

Abstract
Androgens play a major role in the regulation of normal ovarian function; however, they are also involved in the development of ovarian pathologies. These contrasting effects may involve a differential response of granulosa cells to the androgens testosterone (T) and dihydrotestosterone (DHT). To determine the molecular pathways that mediate the distinct effects of T and DHT, we studied the expression of the liver receptor homolog 1 (LRH-1) gene, which is differentially regulated by these steroids. We found that although both T and DHT stimulate androgen receptor (AR) binding to the LRH-1 promoter, DHT prevents T-mediated stimulation of LRH-1 expression. T stimulated the expression of aryl hydrocarbon receptor (AHR) and its interaction with the AR. T also promoted the recruitment of the AR/AHR complex to the LRH-1 promoter. These effects were not mimicked by DHT. We also observed that the activation of extracellular regulated kinases by T is required for AR and AHR interaction. In summary, T, but not DHT, stimulates AHR expression and the interaction between AHR and AR, leading to the stimulation of LRH-1 expression. These findings could explain the distinct response of granulosa cells to T and DHT and provide a molecular mechanism by which DHT negatively affects ovarian function.
INTRODUCTION
Androgens are crucial for normal ovarian function and are involved in the development of ovarian pathologies (1–3). In primates (4, 5) and rodents (6–8), androgens promote the growth of small ovarian follicles via activation of the androgen receptors (AR) in the granulosa cells (3). In large antral follicles, androgens serve as substrates for aromatase, an enzyme that catalyzes the conversion of androgens to estrogens (9). Conversely, androgens increase follicular atresia (10, 11) and inhibit follicle-stimulating hormone (FSH)-induced granulosa cell proliferation (12, 13). Androgens are also thought to be responsible for the halt in follicle growth found in patients with polycystic ovarian syndrome (PCOS), a condition marked by hyperandrogenism (14). The mechanisms that control the balance between the beneficial and harmful effects of androgens in ovarian function remain unknown.
In rat granulosa cells, androgens greatly augment the stimulatory effect of FSH on estradiol (E2) production (15), aromatase expression (16), and tissue plasminogen production (17, 18). Notably, of the two classical androgens, testosterone (T) is more effective than dihydrotestosterone (DHT) in augmenting these actions of FSH (15, 16, 18). Differential effects of T and DHT have been reported for the regulation of the aromatase promoter in bovine granulosa cells (19) and, as we recently reported, for the expression of aromatase in rat granulosa cells (20). In addition, it is known that DHT, but not T, decreases ovulation (11), inhibits gonadotropin-stimulated steroidogenesis (21), and prevents the induction of luteinizing hormone (LH) receptor by FSH (22, 23). Moreover, DHT decreases cell cycle progression by inhibiting the expression of cyclin D2 (13), whereas T potentiates the stimulatory effect of FSH on cyclin D2 (24). Similarly, T treatment of rat granulosa cells stimulates the expression of the transcription factor liver receptor homolog 1 (LRH-1), but DHT treatment has no effect (20). The molecular pathways that mediate the contrasting effects of T and DHT in granulosa cells remain to be investigated.
In this study, we examined the mechanisms involved in the differential response of granulosa cells to T and DHT using the gene for LRH-1 as the reporter gene. In the ovaries, LRH-1 is expressed exclusively in granulosa cells of follicles at all stages of development (25, 26). We initially determined whether the AR is required for the stimulation of LRH-1 by T and the effect of T and DHT on the activation of the AR in granulosa cells. The differential effects of T and DHT on the expression of LRH-1 may be due to the recruitment of alternative cofactors by the AR (27). Therefore, we also tested the hypothesis that the recruitment of specific cofactors by the AR in the presence of T, but not in the presence of DHT, results in the activation and the expression of the LRH-1 gene.
MATERIALS AND METHODS
Animals and granulosa cell culture.
Immature female Sprague-Dawley rats were purchased (Charles River Laboratories Inc., Wilmington, MA) and housed in the Biological Resources Laboratory at the University of Illinois at Chicago. Animals were treated in accordance with the Guide for Care and Use of Laboratory Animals (28), and all protocols were approved by the University of Illinois at Chicago Animal Care Committee. Primary granulosa cells were isolated from immature rats treated with E2 (1.5 mg/day for 3 days) and cultured for 96 h as previously described (20). Culture media and treatments were renewed every 48 h.
RNA quantitation.
Cells were cultured in the presence of steroids for 96 h prior to RNA isolation. Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. mRNA levels were quantified as previously described (20). The relative expression of target genes is expressed in reference to ribosomal L19 expression.
Western blot analysis.
Cytosolic or nuclear extracts were isolated from primary rat granulosa cells as described previously (20). Protein concentration was determined using bovine serum albumin (BSA) as the standard. The protein samples were subjected to SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA) and processed by routine procedures. The primary antibodies used, with their species of origin and the dilutions used in parentheses, were LRH-1 (rabbit, 1:500), lamin B1 (rabbit, 1:500), β-actin (rabbit, 1:500), AHR nuclear translocator (ARNT) (rabbit, 1:300), and aryl hydrocarbon receptor (AHR) (mouse, 1:500), all from Abcam (Cambridge, MA); AR (rabbit, 1:500) from Millipore Corp. (Billerica, MA); and mitogen-activated protein kinase 44/42 (MAPK44/42; rabbit, 1:500) and phosphor-MAPK44/42 (rabbit, 1:500) from Cell Signaling Technology (Danvers, MA). The secondary antibodies used were anti-rabbit IgG–horseradish peroxidase (IgG-HRP) (goat, 1:10,000) from Abcam and anti-mouse IgG–HRP (goat, 1:10,000) from Jackson ImmunoResearch laboratory Inc. (West Grove, PA). Detection was performed with Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore) or Supersignal West Femto maximum-sensitivity substrate (Thermo Scientific, Rockford, IL). Protein expression quantification was performed with ImageJ 1.45S software.
RNA interference.
