gt/gt, while Klf8+ males and Klf8+/+ females are denoted together as Klf8+/+. The observed numbers do not adhere to Mendelian expectance. P < 0.001, χ2 test.
Table 2.
Observed and expected numbers of embryos from crosses between the Klf3−/− and Klf8gt/gt mouse linesa
| Klf8 genotype | No. of embryos (expected, observed) with Klf3 genotype: |
||
|---|---|---|---|
| +/+ | +/− | −/− | |
| +/+ | 7, 10 | 13, 18 | 7, 5 |
| gt/+ | 6, 7 | 12, 9 | 6, 4 |
| gt/gt | 11, 8 | 22, 21 | 11, 12 |
The counts represent genotyping results from embryos up to E12.5 (n = 94). The observed numbers were not found to significantly deviate from Mendelian expectance. P = 0.61, χ2 test.
Derepression of KLF8 in Klf3 null erythroid tissue.
To further investigate the aforementioned repression of the Klf8 gene by KLF3, we analyzed the levels of Klf8 transcripts in a range of tissues from Klf3 null mice (Fig. 2A) (data not shown). In all tissues examined, apart from the brain, we found significant derepression of Klf8 mRNA. However, despite this derepression, in some tissues, such as the liver and the intestine, the level of Klf8 expression remains relatively low. In contrast, we found that in erythroid tissues particularly (that is, the spleen and bone marrow), Klf8 is expressed at comparatively high levels in the absence of KLF3 (Fig. 2A). This trend is observed not only in adult tissues but also in embryonic erythroid tissue: KLF8 protein is barely detected in wild-type and Klf3+/− fetal liver but is considerably upregulated in Klf3 null tissue (Fig. 2B) (11). Taken together, the pronounced upregulation of Klf8 expression in erythroid tissue is consistent with our previous observation that the master erythroid regulator KLF1 directly activates Klf8 transcription in erythroblast cells, particularly in the absence of KLF3 (11).
Fig 2.

Klf8 expression is elevated in erythroid tissue in the absence of KLF3. (A) Klf8 transcript levels were quantified by real-time RT-PCR in whole tissues from adult wild-type (WT; n = 3) and Klf3−/− (Klf3 KO; n = 3) mice. Expression has been normalized to 18S rRNA levels, and the lowest level (wild-type liver) has been set to 1.0. Error bars represent standard errors of the mean. *, P < 0.04 (two-tailed t test for Klf3−/− compared to wild type). (B and C) Western blots of E14.5 (B) and E13.5 (C) fetal liver nuclear extracts using anti-KLF8 (αKLF8), anti-KLF3 (αKLF3), and anti-β-actin sera. Genotypes are indicated, and nuclear extracts from COS cells (mock transfected or overexpressing KLF3 or KLF8) have been included as controls. In panel C, the band corresponding to KLF8 is marked with an asterisk.
Identification of KLF3 and KLF8 target genes in fetal liver.
There is thus a regulatory network within the KLF family in erythroid cells, such that KLF1 drives the expression of KLF3 and KLF8 and KLF3 represses the Klf8 gene. We have previously investigated the function of KLF3 in this network by microarray analysis of Klf3 null E14.5 fetal liver cells (8). We next sought to further unravel the KLF1/KLF3/KLF8 network by analyzing and comparing tissue samples from single mutant (Klf8gt/gt and Klf3−/−) and double mutant (Klf3−/− Klf8gt/gt) animals. As Klf3−/− Klf8gt/gt embryos die at around E14.5, we analyzed Ter119+ (erythroid) fetal liver cells at E13.5. At this developmental stage, Klf3−/− Klf8gt/gt embryos are largely phenotypically normal (see Fig. S3 in the supplemental material) and their livers display no gross morphological abnormalities by histological examination of sections. For this study, total RNA was extracted from cells from four wild-type, four Klf8gt/gt, three Klf3−/−, and four Klf3−/− Klf8gt/gt embryos and was subjected to Affymetrix microarray analysis. In addition, we confirmed that the Klf8 gene trap ablated KLF8 protein expression in Klf3−/− Klf8gt/gt compared to Klf3−/− fetal liver (Fig. 2C).
Comparing Klf8gt/gt embryos with wild-type embryos, there were very few genes that were significantly deregulated (>2-fold; FDR < 0.3), and only one of these encoded an annotated mRNA transcript (H2-Q6; Histocompatibility 2, Q region locus 6) (Fig. 3A). The lack of any considerable gene deregulation in Klf8gt/gt embryos is not unexpected given that KLF8 protein is not readily detectable in wild-type fetal liver (Fig. 2B and C).
Fig 3.

Volcano plots demonstrating gene expression changes in Klf8gt/gt, Klf3−/−, and Klf3−/− Klf8gt/gt Ter119+ E13.5 fetal liver cells. (A) Klf8gt/gt versus wild type (WT). (B) Klf3−/− versus WT. (C) Klf3−/− Klf8gt/gt versus WT. (D) Klf3−/− Klf8gt/gt versus Klf3−/−. Significance thresholds are shown (>2-fold deregulation, FDR < 0.3), and significantly deregulated genes are represented by red dots (derepressed relative to WT [A to C] or Klf3−/− [D]) or green dots (downregulated relative to WT [A to C] or Klf3−/− [D]).
Next we compared Klf3−/− samples with wild-type samples and observed considerable derepression of gene expression (Fig. 3B; see Fig. S4 and Table S1 in the supplemental material). In total, 64 genes were significantly derepressed, while only 4 genes were downregulated in the absence of KLF3, consistent with the notion that KLF3 is primarily a transcriptional repressor in erythroid cells. These results are highly concordant with our previous microarrays performed with E14.5 fetal liver cells, with 57 of these 68 genes being significantly deregulated in both studies (see Table S1). As we had previously observed, Klf8 was one of the most highly derepressed genes in these cells (10.2-fold) (Fig. 3B; see Table S1).