Short hairpin RNAs (shRNAs) under the control of the H1 promoter were used to specifically knock down the expression of the AR, Ahr, Arnt, extracellular signal-regulated kinase 1 (ERK1), and ERK2 genes. shRNA target recognition sequences used were as follows: shAR, CTG CTC CGC AGA CAT TAA AGA CATC; shAhr, GGA GCT CTT CCC AGA TAAT; shArnt, GGA GCT CTT AGG AAA GAAT; shERK1, ACA GAA CAT TCC TAA ATCT; shERK2, GCA GTA TTA TGA CCC AAGT; and shLUC (control), GCC TGA AGT CTC TGA TTA AGT ACA A. Oligonucleotides containing the target recognition sequence and its corresponding antisense sequence separated by a short spacer sequence were chemically synthesized (Integrated DNA Technologies, Inc., Coralville, IA). These oligonucleotides were inserted into a lentivirus shRNA vector (Addgene, Inc., Cambridge, MA). Lentivirus stocks were generated in HEK293 cells (Invitrogen) cotransfected with the shRNA lentiviral vector along with the packaging and envelope plasmids psPAX2 and pMD2G (Addgene). Cell supernatants were concentrated by ultracentrifugation. Viral stocks were titrated in HEK293 cells aided by a green fluorescent protein (GFP) marker present in the shRNA plasmids. Viral stocks carrying shRNA were then added directly to the granulosa cells 2 h after plating at a multiplicity of infection of 20, followed by incubation for 96 h in the presence of the treatments described below for each figure. Culture medium was changed 48 h after plating but without the addition of virus.
Plasmid and reporter constructs and cell transfection.
The promoter region, from −507 to +1, of the rat LRH-1 gene was cloned from genomic DNA using PCR and standard cloning techniques. PCR products were cloned into the pGL3-Basic luciferase reporter vector (Promega Corp., Madison, WI) by using XhoI and HindIII restriction sites. All cloning was confirmed by bidirectional sequencing. The region from −507 to +1 was analyzed to predict transcription factor binding sites using online software (http://jaspar.cgb.ki.se and http://www.gene-regulation.com/pub/programs.html#match) as previously described (29). Reporter vectors with serial deletions of the 5′ end of LRH-1 promoter were generated. These LRH-1 promoter luciferase reporter constructs were transfected into granulosa cells 48 h after plating using FugeneHD (Roche, Indianapolis, IN). Ninety-six hours after the initiation of cultures, luciferase activity was assessed using the dual-luciferase assay kit (Promega) and reported as relative luciferase activity. The AR response elements found at positions −346 and −61 of the LRH-1 promoter were mutated using QuikChange II XL site-directed mutagenesis (Agilent, Santa Clara, CA).
Expression plasmid encoding wild-type (WT) AHR or the Y9F AHR mutant were kindly provided by Tom Gasiewicz and Chris Bradfield. A FLAG sequence (GAT TAC AAG GAT GAC GAT GAC AAG) was fused to the 5′ end of the WT AHR and Y9F AHR coding regions and subcloned into the pCDH vector (System Biosciences, Mountain View, CA). Viruses were produced in HEK293 cells as described for shRNAs. To confirm the expression and the DNA binding capacity of WT AHR and Y9F AHR, both proteins were overexpressed in HEK293 cells and the protein extracts were used to perform electrophoretic mobility shift assays (EMSAs) and Western blotting.
EMSAs.
Nuclear protein extracts from approximately 6 × 106 granulosa cells cultured in 100-mm plates in the absence or presence of steroids were prepared as described previously (20). Radioactively labeled double-stranded oligonucleotide probes containing ARE346 or ARE61 were used. For competition experiments, increasing concentrations of unlabeled ARE oligonucleotide probes were used. For supershift experiments, 2 μl of the AR antibody (rabbit, PG21; Millipore Corp.) or 2 μl of normal rabbit serum was added to the binding reaction 20 min before the addition of the labeled probe.
ChIP assay.
Granulosa cells (10 × 106) were cultured in 100-mm plates and treated as indicated below. Chromatin immunoprecipitation (ChIP) assays were performed using the MAGNA ChIP kit (Millipore Corp) according to the manufacturer's protocol. For each 100-mm plate, immunoprecipitations (IP) were carried out using 6 μg of anti-AR antibody, anti-Ahr antibody, or IgG. Immunoprecipitated DNA (ChIP DNA) and input DNA were amplified by PCR using primers spanning the ARE346 or ARE61 region found in the LRH-1 promoter.
Co-IP assays.
Coimmunoprecipitation (Co-IP) experiments were performed on whole-cell protein extracts obtained from 10 × 106 rat granulosa cells cultured in 100-mm plates using a Pierce cross-link immunoprecipitation kit (Thermo Scientific; catalog number 26147) according to the manufacturer's instructions. Antibodies used for IP were anti-AR, anti-AHR, normal rabbit serum, or normal mouse serum (IgG). The precipitated protein complexes were then used for Western blot analysis as described above.
Nuclear receptor array.
PCR arrays were performed using the Rat Nuclear Receptors & Coregulators RT2 Profiler PCR array (catalog number PARN-056; Qiagen, Germantown, MD), which profiles the expression of 84 genes encoding nuclear receptors and their coregulators. The array was performed according to the manufacturer's protocol. Briefly, total RNA was isolated from granulosa cells treated with a vehicle, T, or DHT as described above, and cDNA was generated using the RT2 first-strand kit included in the PCR array kit; 5 μg of RNA was used in each reverse transcription (RT) reaction. cDNA was loaded in the plate provided in the array kit and amplified using real-time PCR. The threshold cycle (CT) values were analyzed by PCR array data analysis Web-based software (Qiagen).
Statistical analysis.
One-way or two-way analysis of variance (ANOVA) was used for the statistical analysis of data using Prism software (GraphPad Software, Inc., San Diego, CA). The tests used are indicated in the figure legends. Values were considered statistically significant at a P value of <0.05.
RESULTS
Testosterone-specific stimulation of LRH-1 in granulosa cells.
Treatment of granulosa cells with increasing concentrations of T induced LRH-1 expression in a concentration-dependent manner (Fig. 1A). T also increased LRH-1 protein levels 6-fold (Fig. 1B) and stimulated LRH-1 promoter activity 2-fold (Fig. 1C). In contrast, DHT had no effect on LRH-1 expression, protein level, or promoter activity.
Fig 1.