Klf3−/− Klf8gt/gt cells exhibited even greater deregulation of gene expression than Klf3−/− cells, with 112 genes being upregulated and 74 downregulated compared to the wild type (Fig. 3C and Fig. 4). Strikingly, almost all of the 64 genes that were derepressed in Klf3−/− cells were also significantly derepressed in Klf3−/− Klf8gt/gt tissue (see Fig. S4 and Table S1 in the supplemental material). Only four of the derepressed genes did not meet these criteria (Frrs1, Cmpk2, Acot9, and Treml2) but nonetheless displayed upregulation in Klf3−/− Klf8gt/gt cells (between 1.8- and 1.9-fold), albeit not significantly.
Fig 4.

Heat map showing the relative expression of the genes that are deregulated in Klf3−/− Klf8gt/gt Ter119+ E13.5 fetal liver cells compared to the wild type (WT). Genes that are significantly upregulated (group I) and downregulated (group II) in Klf3−/− Klf8gt/gt cells are represented, and their relative expression across the four genotypes (wild type, Klf8gt/gt, Klf3−/−, and Klf3−/− Klf8gt/gt) is shown.
The genes that are derepressed in the absence of KLF3 are, by and large, not further derepressed upon ablation of KLF8 (Fig. 4; see Fig. S4 and Table S1 in the supplemental material). This suggests that these genes are largely regulated by KLF3 and that KLF8 may play little or no role in their regulation in the absence of KLF3. Nonetheless, Klf3−/− Klf8gt/gt cells did indeed display a greater deregulation of gene expression than Klf3−/− cells (Fig. 3C and Fig. 4). These additional genes displayed both upregulation and downregulation consistent with the dual role of KLF8 as both an activator and a repressor of transcription. These genes represent potential target genes of KLF8, which KLF8 may redundantly regulate with KLF3 or which may be uniquely regulated by KLF8.
To identify likely KLF8 candidate target genes, we compared expression profiles between Klf3−/− Klf8gt/gt and Klf3−/− cells and compiled a list of 30 significantly differentially expressed genes, 17 of which were upregulated and 13 of which were downregulated (Fig. 3D; see Fig. S5 in the supplemental material). Notably, several of these genes, including Itgb7 (78), Vegfa (79, 80), Periostin (81), Parm1 (82, 83), Gbp1 (84, 85), Tcfl5 (86, 87), Rps3a (88), and Fpr1 (89, 90), are regulators of cell adherence, migration, and invasiveness, consistent with the role of KLF8 in oncogenesis. In addition, the Klf8 gene itself is significantly downregulated in Klf3−/− Klf8gt/gt compared to Klf3−/− cells, as anticipated (Fig. 3D; see Fig. S5).
Embryonic globin genes are derepressed in the absence of KLF3 and KLF8.
The most highly upregulated gene within this list was that encoding Hbb-bh1 (3.8-fold), an embryonic β-globin (Fig. 3D). This was of interest given that KLF1 plays a crucial role in the activation of adult β-globin and the silencing of embryonic and fetal globins in definitive erythroid cells. This raised the possibility of an elegant system whereby KLF1 drives adult β-globin and also upregulates KLF3 and KLF8, which then function to repress the expression of embryonic globins. Indeed, inspection of globin gene expression in Klf3−/− Klf8gt/gt cells compared to Klf3−/− cells revealed that the other murine embryonic genes, Hba-x and Hbb-y, are elevated 2.8-fold and 1.5-fold, respectively (Fig. 3D), while the adult globin genes, Hbb-b1 and Hba-a1, are unaltered.
We next sought to validate the derepression of embryonic globins by qRT-PCR analysis of Ter119+ E13.5 fetal liver cell samples independently purified from those used in the arrays. We consistently observed significant upregulation of all three embryonic globin genes in Klf3−/− cells compared to wild-type cells (4.07-, 2.66-, and 1.62-fold, respectively, for Hba-x, Hbb-y, and Hbb-bh1) (Fig. 5A to C). We also tested the hypothesis that these embryonic globin genes are further derepressed in Klf3−/− Klf8gt/gt cells and found this to be the case (1.60-, 1.68-, and 1.67-fold, respectively, compared to Klf3−/−; P = 0.04, P = 0.07, and P = 0.03, one-tailed t tests) (Fig. 5A to C). Adult globin expression was unaffected in both Klf3−/− and Klf3−/− Klf8gt/gt cells compared to wild-type cells (Fig. 5D and E).
Fig 5.

Embryonic, but not adult, globin genes are derepressed in Klf3−/− and Klf3−/− Klf8gt/gt Ter119+ E13.5 fetal liver cells. Transcript levels for Hba-x (A), Hbb-y (B), Hbb-bh1 (C), Hba-a1 (D), and Hbb-b1 (E) were determined by qRT-PCR analysis of total RNA from three wild-type (WT), five Klf3−/−, seven Klf8gt/gt, and four Klf3−/− Klf8gt/gt embryos. Embryonic globin genes are shown in light gray, and adult globin genes are in dark gray. Expression has been normalized to 18S rRNA levels, and wild-type samples have been set to 1.0 for each gene. Error bars indicate standard errors of the mean. *, P ≤ 0.05 (two-tailed t test compared to wild type).
The embryonic lethality of Klf3−/− Klf8gt/gt mice precluded analysis of later developmental stages; however, examination of Klf3−/− cells revealed elevated expression of embryonic but not adult globins at both E14.5 (see Fig. S6 in the supplemental material) and E16.5 (see Fig. S7 in the supplemental material). Cytospins of peripheral blood and fetal liver tissue at E14.5 revealed no difference in the numbers of primitive erythrocytes between wild-type and Klf3−/− animals that might explain the elevated expression of embryonic globins (see Fig. S8 in the supplemental material). Furthermore, we observed no deregulation of either embryonic or adult globin expression in Klf3−/− E10.5 yolk sac, a source of primitive erythroid cells (see Fig. S9A to E in the supplemental material). Similarly, both embryonic and adult globin transcript levels were comparable in Klf3−/− Klf8gt/gt and litter-matched Klf3+/+ Klf8gt/gt yolk sacs (see Fig. S9F to J). Taken together, these results suggest that KLF3 in particular, represses the expression of the embryonic globin genes in definitive erythroid cells and that KLF8 partially compensates in its absence.