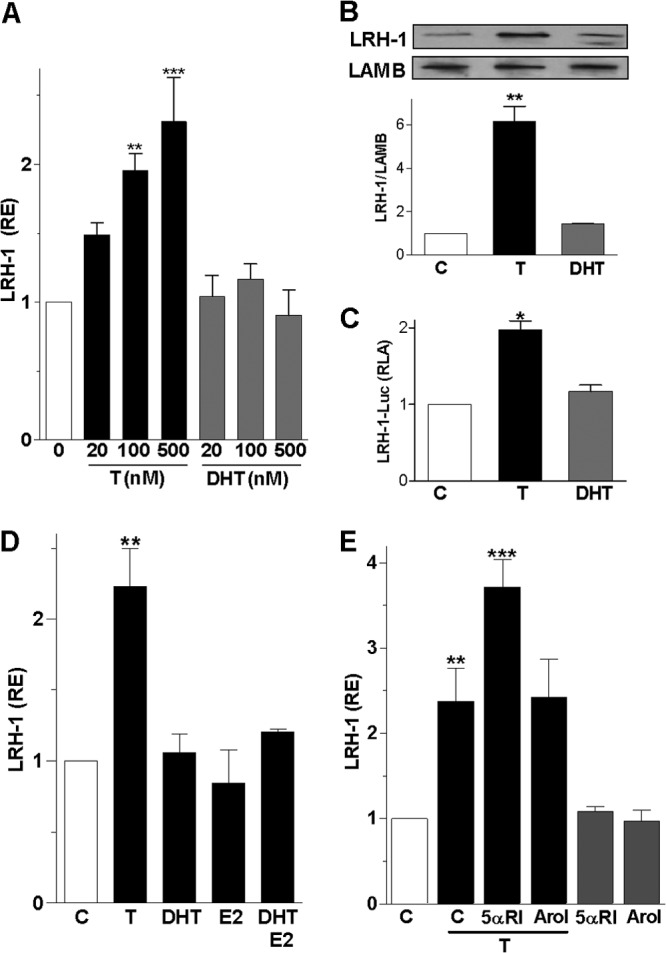
Testosterone but not dihydrotestosterone stimulates LRH-1 expression. (A) LRH-1 mRNA expression in granulosa cells cultured for 96 h in the presence of increasing concentrations of T or DHT ranging from 20 to 500 nM. RE, relative expression. (B) LRH-1 and lamin B (LAMB, loading control) protein expression in granulosa cells treated with vehicle (C), T (100 nM), or DHT (100 nM) for 96 h. Top, representative Western blot; bottom, quantification of three blots. (C) LRH-1 promoter activity in cells treated with a vehicle, T, or DHT. RLA, relative luciferase activity. (D) LRH-1 expression in cells incubated in the presence of 100 nM T, DHT, E2, or DHT plus E2. (E) LRH-1 expression in cells treated with a vehicle (C) or T in the presence or absence of the 5α-reductase inhibitor finasteride (5 μM) or the aromatase inhibitor AroI; (5 μM). All experiments were repeated at least four times, and the results are shown as the means ± SEMs. In panel A, P < 0.001 (***) and P < 0.01 (**) versus the control (0 nM) (two-way ANOVA, Bonferroni test). In panels B, C, D, and E, P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
Granulosa cells convert T into either estradiol (E2) or DHT by action of aromatase or 5α-reductase, respectively (25). To determine whether T effects are mediated by its conversion into E2 or DHT, we treated cells with E2, DHT, or a combination of the two. The results suggested that E2 and DHT, alone or combined, had no effect on LRH-1 expression (Fig. 1D). To examine if the conversion of T to DHT or E2 affects LRH-1 expression, we treated cells with T in the presence of the 5α-reductase inhibitor finasteride (5αRI) or the aromatase inhibitor formestane (AroI). Remarkably, finasteride enhanced the stimulatory effect of T on LRH-1 expression by 57%; in contrast, formestane had no effect (Fig. 1E). We previously demonstrated that in granulosa cells, finasteride and formestane block DHT and E2 synthesis, respectively (20). These results confirm the specific effect of T on LRH-1 expression and suggest that 5α-reductase activity may decrease LRH-1 expression by facilitating the conversion of T to DHT.
The AR mediates T stimulation of LRH-1 expression.
To examine if the androgen receptor (AR) mediates the stimulation of LRH-1 expression by T, granulosa cells were treated with hydroflutamine (HOF) or bicalutamide (CDX), two AR antagonists. We observed that both compounds prevented the stimulation of LRH-1 by T (Fig. 2A). To confirm this finding, AR expression was silenced using a virus carrying small hairpin RNA (shRNA) against the AR. We observed that the stimulation of LRH-1 gene expression (Fig. 2B) and promoter activity (Fig. 2C) in response to T was prevented by exposing granulosa cells to a virus carrying anti-AR shRNAs. These results suggest that the AR is required for T stimulation of LRH-1 expression.
Fig 2.
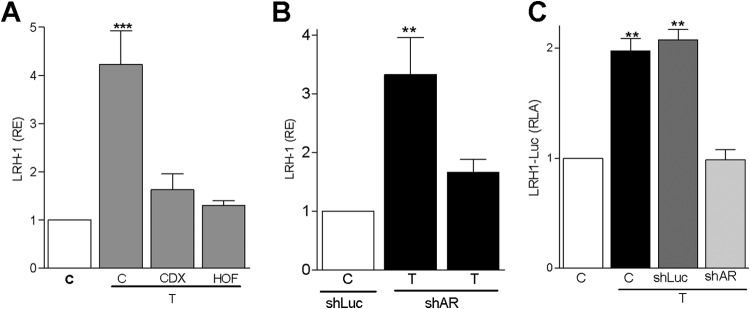
The androgen receptor mediates the stimulation of LRH-1 expression by T. (A) LRH-1 expression in cells incubated in the presence of T and AR antagonists, hydroflutamine (HOF) and bicalutamide (CDX), both at 5 μM. (B and C) LRH-1 expression (B) and promoter activity (C) in cells exposed to virus carrying shRNA anti-AR (shAR) or control (shLuc). Bars represent means ± SEMs (n = 3). P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
After we established that the AR mediates the effects of T on LRH-1 expression, we examined whether the differential effects of T and DHT on LRH-1 may correlate with differences in AR expression or subcellular localization. We observed that treatment with T or DHT increased the levels of AR in the nucleus, whereas the amount of AR protein in total cell lysates was not affected (Fig. 3A). These findings indicate that both T and DHT promote the translocation of the AR to the nucleus without altering AR protein levels.