Other KLFs, namely, KLF1 and KLF2, have previously been shown to directly bind to the promoters of the embryonic globin genes in erythroid cells (50, 52). To explore the possibility that KLF3 represses embryonic globin expression by a similar mechanism, we conducted chromatin immunoprecipitation (ChIP) studies using differentiated MEL cells. These erythroid cells routinely yield robust immunoprecipitation using antibodies for KLF3, as evidenced by significant enrichment at a positive-control region (Klf8 promoter 1a) (8, 11) (Fig. 6). We observed no enrichment at the promoters of the embryonic globin genes (Hba-x and Hbb-y), but unexpectedly, we found significant KLF3 occupancy at the promoters of the adult globin genes, particularly at Hba-a1 and to a lesser extent at Hbb-b1. We also observed significant binding at upstream DNase-hypersensitive sites (HS26 and HS2) known to play a role in the regulation of the α- and β-globin loci, respectively. These data raise the possibility that KLF3 influences embryonic globin gene expression not by direct binding to their promoters but through distal sites within the globin loci.
Fig 6.
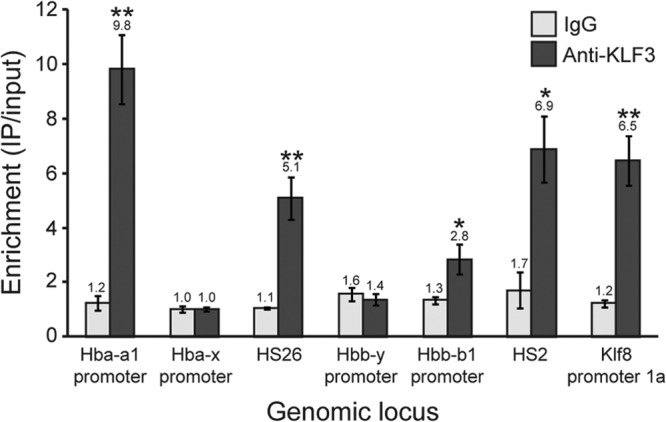
Chromatin immunoprecipitation analysis of KLF3 occupancy at α- and β-globin loci in induced MEL cells. ChIP assays were conducted in triplicate, and enrichment has been determined by quantitative real-time PCR and has been normalized to input. The lowest values for both IgG and anti-KLF3 have been set to 1.0. Klf8 promoter 1a has been included as a positive control. Error bars represent standard errors of the mean. *, P < 0.05; **, P < 0.005 (one-tailed t test compared to IgG). HS, DNase-hypersensitive site.
DISCUSSION
Deregulation of KLF8 has been observed in a wide variety of cancers; however, the normal physiological roles of this transcription factor have remained largely unknown. To this end, we have generated a mouse line with disrupted KLF8 expression. Klf8gt/gt mice are viable and fertile but have a shortened life span, the cause of which could not be identified despite extensive histological characterization. Mice that are deficient in the related family member KLF3 are also viable, yet Klf3−/− Klf8gt/gt double mutant animals die at around E14.5. This suggests that KLF3 and KLF8 have overlapping roles in vivo and can at least partially compensate in each other's absence.
Analysis of Klf3−/− mice revealed marked upregulation of KLF8 expression in several tissues, but particularly erythroid tissue. Given that both the Klf3 and Klf8 genes are activated by the erythroid factor KLF1, we hypothesized that these three factors operate in a regulatory network to control gene expression. Microarray analysis of Ter119+ fetal liver cells from single mutant (Klf3−/− and Klf8gt/gt) and double mutant (Klf3−/− Klf8gt/gt) embryos revealed that this was indeed the case. We observed more extensive deregulation of gene expression in Klf3−/− Klf8gt/gt cells compared to either of the single knockouts. It appears that KLF3 plays a somewhat nonredundant role in this network in that 64 genes are upregulated in the absence of KLF3 alone, and these are not further significantly elevated in Klf3−/− Klf8gt/gt embryos. Klf3−/− Klf8gt/gt embryos did, however, display greater dysregulation of gene expression than Klf3−/− embryos, with 186 genes being significantly differentially expressed relative to the wild type. These genes were both up- and downregulated, consistent with the dual role of KLF8 as both an activator and a repressor of transcription. By comparing Klf3−/− Klf8gt/gt gene expression profiles with Klf3−/−, we refined a list of the most likely KLF8 targets. Included in this list were several genes involved in cell cycle regulation, adherence, and invasiveness, in agreement with the role of KLF8 in oncogenesis.
We also confirmed by qRT-PCR that embryonic globin expression is derepressed in Klf3−/− and Klf3−/− Klf8gt/gt cells, while adult globin expression is unchanged. This was observed not only at E13.5 but also at later stages of fetal liver development (E14.5 and E16.5) in Klf3−/− embryos. The simplest interpretation of these results is that KLF3, which is highly expressed in fetal liver, is the primary repressor of embryonic globin expression, but in its absence, KLF8 is able to partially compensate. While the effects on embryonic globin expression are indeed modest, we suggest that KLF3 and KLF8 together participate in the silencing of embryonic globins with other proposed repressors, such as SOX6, GATA1, YY1, COUP-TF, and DRED (91–94). The expression levels of these repressors were not significantly altered in the microarrays presented here in either Klf3−/− or Klf3−/− Klf8gt/gt cells (all less than 1.12- and 1.35-fold, respectively, compared to the wild type). Similarly, the expression of BCL11A, a transcriptional repressor of the murine embryonic and human fetal globin genes (48, 49), was found to be unchanged in Klf3−/− and Klf3−/− Klf8gt/gt cells compared to the wild type. Together, this suggests that the repression of embryonic globin genes by KLF3 and KLF8 is not indirectly achieved by altering the transcription of these known regulators in erythroid cells.