Fig 3.
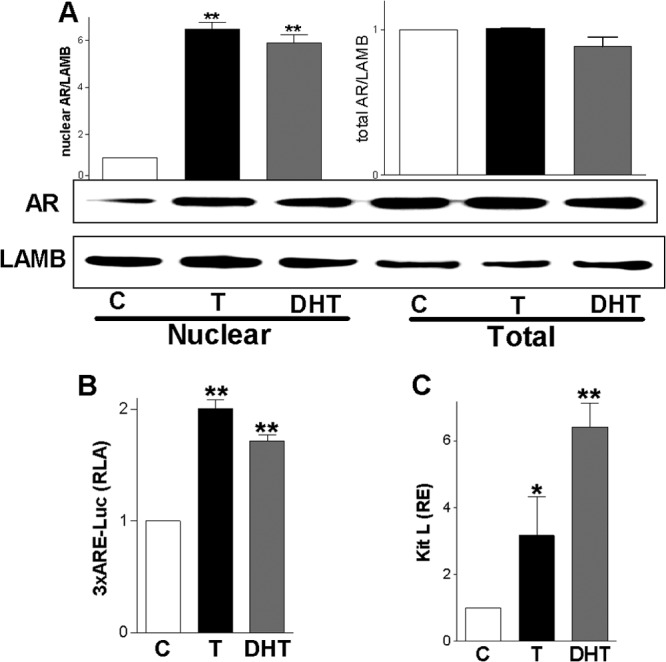
Testosterone and DHT stimulate androgen receptor nuclear translocation. (A) AR expression in the nuclear fraction or total cell lysates of granulosa cells treated with T or DHT. Lamin B (LAMB) was used as loading control. A representative blot is shown; the graphs on top represent the means ± SEMs of three different experiments. (B) Activity of the AR reporter 3×ARE-Luc in cells incubated in the presence of a vehicle (C), T, or DHT. (C) KitL expression in cells cultured in the presence of T or DHT. Experiments were repeated at least three times. P < 0.01 (**) and P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
Next, we examined if T and DHT regulate the transcriptional activity of the AR. For this purpose, a reporter construct consisting of three tandem repeats of the consensus AR response element (ARE) GGTACANNNTGTTCT upstream of the c-Fos minimal promoter and the luciferase coding sequence (3×ARE-Luc) was used. Granulosa cells were transfected with 3×ARE-Luc and incubated in the presence of T or DHT. The results indicated that both T and DHT increased 3×ARE-Luc activity (Fig. 3B), which suggests that both androgens can stimulate the transcriptional activity of the AR. We also examined the effect of DHT on the expression of Kit ligand (KitL), the gene for which is highly expressed in the ovaries of DHT-treated mice (1). The results showed that DHT stimulates KitL expression 6-fold, despite the fact that DHT is unable to stimulate LRH-1 expression (Fig. 3C). Testosterone treatment also stimulated KitL expression but to a lesser extent (3-fold) than DHT. These results suggest that both DHT and T have the potential to activate the AR and stimulate KitL expression in granulosa cells; however, only T increases LRH-1 expression.
DHT inhibits testosterone stimulation of LRH-1.
Knowing that inhibition of the conversion of T into DHT potentiates the effect of T (Fig. 1) and that both T and DHT stimulate AR nuclear localization and transcriptional activity (Fig. 3), we explored the possibility that DHT competes with T for binding to the AR and prevent LRH-1 expression. For this purpose, granulosa cells were incubated in the presence of 100 nM T and increasing concentrations (1 to 100 nM) of DHT. The results showed that DHT inhibited T stimulation of LRH-1 mRNA expression in a concentration-dependent manner where 50 nM DHT prevented the stimulation of LRH-1 expression by 100 nM T (Fig. 4A). Cotreatment with T and R1881, a nonmetabolizable AR agonist, also resulted in the inhibition of T stimulation of LRH-1 (Fig. 4B). It is noteworthy that the inhibitory effect of DHT on T-induced LRH-1 expression was markedly enhanced in the presence of aromatase and 5α-reductase inhibitors (Fig. 4C); however, both steroids synergized on the stimulation of 3×ARE-Luc activity (Fig. 4D) and on the expression of KitL (data not shown). These findings suggest that DHT is able to displace T from the AR, preventing the stimulation of LRH-1 expression.
Fig 4.
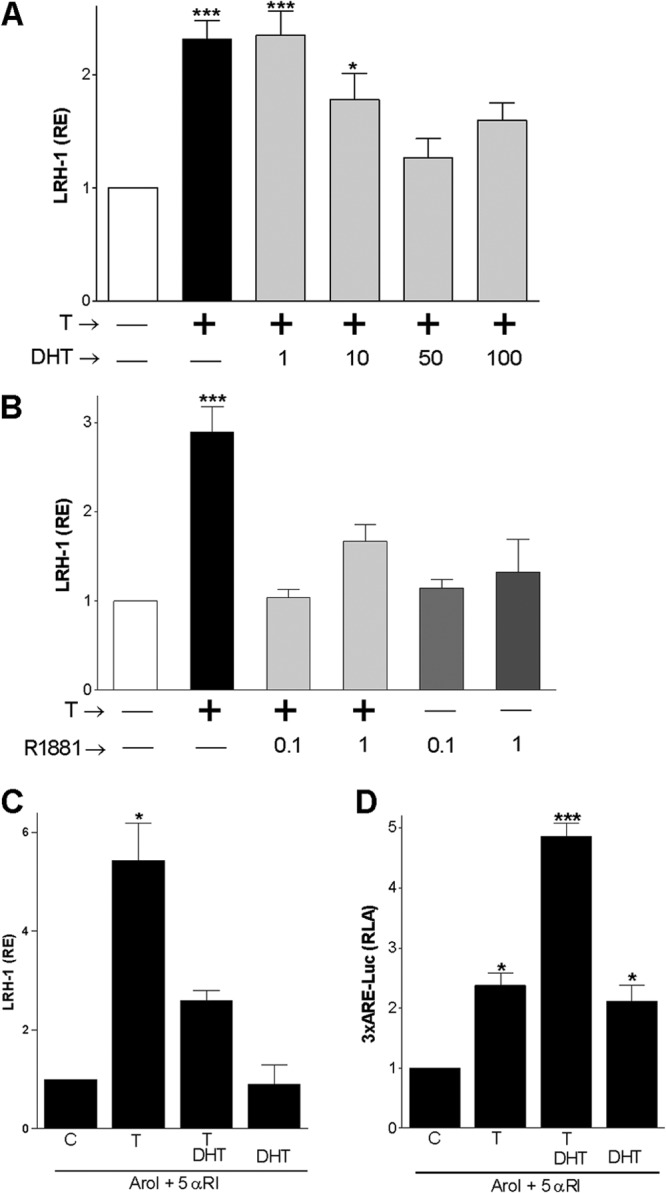
DHT or R1881 inhibit T stimulation of LRH-1 expression. (A and B) LRH-1 expression in the presence of T (100 nM) and increasing concentrations of DHT (1 to 100 nM) or R1881, an AR agonist (0.1 to 1 μM). (C) LRH-1 expression in cells treated with a vehicle (C), T, or T plus DHT in the presence of 5αRI (5 μM) and AroI (5 μM). (D) 3×ARE reporter activity in cells treated with a vehicle (C), T, or T plus DHT in the presence of 5αRI and AroI. All experiments were repeated at least three times, and the results are shown as the means ± SEMs. P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
The LRH-1 promoter contains an androgen response element that responds selectively to testosterone.