Interestingly, an analogous system of embryonic globin gene regulation and functional compensation has been observed for the activator KLFs, KLF1 and KLF2. The Klf1−/− and Klf2−/− single knockout mice die at around E14.5 and E12.5 to E14.5, respectively, whereas Klf1−/− Klf2−/− double knockout animals die before E11.5 (43, 44, 95–97). Expression of the β-like embryonic globins (Hbb-y and Hbb-bh1) is significantly reduced in the primitive erythroid cells of both Klf1−/− and Klf2−/− single knockout embryos (50, 51, 98) and is further depleted in Klf1−/− Klf2−/− double knockout embryos (97). Thus, in primitive erythroid cells, KLF1 and KLF2 drive transcription of embryonic globin genes, while in definitive erythroid cells, KLF3 and KLF8 serve to repress embryonic globin expression.
In addition to the β-like embryonic globins, we also observed upregulated Hba-x expression in Klf3−/− and Klf3−/− Klf8gt/gt fetal liver cells. The Hba-x gene has a functional CACCC box in its promoter (99) and like Hbb-y and Hbb-bh1 is also downregulated in Klf1−/− and Klf1−/− Klf2−/− embryos (97). Hba-x expression is also strongly upregulated, more so than Hbb-y and Hbb-bh1, upon inducible restoration of KLF1 activity induced in Klf1−/− erythroblast cells (32). Taken together, these results implicate the KLFs in the regulation of the α-globin locus in addition to the β-globin locus.
The concomitant upregulation of both α-like and β-like embryonic globin genes has also been observed in other mouse models. Perhaps the best characterized repressor of embryonic globin expression is the SOX6 transcription factor (100). SOX6 directly binds to the Hbb-y promoter and silences its expression in definitive erythroid cells. As such, mice lacking SOX6 exhibit persistent expression of Hbb-y in the fetal liver. In addition, Hba-x and Hbb-bh1 transcript levels are also considerably elevated in Sox6−/− fetal liver cells, albeit not to the same extent (100). Thus, the coinciding deregulation of embryonic globin genes is a feature that is shared by the KLF3/8, KLF1/2, and SOX6 mouse models.
It is likely that this network of cross-regulation and functional compensation within the KLF family serves to fine-tune the expression of globin genes and other genes during development. Such regulatory circuitries have indeed been identified for other transcription factor families, such as the myogenic basic helix-loop-helix factors (MYOD, MYF5, myogenin, and MRF4) and the paired box (PAX) factors. These families are subject to transcriptional cross-regulation, and family members display overlapping physiological roles, such that single factor knockouts often exhibit mild or no phenotype, while combinatorial knockouts can result in severe physiological perturbations (101, 102). Correct temporal expression of globins is thus likely to be sensitive to the levels of both activating and repressing KLFs. Indeed a dose effect has previously been observed for both KLF1 and KLF2. Hbb-y and Hbb-bh1 are both downregulated in Klf2+/− primitive erythroid cells compared to wild-type cells (98), while Klf1+/− fetal livers show a reduction in β-globin transcript levels (103). In addition, haploinsufficiency of KLF1 results in delayed globin switching (103, 104), while overexpression of KLF1 causes premature switching (105).
The network discussed here, in which KLF1 activates the expression of Klf3 and Klf8, while KLF3 represses Klf8, is known as an incoherent type 1 feed forward network. Such networks are able to effect transient pulses in expression of their target genes and can accelerate the response of these genes to upstream signaling (106). They can also serve to sensitize transcriptional programs to various amplitudes of input signals from external stimuli (107). From the work presented here and previously (8, 32, 33, 36), it is evident that disruption of the KLF1/KLF3/KLF8 network, by removal of single factors or combinations thereof, results in altered transcriptional profiles, suggesting that the network operates to ensure the correct developmental control of gene expression required for normal erythropoiesis.
Lastly, it should be noted that although KLF1 and KLF2 have both been shown to directly bind to the promoters of the embryonic globin genes, we have thus far not detected KLF3 occupancy at these sites (Fig. 6). Unexpectedly, we observed binding at the promoters of the adult globin genes and also at upstream DNase-hypersensitive sites. The mechanism by which KLF3 regulates globin expression thus remains unclear and may involve distal binding and the formation or disruption of long-range interactions, as has been shown to be the case for BCL11A (48, 108). Alternatively, it is possible that KLF3 does bind the proximal promoter CACCC boxes of the embryonic globin genes during a particular window of development or stage of cellular maturation that we have not yet examined.
Taken together, these results establish KLF3 and KLF8 as a pair of transcriptional regulators that operate in an erythroid transcriptional network downstream of KLF1. Of these two factors, KLF3 plays the dominant role in regulating gene expression owing to its comparatively higher abundance. KLF8 is able to partially compensate at some loci, however, and indeed ablation of both KLF3 and KLF8 results in more widespread gene dysregulation than knockout of KLF3 alone. Among the most significantly affected genes, we identified the embryonic globin genes, suggesting that KLF3 and KLF8 participate in their developmental silencing together with other repressors, such as SOX6, GATA1, YY1, COUP-TF, and DRED. Thus, in addition to BCL11A, KLF3 and KLF8 represent two examples of transcriptional repressors downstream of KLF1 that cooperate to achieve normal globin regulation during ontogeny.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by funding from the National Health and Medical Research Council and the Australian Research Council.
Authorship contributions are as follows. A.P.W.F. designed and performed research, analyzed data, and wrote the paper. K.S.M., L.J.N., G.J.P., T.R., and M.P. performed research and analyzed data. N.A.T., M.R.W., and K.S.B.-A. analyzed data. S.T.F., A.C.P., and P.P.T. designed research and analyzed data. R.C.M.P. designed research, analyzed data, and wrote the paper. M.C. devised the concept and research, analyzed data, and wrote the paper.