To determine the molecular mechanisms that mediate the effects of T on LRH-1 expression, the LRH-1 promoter region was analyzed to identify potential AR response elements (AREs). Two AREs were found within the promoter at 346 (ARE346) and 61 (ARE61) bp upstream of the transcription start site (30). To test the functionality of these elements, luciferase reporter constructs containing serial deletions of the LRH-1 promoter (LRH-1Luc) were made. T stimulated the activity of the reporter constructs containing the ARE346 element (507LRH-1Luc and 348LRH-1Luc), whereas the activity of the constructs lacking this element (266LRH-1Luc, −172LRH-1Luc, and the empty pGL3 plasmid) was not affected by treatment with T. As expected, DHT had no effect on the activity of any of the reporter constructs tested (Fig. 5A). To confirm the participation of the ARE346 element in the regulation of LRH-1 by T, we created reporter constructs carrying mutations at ARE346, ARE61, or both sites. The mutation at ARE346 or both ARE346 and ARE61 eliminated T stimulation of LRH-1, whereas mutation of ARE61 alone had no effect (Fig. 5B). These findings suggest that the AR response element found at position −346 of the LRH-1 promoter may mediate the increase in LRH-1 expression induced by T in granulosa cells.
Fig 5.

Testosterone stimulates LRH-1 promoter activity. (A) Activity of luciferase reporters carrying serial deletions of the LRH-1 promoter in the presence of T or DHT. Empty pGL3 vector was used as a control. (B) Effect of T or DHT on the activity of the wild-type (WT) 348-bp LRH-1luc reporter or the mutants ARE346 (346mut), ARE61 (61mut), or both (346/61mut). All experiments were repeated at least four times, and the results are shown as the mean ± SEMs. For each construct, P < 0.005 (***), P < 0.01 (**), and P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
Testosterone and DHT stimulate AR binding to the LRH-1 promoter.
To determine whether the AR binds to the LRH-1 promoter, we performed electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) analyses. Nuclear extracts of T- or DHT-treated granulosa cells formed a shift band in the presence of a radiolabeled oligonucleotide probe containing the ARE346 element (Fig. 6A). The formation of this band was prevented by the addition of an excess of an unlabeled probe. The addition of an anti-AR antibody to the binding reaction caused the formation of a supershift band. In contrast, no shift bands were observed using a probe containing the sequence of ARE61 (data not shown).
Fig 6.

Testosterone induces AR recruitment to the LRH-1 promoter. (A) Left gel, EMSA performed using nuclear extracts from granulosa cells cultured in the presence of vehicle (C), T, androstenedione (Δ4A), or DHT and a probe containing the ARE sequence found at position −346 of LRH-1 promoter. Middle gel, EMSA carried out in the presence of increasing concentrations of an unlabeled probe. Right gel, supershift analysis performed by adding normal antiserum (NS) or anti-AR antibody (αAR) to the EMSA reaction. (B) Top, primers used to amplify the region surrounding ARE346 or ARE61. Middle, representative gel of PCR product resulting from the amplification of total (input) or anti-AR immunoprecipitated (αAR IP) chromatin obtained from cells cultured in the presence of a vehicle (C), T, or DHT. Bottom, αAR IP/input ratio after real-time PCR quantification of total or αAR IP DNA. These experiments were repeated five times. In panel A, a representative gel is shown. In panel B, bars represent the mean ± SEMs. P < 0.01 (**) versus the control (one-way ANOVA, Tukey test).
ChIP performed in T- or DHT-treated granulosa cells revealed that there is an enrichment of the region containing the ARE346 element after chromatin precipitation using an anti-AR antibody (Fig. 6B). AR binding to the region containing ARE61 was not observed. Quantification of ChIP analyses indicated that both T and DHT increase recruitment of the AR to the LRH-1 promoter greater than 3.5-fold (Fig. 6B). These findings suggest that in the presence of T or DHT, the AR is recruited to the promoter of the LRH-1 gene at or close to the ARE found at position −346.
Cofactors differentially regulated by T and DHT in granulosa cells.
The finding that both T and DHT stimulate AR recruitment to the LRH-1 promoter suggested that the ability of the AR to stimulate LRH-1 expression in the presence of T but not in the presence of DHT may rest on the recruitment of different cofactors in response to the two treatments. To address this possibility, a PCR array of nuclear receptors and coregulators was carried out. This analysis revealed that T and DHT differentially regulate the expression of several transcription factors and nuclear receptors (Table 1). Of these factors, the expression of aryl hydrocarbon receptor (AHR) and nuclear receptor 1D1 was stimulated by T, whereas nuclear receptor 1H3 expression increased in T-treated cells but decreased in DHT-treated cells. The levels of expression of AHR nuclear translocator (ARNT), estrogen receptors 1 and 2, K (lysine)-acetyltransferase 5, nuclear receptors Nr0b2, Nr1d2, and Nr1i2, and nuclear coactivator 6 were decreased by DHT but not affected by T.