We declare that no competing financial interests exist.
Footnotes
Published ahead of print 28 May 2013
A.P.W.F. and K.S.M. contributed equally to this article.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00074-13.
REFERENCES
- 1.Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. 2008. Kruppel-like transcription factors: a functional family. Int. J. Biochem. Cell Biol. 40:1996–2001 [DOI] [PubMed] [Google Scholar]
- 2.McConnell BB, Yang VW. 2010. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 90:1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng WC, Southwood CM, Bieker JJ. 1994. Analyses of beta-thalassemia mutant DNA interactions with erythroid Kruppel-like factor (EKLF), an erythroid cell-specific transcription factor. J. Biol. Chem. 269:1493–1500 [PubMed] [Google Scholar]
- 4.Klevit RE. 1991. Recognition of DNA by Cys2,His2 zinc fingers. Science 253:1367–1393 [DOI] [PubMed] [Google Scholar]
- 5.Shields JM, Yang VW. 1998. Identification of the DNA sequence that interacts with the gut-enriched Kruppel-like factor. Nucleic Acids Res. 26:796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tallack MR, Whitington T, Yuen WS, Wainwright EN, Keys JR, Gardiner BB, Nourbakhsh E, Cloonan N, Grimmond SM, Bailey TL, Perkins AC. 2010. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 20:1052–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuetz A, Nana D, Rose C, Zocher G, Milanovic M, Koenigsmann J, Blasig R, Heinemann U, Carstanjen D. 2011. The structure of the Klf4 DNA-binding domain links to self-renewal and macrophage differentiation. Cell. Mol. Life Sci. 68:3121–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Funnell AP, Norton LJ, Mak KS, Burdach J, Artuz CM, Twine NA, Wilkins MR, Power CA, Hung TT, Perdomo J, Koh P, Bell-Anderson KS, Orkin SH, Fraser ST, Perkins AC, Pearson RC, Crossley M. 2012. The CACCC-binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol. Cell. Biol. 32:3281–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. 2008. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 10:353–360 [DOI] [PubMed] [Google Scholar]
- 10.Funnell AP, Maloney CA, Thompson LJ, Keys J, Tallack M, Perkins AC, Crossley M. 2007. Erythroid Kruppel-like factor directly activates the basic Kruppel-like factor gene in erythroid cells. Mol. Cell. Biol. 27:2777–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eaton SA, Funnell AP, Sue N, Nicholas H, Pearson RC, Crossley M. 2008. A network of Kruppel-like factors (Klfs): Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J. Biol. Chem. 283:26937–26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adam PJ, Regan CP, Hautmann MB, Owens GK. 2000. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J. Biol. Chem. 275:37798–37806 [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Sinha S, Owens G. 2003. A transforming growth factor-beta control element required for SM alpha-actin expression in vivo also partially mediates GKLF-dependent transcriptional repression. J. Biol. Chem. 278:48004–48011 [DOI] [PubMed] [Google Scholar]
- 14.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. 2004. Kruppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene 23:3404–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piccinni SA, Bolcato-Bellemin AL, Klein A, Yang VW, Kedinger M, Simon-Assmann P, Lefebvre O. 2004. Kruppel-like factors regulate the Lama1 gene encoding the laminin alpha1 chain. J. Biol. Chem. 279:9103–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. 2000. Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 28:2969–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaczynski J, Cook T, Urrutia R. 2003. Sp1- and Kruppel-like transcription factors. Genome Biol. 4:206. 10.1186/gb-2003-4-2-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller IJ, Bieker JJ. 1993. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell. Biol. 13:2776–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner J, Crossley M. 1998. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Kruppel-like factor and other mammalian transcriptional regulators. EMBO J. 17:5129–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Vliet J, Turner J, Crossley M. 2000. Human Kruppel-like factor 8: a CACCC-box binding protein that associates with CtBP and represses transcription. Nucleic Acids Res. 28:1955–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. 2003. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell 11:1503–1515 [DOI] [PubMed] [Google Scholar]
- 22.Wei H, Wang X, Gan B, Urvalek AM, Melkoumian ZK, Guan JL, Zhao J. 2006. Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J. Biol. Chem. 281:16664–16671 [DOI] [PubMed] [Google Scholar]
- 23.Urvalek AM, Wang X, Lu H, Zhao J. 2010. KLF8 recruits the p300 and PCAF co-activators to its amino terminal activation domain to activate transcription. Cell Cycle 9:601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urvalek AM, Lu H, Wang X, Li T, Yu L, Zhu J, Lin Q, Zhao J. 2011. Regulation of the oncoprotein KLF8 by a switch between acetylation and sumoylation. Am. J. Transl. Res. 3:121–132 [PMC free article] [PubMed] [Google Scholar]