Table 1.
Cofactors differentially regulated by T and DHT in ovarian granulosa cellsa
| Gene | Ratio |
GenBank accession no. | Name | ||
|---|---|---|---|---|---|
| T | DHT | T/DHT | |||
| Ahr | 2.2 | 1.0 | 2.1 | NM_013149 | Aryl hydrocarbon receptor |
| Arnt | 1.6 | 0.1 | 21.5 | NM_012780 | Aryl hydrocarbon receptor nuclear translocator |
| Esr1 | 0.6 | 0.4 | 1.7 | NM_012689 | Estrogen receptor 1 |
| Esr2 | 0.7 | 0.0 | 72.0 | NM_012754 | Estrogen receptor 2 |
| Kat5 | 1.5 | 0.5 | 3.3 | NM_001005872 | K (lysine) acetyltransferase 5 |
| Ncoa6 | 1.1 | 0.3 | 3.2 | XM_342552 | Nuclear receptor coactivator 6 |
| Nr0b2 | 1.4 | 0.3 | 5.4 | NM_057133 | Nuclear receptor subfamily 0, group B, member 2 |
| Nr1d1 | 2.3 | 0.8 | 3.1 | NM_145775 | Nuclear receptor subfamily 1, group D, member 1 |
| Nr1d2 | 1.1 | 0.2 | 5.5 | NM_147210 | Nuclear receptor subfamily 1, group D, member 2 |
| Nr1h3 | 3.1 | 0.0 | 233.4 | NM_031627 | Nuclear receptor subfamily 1, group H, member 3 |
| Nr1i2 | 1.7 | 0.4 | 5.0 | NM_052980 | Nuclear receptor subfamily 1, group I, member 2 |
Cells were cultured in the presence of a vehicle (control), T, or DHT for 96 h prior to the determination of gene expression using the Rat Nuclear Receptors & Coregulators RT2 Profiler PCR Array as described in Materials and Methods. The results are expressed as the ratio between control and T (T column), control and DHT (DHT column), or T and DHT (T/DHT column).
AHR and ARNT are known to interact with the AR (31); therefore, we evaluated the effects of T and DHT on their expression using real-time PCR and Western blotting. Confirming the PCR array results, T stimulated the expression of Ahr mRNA 4-fold and Arnt mRNA levels 3-fold, whereas DHT had no effect (Fig. 7A). In addition, T stimulated the expression of AHR and ARNT protein, but DHT had no effect (Fig. 7B). These findings suggest that T and DHT differently regulate the expression of AHR and ARNT, two factors known to interact with and regulate AR activity (31).
Fig 7.

Testosterone induces AHR and ARNT expression. (A) AHR and ARNT mRNA expression in granulosa cells treated with a vehicle (C), T, or DHT. (B) Representative blot showing AHR and ARNT protein levels in cells incubated in the presence of a vehicle (C), T, or DHT. β-Actin (BACT) was used as loading control. Graphs show the means ± SEMs of three different experiments. (C) LRH-1, AHR, or ARNT expression in cells cultured in the presence of a vehicle (C) or T plus the addition of the AHR antagonists α-naphthoflavone (Naph) (10 μM) and 6,2′,4′-trimethoxyflavone (TMF) (10 nM). (D) LRH-1, AHR, or ARNT expression in granulosa cells infected with virus carrying control shRNA (shLuc), anti-AHR shRNA (shAhr), or anti-ARNT shRNA (shArnt). All the experiments were repeated at least four times, and the results are shown as the means ± SEMs. P < 0.05 (*) versus the control and DHT. Columns with different letters differ significantly as follows: a-b, P < 0.05; a-c and b-c, P < 0.01 (one-way ANOVA, Tukey test).
Next, we examined if AHR activity is necessary for the upregulation of LRH-1 expression in response to T by using two AHR antagonists, α-naphthoflavone and 6,2′,4′-trimethoxyflavone. We observed that T-induced LRH-1 expression was prevented in the presence of the inhibitors. The inhibitors had no effect on the stimulation of Ahr or Arnt expression by T (Fig. 7C). To confirm the effect of the AHR inhibitors and to determine the role of ARNT in the stimulation of LRH-1 expression, we used shRNA to decrease the expression of Ahr or Arnt. The results revealed that T is unable to stimulate LRH-1 expression in granulosa cells with reduced expression of either AHR or ARNT (Fig. 7D). Taken together, these findings suggest that AHR and ARNT are both required for T induction of LRH-1 expression.
The AR interacts with AHR and ARNT in a testosterone-dependent manner.
The finding that a lack of AHR abolished the effect of T on LRH-1 expression prompted us to investigate if AHR interacts with AR in granulosa cells and to examine if this interaction is regulated by T. For this purpose, we immunoprecipitated lysates from granulosa cells treated with a vehicle, T, or DHT using anti-AR or anti-AHR antibodies. Immunoprecipitates were analyzed by Western blotting using anti-AR, anti-AHR, or anti-ARNT antibodies. The results showed that the anti-AR antibody pulled down AHR and ARNT only in cells treated with T (Fig. 8A). Similarly, AR and ARNT were immunoprecipitated together with AHR in lysates of T-treated cells but not in vehicle- or DHT-treated cells (Fig. 8B). These results suggest that T not only increases the expression of AHR but also stimulates the interaction between AR and AHR.
Fig 8.

Testosterone induces AR-AHR-ARNT complex formation. (A) AHR, ARNT, and AR expression in IP fractions obtained using an anti-AR antibody (αAR) or normal rabbit serum (IgG) and total lysate of cells treated with a vehicle (C), T, or DHT. (B) AR, ARNT, and AHR expression in IP fractions obtained using an anti-AHR antibody (αAHR) or normal mouse serum (IgG) and total lysate of cells treated with a vehicle (C), T, or DHT. (C) LRH-1 promoter enrichment in genomic DNA immunoprecipitated using an anti-AHR antibody (αAhr). DNA was obtained from cells cultured in the presence of a vehicle (C), T, DHT, T plus shLuc, or T plus shAR. The graph represents the means ± SEMs of three different experiments. P < 0.05 (*) versus the control (one-way ANOVA, Tukey test). (D) LRH-1 expression in granulosa cells infected with an empty virus or viruses carrying DNA encoding wild-type AHR (WT) or Y9F AHR (Mut). EMSA was performed using total lysate of HEK293 cells transfected with empty, WT, or Mut expression vectors and a DRE probe. “WT*” indicates a reaction in which binding was competed with an excess of unlabeled probe. GC, nuclear extract of granulosa cells was used as a positive control for AHR binding; WB, Western blot against the FLAG epitope on total lysate obtained from HEK293 cells transfected with WT or Mut expression vectors. All experiments were repeated at least three times. The graph represents the means ± SEMs of three different experiments. P < 0.05 (*) versus the control (one-way ANOVA, Tukey test).
Knowing that AR binds to the LRH-1 promoter and that AR and AHR interact in a T-dependent manner, we sought to determine whether AHR is recruited to the LRH-1 promoter in an AR-dependent manner. ChIP analysis of the LRH-1 promoter indicated that only chromatin from T-treated cells immunoprecipitated with the anti-AHR antibody showed an enrichment of the LRH-1 promoter region containing the ARE346 element (Fig. 8C). To determine if the AR is required for AHR recruitment to the LRH-1 promoter, the same analysis was performed in cells transfected with control shRNA (shLuc) or with anti-AR shRNA (shAR). The results showed that enrichment of the LRH-1 promoter by the anti-AHR antibody was reduced in granulosa cells expressing lower levels of AR (Fig. 8C, 4th and 5th lanes). Quantification of these findings using real-time PCR indicated that silencing of the AR caused a 90% decrease of the enrichment of AHR recruitment to the LRH-1 promoter compared with shLuc-transfected cells. These findings suggest that AHR is recruited to the LRH-1 promoter in the presence of T in an AR-dependent manner, further suggesting that AHR could function as an AR cofactor in the regulation of LRH-1 expression.
Because the LRH-1 promoter lacks AHR/dioxin response elements (DRE) (data not shown) and the mutation of the ARE346 response element is enough to completely prevent LRH-1 promoter activation by T (Fig. 5B), the role of AHR in LRH-1 expression seems to occur in the absence of AHR interaction with DNA. To investigate this possibility, we overexpressed a wild-type (WT) or a DNA-binding mutant AHR in granulosa cells. Mutation of tyrosine 9 to phenylalanine (Y9F AHR) in AHR abolishes the binding of this protein to all DRE and abrogates DRE-driven gene induction mediated by the AHR (32). We observed that both WT AHR and Y9F AHR were able to synergize with T to stimulate LRH-1 expression (Fig. 8D). Western blotting and EMSA analysis confirmed the expression of both proteins and the ability of WT AHR but not Y9F AHR to bind DNA (Fig. 8D, left). These findings suggest that AHR acts mainly as a cofactor of the AR and that the effects of AHR on LRH-1 expression are unlikely to be mediated by its interaction with DNA.
ERK1/2 is needed for the stimulation of AHR and ARNT by testosterone.
Extracellular signal-regulated kinases (ERK) affect the transactivation potential of the AHR (33, 34); therefore, we examined if T regulates the activity of these kinases in granulosa cells. The results showed that T stimulates ERK1 and ERK2 phosphorylation; in contrast, DHT treatment had no effect. Moreover, inhibition of 5α-reductase and aromatase enhanced the stimulatory effect of T on ERK phosphorylation (Fig. 9A).
Fig 9.

ERK1 and ERK2 are required for the interaction of AHR and AR and for LRH-1 expression induced by testosterone. (A) Top, representative blot showing phosphorylated ERK1/2 (P-ERK1/2) and total ERK1/2 (t-ERK1/2) in granulosa cells cultured with a vehicle (C), T, or DHT in the presence or absence of 5αRI or AroI. Graphs show the means ± SEMs of the P-ERK/t-ERK ratio from three different experiments. P < 0.05 (*) versus the control (one-way ANOVA, Tukey test). (B) LRH-1 expression in cells cultured in the presence of a vehicle (C) or T infected with virus carrying control shRNA (shLUC) or shRNAs against ERK1 and ERK2 (shERK1/2). Bars represent the means ± SEMs (n = 4). P < 0.01 (**) versus the control (two-way ANOVA, Tukey test). (C) AHR and AR expression in IP fractions obtained using an anti-AR antibody (αAR) and total lysate of cells treated with a vehicle (C) or T in the presence or absence of virus carrying control shRNA (shLuc) or shRNAs against ERK1 and ERK2 (shERK1/2). The blots are representative results of three different experiments.
Next, we examined if ERK1/2 activity is required for the induction of LRH-1 expression by T. Because the two most commonly used inhibitors of ERK1/2 activation, U0126 and PD98059, are also potent antagonists of AHR (35–37), we examined if ERK1/2 activity is needed for the stimulation of LRH-1 by downregulating the expression of these kinases using shRNA. We observed that the induction of LRH-1 expression by T was abolished in the presence of anti-ERK1 and anti-ERK2 shRNAs (Fig. 9B). Moreover, we found that in cells transfected with anti-ERK1 and anti-ERK2 shRNAs, the interaction between AR and AHR was abolished (Fig. 9C). These results suggest that normal expression levels of ERK1/2 are required for T to induce the interaction between AR and AHR and the expression of LRH-1 in ovarian granulosa cells.
DISCUSSION
Our findings reveal a unique mechanism by which T and DHT may elicit differential responses in granulosa cells. The results suggest that T increases LRH-1 expression in an AR-dependent manner and stimulates, through the activation of ERK1/2, the interaction of between AR and AHR. The AR/AHR complex is, in turn, recruited to the promoter of the LRH-1 gene, leading to increased expression of this transcription factor. In contrast, DHT has no effect on AHR expression, ERK1/2 activation, or LRH-1 expression (Fig. 10).
Fig 10.