- 25.Siatecka M, Xue L, Bieker JJ. 2007. Sumoylation of EKLF promotes transcriptional repression and is involved in inhibition of megakaryopoiesis. Mol. Cell. Biol. 27:8547–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Bieker JJ. 2001. Unanticipated repression function linked to erythroid Kruppel-like factor. Mol. Cell. Biol. 21:3118–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Bieker JJ. 2004. Stage-specific repression by the EKLF transcriptional activator. Mol. Cell. Biol. 24:10416–10424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pilon AM, Ajay SS, Kumar SA, Steiner LA, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, Margulies EH, Bodine DM. 2011. Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood 118:e139–e148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin SH. 1996. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol. 16:1695–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Himeda CL, Ranish JA, Pearson RC, Crossley M, Hauschka SD. 2010. KLF3 regulates muscle-specific gene expression and synergizes with serum response factor on KLF binding sites. Mol. Cell. Biol. 30:3430–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drissen R, von Lindern M, Kolbus A, Driegen S, Steinlein P, Beug H, Grosveld F, Philipsen S. 2005. The erythroid phenotype of EKLF-null mice: defects in hemoglobin metabolism and membrane stability. Mol. Cell. Biol. 25:5205–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodge D, Coghill E, Keys J, Maguire T, Hartmann B, McDowall A, Weiss M, Grimmond S, Perkins A. 2006. A global role for EKLF in definitive and primitive erythropoiesis. Blood 107:3359–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pilon AM, Arcasoy MO, Dressman HK, Vayda SE, Maksimova YD, Sangerman JI, Gallagher PG, Bodine DM. 2008. Failure of terminal erythroid differentiation in EKLF-deficient mice is associated with cell cycle perturbation and reduced expression of E2F2. Mol. Cell. Biol. 28:7394–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pilon AM, Nilson DG, Zhou D, Sangerman J, Townes TM, Bodine DM, Gallagher PG. 2006. Alterations in expression and chromatin configuration of the alpha hemoglobin-stabilizing protein gene in erythroid Kruppel-like factor-deficient mice. Mol. Cell. Biol. 26:4368–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tallack MR, Perkins AC. 2010. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. IUBMB Life 62:886–890 [DOI] [PubMed] [Google Scholar]
- 36.Tallack MR, Magor GW, Dartigues B, Sun L, Huang S, Fittock JM, Fry SV, Glazov EA, Bailey TL, Perkins AC. 2012. Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq. Genome Res. 22:2385–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yien YY, Bieker JJ. 2012. Functional interactions between erythroid Kruppel-like factor (EKLF/KLF1) and protein phosphatase PPM1B/PP2Cbeta. J. Biol. Chem. 287:15193–15204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raich N, Romeo PH. 1993. Erythroid regulatory elements. Stem Cells 11:95–104 [DOI] [PubMed] [Google Scholar]
- 39.Trimborn T, Gribnau J, Grosveld F, Fraser P. 1999. Mechanisms of developmental control of transcription in the murine alpha- and beta-globin loci. Genes Dev. 13:112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dzierzak E, Speck NA. 2008. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 9:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shyu YC, Wen SC, Lee TL, Chen X, Hsu CT, Chen H, Chen RL, Hwang JL, Shen CK. 2006. Chromatin-binding in vivo of the erythroid Kruppel-like factor, EKLF, in the murine globin loci. Cell Res. 16:347–355 [DOI] [PubMed] [Google Scholar]
- 42.Im H, Grass JA, Johnson KD, Kim SI, Boyer ME, Imbalzano AN, Bieker JJ, Bresnick EH. 2005. Chromatin domain activation via GATA-1 utilization of a small subset of dispersed GATA motifs within a broad chromosomal region. Proc. Natl. Acad. Sci. U. S. A. 102:17065–17070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perkins AC, Sharpe AH, Orkin SH. 1995. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature 375:318–322 [DOI] [PubMed] [Google Scholar]
- 44.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. 1995. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature 375:316–318 [DOI] [PubMed] [Google Scholar]
- 45.Orkin SH, Kazazian HH, Jr, Antonarakis SE, Goff SC, Boehm CD, Sexton JP, Waber PG, Giardina PJ. 1982. Linkage of beta-thalassaemia mutations and beta-globin gene polymorphisms with DNA polymorphisms in human beta-globin gene cluster. Nature 296:627–631 [DOI] [PubMed] [Google Scholar]
- 46.Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. 2010. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat. Genet. 42:742–744 [DOI] [PubMed] [Google Scholar]
- 47.Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, Cassar W, Galdies R, van Ijcken W, Ozgur Z, Gillemans N, Hou J, Bugeja M, Grosveld FG, von Lindern M, Felice AE, Patrinos GP, Philipsen S. 2010. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 42:801–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. 2008. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322:1839–1842 [DOI] [PubMed] [Google Scholar]
- 49.Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, Tucker PW, Orkin SH. 2009. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460:1093–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alhashem YN, Vinjamur DS, Basu M, Klingmuller U, Gaensler KM, Lloyd JA. 2011. Transcription factors KLF1 and KLF2 positively regulate embryonic and fetal beta-globin genes through direct promoter binding. J. Biol. Chem. 286:24819–24827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isern J, Fraser ST, He Z, Zhang H, Baron MH. 2010. Dose-dependent regulation of primitive erythroid maturation and identity by the transcription factor Eklf. Blood 116:3972–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou D, Pawlik KM, Ren J, Sun CW, Townes TM. 2006. Differential binding of erythroid Krupple-like factor to embryonic/fetal globin gene promoters during development. J. Biol. Chem. 281:16052–16057 [DOI] [PubMed] [Google Scholar]
- 53.Pearson RC, Funnell AP, Crossley M. 2011. The mammalian zinc finger transcription factor Kruppel-like factor 3 (KLF3/BKLF). IUBMB Life 63:86–93 [DOI] [PubMed] [Google Scholar]
- 54.Lomberk G, Urrutia R. 2005. The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem. J. 392:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lahiri SK, Zhao J. 2012. Kruppel-like factor 8 emerges as an important regulator of cancer. Am. J. Transl. Res. 4:357–363 [PMC free article] [PubMed] [Google Scholar]
- 56.He HJ, Gu XF, Xu WH, Yang DJ, Wang XM, Su Y. 2012. Kruppel-like factor 8 is a novel androgen receptor co-activator in human prostate cancer. Acta Pharmacol. Sin. 34:282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen G, Yang W, Jin W, Wang Y, Tao C, Yu Z. 2012. Lentivirus-mediated gene silencing of KLF8 reduced the proliferation and invasion of gastric cancer cells. Mol. Biol. Rep. 39:9809–9815 [DOI] [PubMed] [Google Scholar]
- 58.Liu L, Liu N, Xu M, Liu Y, Min J, Pang H, Zhang N, Zhang H. 2012. Lentivirus-delivered Kruppel-like factor 8 small interfering RNA inhibits gastric cancer cell growth in vitro and in vivo. Tumour Biol. 33:53–61 [DOI] [PubMed] [Google Scholar]
- 59.Li JC, Yang XR, Sun HX, Xu Y, Zhou J, Qiu SJ, Ke AW, Cui YH, Wang ZJ, Wang WM, Liu KD, Fan J. 2010. Up-regulation of Kruppel-like factor 8 promotes tumor invasion and indicates poor prognosis for hepatocellular carcinoma. Gastroenterology 139:2146–2157 [DOI] [PubMed] [Google Scholar]
- 60.Yang T, Cai SY, Zhang J, Lu JH, Lin C, Zhai J, Wu MC, Shen F. 2012. Kruppel-like factor 8 is a new Wnt/beta-catenin signaling target gene and regulator in hepatocellular carcinoma. PLoS One 7:e39668. 10.1371/journal.pone.0039668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schnell O, Romagna A, Jaehnert I, Albrecht V, Eigenbrod S, Juerchott K, Kretzschmar H, Tonn JC, Schichor C. 2012. Kruppel-like factor 8 (KLF8) is expressed in gliomas of different WHO grades and is essential for tumor cell proliferation. PLoS One 7:e30429. 10.1371/journal.pone.0030429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. 2007. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 67:7184–7193 [DOI] [PubMed] [Google Scholar]
- 63.Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ, Zhao J. 2011. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene 30:1901–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu WJ, Li JC, Wu XY, Yang ZB, Mo ZN, Huang JW, Xia GW, Ding Q, Liu KD, Zhu HG. 2010. Small interference RNA targeting Kruppel-like factor 8 inhibits the renal carcinoma 786-0 cells growth in vitro and in vivo. J. Cancer Res. Clin. Oncol. 136:1255–1265 [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Zhao J. 2007. KLF8 transcription factor participates in oncogenic transformation. Oncogene 26:456–461 [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Urvalek AM, Liu J, Zhao J. 2008. Activation of KLF8 transcription by FAK in human ovarian epithelial and cancer cells. J. Biol. Chem. 283:13934–13942 [DOI] [PubMed] [Google Scholar]
- 67.Wan W, Zhu J, Sun X, Tang W. 2012. Small interfering RNA targeting Kruppel-like factor 8 inhibits U251 glioblastoma cell growth by inducing apoptosis. Mol. Med. Rep. 5:347–350 [DOI] [PubMed] [Google Scholar]
- 68.Lu H, Hu L, Li T, Lahiri S, Shen C, Wason MS, Mukherjee D, Xie H, Yu L, Zhao J. 2012. A novel role of Kruppel-like factor 8 in DNA repair in breast cancer cells. J. Biol. Chem. 287:43720–43729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sue N, Jack BH, Eaton SA, Pearson RC, Funnell AP, Turner J, Czolij R, Denyer G, Bao S, Molero-Navajas JC, Perkins A, Fujiwara Y, Orkin SH, Bell-Anderson K, Crossley M. 2008. Targeted disruption of the basic Kruppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol. Cell. Biol. 28:3967–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hancock D, Funnell A, Jack B, Johnston J. 2010. Introducing undergraduate students to real-time PCR. Biochem. Mol. Biol. Educ. 38:309–316 [DOI] [PubMed] [Google Scholar]
- 71.Perdomo J, Verger A, Turner J, Crossley M. 2005. Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol. Cell. Biol. 25:1549–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Funnell AP, Wilson MD, Ballester B, Mak KS, Burdach J, Magan N, Pearson RC, Lemaigre FP, Stowell KM, Odom DT, Flicek P, Crossley M. 2013. A CpG mutational hotspot in a ONECUT binding site accounts for the prevalent variant of hemophilia B Leyden. Am. J. Hum. Genet. 92:460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. 2009. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 48:240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vernimmen D, De Gobbi M, Sloane-Stanley JA, Wood WG, Higgs DR. 2007. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 26:2041–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vu TT, Gatto D, Turner V, Funnell AP, Mak KS, Norton LJ, Kaplan W, Cowley MJ, Agenes F, Kirberg J, Brink R, Pearson RC, Crossley M. 2011. Impaired B cell development in the absence of Kruppel-like factor 3. J. Immunol. 187:5032–5042 [DOI] [PubMed] [Google Scholar]
- 76.Turchinovich G, Vu TT, Frommer F, Kranich J, Schmid S, Alles M, Loubert JB, Goulet JP, Zimber-Strobl U, Schneider P, Bachl J, Pearson R, Crossley M, Agenes F, Kirberg J. 2011. Programming of marginal zone B-cell fate by basic Kruppel-like factor (BKLF/KLF3). Blood 117:3780–3792 [DOI] [PubMed] [Google Scholar]
- 77.Copp AJ. 1995. Death before birth: clues from gene knockouts and mutations. Trends Genet. 11:87–93 [DOI] [PubMed] [Google Scholar]
- 78.Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, Lin C, Duggan P, Tassone P, Mansoor A, Stewart DA, Boise LH, Ghobrial IM, Bahlis NJ. 2011. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 117:6202–6213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. 2008. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456:814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hicklin DJ, Ellis LM. 2005. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23:1011–1027 [DOI] [PubMed] [Google Scholar]
- 81.Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. 2012. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481:85–89 [DOI] [PubMed] [Google Scholar]
- 82.Fladeby C, Gupta SN, Barois N, Lorenzo PI, Simpson JC, Saatcioglu F, Bakke O. 2008. Human PARM-1 is a novel mucin-like, androgen-regulated gene exhibiting proliferative effects in prostate cancer cells. Int. J. Cancer 122:1229–1235 [DOI] [PubMed] [Google Scholar]
- 83.Cornet AM, Hanon E, Reiter ER, Bruyninx M, Nguyen VH, Hennuy BR, Hennen GP, Closset JL. 2003. Prostatic androgen repressed message-1 (PARM-1) may play a role in prostatic cell immortalisation. Prostate 56:220–230 [DOI] [PubMed] [Google Scholar]
- 84.Yu CJ, Chang KP, Chang YJ, Hsu CW, Liang Y, Yu JS, Chi LM, Chang YS, Wu CC. 2011. Identification of guanylate-binding protein 1 as a potential oral cancer marker involved in cell invasion using omics-based analysis. J. Proteome Res. 10:3778–3788 [DOI] [PubMed] [Google Scholar]
- 85.Li M, Mukasa A, Inda M, Zhang J, Chin L, Cavenee W, Furnari F. 2011. Guanylate binding protein 1 is a novel effector of EGFR-driven invasion in glioblastoma. J. Exp. Med. 208:2657–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Silveira VS, Scrideli CA, Moreno DA, Yunes JA, Queiroz RG, Toledo SC, Lee ML, Petrilli AS, Brandalise SR, Tone LG. 2013. Gene expression pattern contributing to prognostic factors in childhood acute lymphoblastic leukemia. Leuk. Lymphoma 54:310–314 [DOI] [PubMed] [Google Scholar]
- 87.Dardousis K, Voolstra C, Roengvoraphoj M, Sekandarzad A, Mesghenna S, Winkler J, Ko Y, Hescheler J, Sachinidis A. 2007. Identification of differentially expressed genes involved in the formation of multicellular tumor spheroids by HT-29 colon carcinoma cells. Mol. Ther. 15:94–102 [DOI] [PubMed] [Google Scholar]
- 88.Lim KH, Kim KH, Choi SI, Park ES, Park SH, Ryu K, Park YK, Kwon SY, Yang SI, Lee HC, Sung IK, Seong BL. 2011. RPS3a over-expressed in HBV-associated hepatocellular carcinoma enhances the HBx-induced NF-kappaB signaling via its novel chaperoning function. PLoS One 6:e22258. 10.1371/journal.pone.0022258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang J, Chen K, Chen J, Gong W, Dunlop NM, Howard OM, Gao Y, Bian XW, Wang JM. 2010. The G-protein-coupled formylpeptide receptor FPR confers a more invasive phenotype on human glioblastoma cells. Br. J. Cancer 102:1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Otani T, Ikeda S, Lwin H, Arai T, Muramatsu M, Sawabe M. 2011. Polymorphisms of the formylpeptide receptor gene (FPR1) and susceptibility to stomach cancer in 1531 consecutive autopsy cases. Biochem. Biophys. Res. Commun. 405:356–361 [DOI] [PubMed] [Google Scholar]
- 91.Stamatoyannopoulos G. 2005. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 33:259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanabe O, Katsuoka F, Campbell AD, Song W, Yamamoto M, Tanimoto K, Engel JD. 2002. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 21:3434–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Filipe A, Li Q, Deveaux S, Godin I, Romeo PH, Stamatoyannopoulos G, Mignotte V. 1999. Regulation of embryonic/fetal globin genes by nuclear hormone receptors: a novel perspective on hemoglobin switching. EMBO J. 18:687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tanimoto K, Liu Q, Grosveld F, Bungert J, Engel JD. 2000. Context-dependent EKLF responsiveness defines the developmental specificity of the human epsilon-globin gene in erythroid cells of YAC transgenic mice. Genes Dev. 14:2778–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. 1997. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 11:2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wani MA, Means RT, Jr, Lingrel JB. 1998. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res. 7:229–238 [DOI] [PubMed] [Google Scholar]
- 97.Basu P, Lung TK, Lemsaddek W, Sargent TG, Williams DC, Jr, Basu M, Redmond LC, Lingrel JB, Haar JL, Lloyd JA. 2007. EKLF and KLF2 have compensatory roles in embryonic beta-globin gene expression and primitive erythropoiesis. Blood 110:3417–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Basu P, Morris PE, Haar JL, Wani MA, Lingrel JB, Gaensler KM, Lloyd JA. 2005. KLF2 is essential for primitive erythropoiesis and regulates the human and murine embryonic beta-like globin genes in vivo. Blood 106:2566–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sabath DE, Koehler KM, Yang WQ, Phan V, Wilson J. 1998. DNA-protein interactions in the proximal zeta-globin promoter: identification of novel CCACCC- and CCAAT-binding proteins. Blood Cells Mol. Dis. 24:183–198 [DOI] [PubMed] [Google Scholar]
- 100.Yi Z, Cohen-Barak O, Hagiwara N, Kingsley PD, Fuchs DA, Erickson DT, Epner EM, Palis J, Brilliant MH. 2006. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet. 2:e14. 10.1371/journal.pgen.0020014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yokoyama S, Asahara H. 2011. The myogenic transcriptional network. Cell. Mol. Life Sci. 68:1843–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buckingham M, Relaix F. 2007. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell Dev. Biol. 23:645–673 [DOI] [PubMed] [Google Scholar]
- 103.Wijgerde M, Gribnau J, Trimborn T, Nuez B, Philipsen S, Grosveld F, Fraser P. 1996. The role of EKLF in human beta-globin gene competition. Genes Dev. 10:2894–2902 [DOI] [PubMed] [Google Scholar]
- 104.Perkins AC, Gaensler KM, Orkin SH. 1996. Silencing of human fetal globin expression is impaired in the absence of the adult beta-globin gene activator protein EKLF. Proc. Natl. Acad. Sci. U. S. A. 93:12267–12271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tewari R, Gillemans N, Wijgerde M, Nuez B, von Lindern M, Grosveld F, Philipsen S. 1998. Erythroid Kruppel-like factor (EKLF) is active in primitive and definitive erythroid cells and is required for the function of 5′HS3 of the beta-globin locus control region. EMBO J. 17:2334–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mangan S, Itzkovitz S, Zaslaver A, Alon U. 2006. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 356:1073–1081 [DOI] [PubMed] [Google Scholar]
- 107.Goentoro L, Shoval O, Kirschner MW, Alon U. 2009. The incoherent feed forward loop can provide fold-change detection in gene regulation. Mol. Cell 36:894–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, Orkin SH. 2010. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 24:783–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.