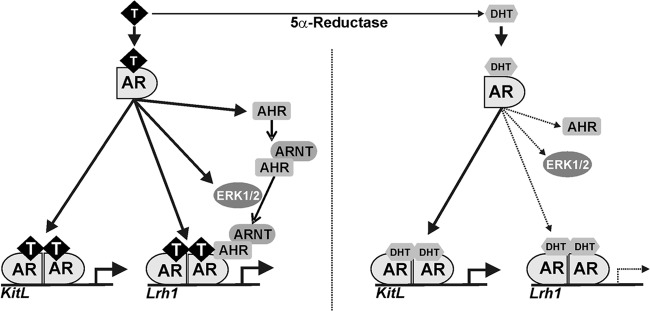
Potential mechanism of LRH-1 upregulation by testosterone in rat granulosa cells. Both T and DHT induce the binding of the AR to the promoter region of the LRH-1 gene. T specifically stimulates the expression of AHR and its association with the AR. The interaction between AHR and AR requires the activation of ERK1/2 by T. In contrast, DHT does not stimulate AHR or ERK1/2 activity; consequently, it has no effect on the expression of LRH-1. However, DHT stimulates Kit ligand (KitL) expression more efficiently than T. Solid lines indicate stimulation. Dashed lines indicate no effect. The thickness of the lines represents the extent of the effect.
As the two major physiological androgens, T and DHT bind to the AR with high affinity (7, 15). This is supported by crystallographic studies showing nearly identical interactions of T and DHT with the AR ligand-binding pocket (15). However, the effects of T and DHT on gene expression are not always equivalent, as they differentially regulate the expression of a subset of AR target genes in the prostate (38–40) and the ovaries (5, 15, 20, 41). The mechanisms of the differential response of ovarian granulosa cells to T or DHT have not been previously examined. Our results suggest that the negative effects of DHT on ovarian function may be explained by the displacement of T from the AR and the consequent decrease in the regulation of a specific set of ovarian genes.
Our findings suggest that in granulosa cells, the AR uses AHR and ARNT as cofactors. AHR binds with high affinity to a wide variety of hydrophobic environmental contaminants, such as dioxins (42). ARNT is an AHR-interacting protein found in the nucleus that facilitates AHR regulation of gene expression (43). Both AHR and ARNT are expressed in granulosa cells of several species (44, 45). AHR knockout mice are subfertile due to folliculogenesis defects, impaired aromatase expression (46), and slow growth of early antral follicles (47). Follicle growth is also impaired in the absence of AR expression in granulosa cells (3). Because estradiol restores follicle growth in AHR-null animals (48) and T stimulates aromatase expression (20), the interaction between AR and AHR may support folliculogenesis by increasing estradiol synthesis and maintaining normal follicle growth.
Cofactor recruitment is a crucial regulatory step in AR signal transduction and function (49, 50). The interaction between AHR and AR modulates the transcriptional activity of both proteins (51); however, the factors or signals that regulate the interaction between AHR and AR remain unclear. Our findings suggest that, at least in granulosa cells, AHR and AR association takes place only in the presence of T and requires the activation of ERK1/2. T increases ERK1/2 activation not only in granulosa cells, as shown here, but also in Sertoli cells (52). The effectiveness of the different androgens on ERK1/2 activation has not previously been examined. Our results indicate that ERK1/2 activation occurs in granulosa cells cultured in the presence of T but not DHT. Similar findings have been described for Xenopus oocytes (30). In addition, T is also able to stimulate AHR expression in granulosa cells, suggesting that it has a dual effect on the AR and AHR interaction. We propose that T regulates a set of specific genes needed for normal follicle growth by controlling the expression of AHR and its interaction with the AR.
The detrimental effect of DHT in ovarian gene expression may play a key role in the development of ovarian diseases associated with high levels of androgens such as PCOS. PCOS patients have increased 5α-reductase activity, as evidenced by higher conversion rates of oral dehydroepiandrosterone into DHT (53), augmented levels of 5α-reduced steroids in urine, serum, and follicular fluid (54, 55), and high 5α-reductase activity in peripheral tissues (53) and granulosa cells (56). Moreover, the incidence of PCOS decreases in people carrying inactivating mutations of 5α-reductase (57, 58). These findings indicate that high levels of 5α-steroids, including DHT, may contribute to the pathophysiology of PCOS by preventing T actions in granulosa cells, leading to a halt in follicle maturation.
In summary, we have found that T stimulation of AHR expression and AHR interaction with AR leads to the recruitment of both AR and AHR to the promoter region of the LRH-1 gene resulting in an increase in LRH-1 expression. Our findings can be used to explain the differential response of granulosa cells to T and DHT and provide a molecular mechanism by which DHT may negatively affect ovarian function.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01HD057110 and R21HD066233.
We are grateful to Tom Gasiewicz and Chris Bradfield for sharing the wild-type and mutant AHR expression vectors.
Footnotes
Published ahead of print 20 May 2013
REFERENCES
- 1.Shiina H, Matsumoto T, Sato T, Igarashi K, Miyamoto J, Takemasa S, Sakari M, Takada I, Nakamura T, Metzger D, Chambon P, Kanno J, Yoshikawa H, Kato S. 2006. Premature ovarian failure in androgen receptor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 103:224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu YC, Wang PH, Yeh S, Wang RS, Xie C, Xu Q, Zhou X, Chao HT, Tsai MY, Chang C. 2004 Subfertility and defective folliculogenesis in female mice lacking androgen receptor. Proc. Natl. Acad. Sci. U. S. A. 101:11209–11214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen A, Hammes SR. 2010. Granulosa cell-specific androgen receptors are critical regulators of ovarian development and function. Mol. Endocrinol. 24:1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vendola K, Zhou J, Wang J, Famuyiwa OA, Bievre M, Bondy CA. 1999. Androgens promote oocyte insulin-like growth factor I expression and initiation of follicle development in the primate ovary. Biol. Reprod. 61:353–357 [DOI] [PubMed] [Google Scholar]
- 5.Vendola KA, Zhou J, Adesanya OO, Weil SJ, Bondy CA. 1998. Androgens stimulate early stages of follicular growth in the primate ovary. J. Clin. Invest. 101:2622–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Andoh K, Hagiwara H, Xiaowei L, Kikuchi N, Abe Y, Yamada K, Fatima R, Mizunuma H. 2001. Effect of adrenal and ovarian androgens on type 4 follicles unresponsive to FSH in immature mice. Endocrinology 142:4930–4936 [DOI] [PubMed] [Google Scholar]
- 7.Xue K, Liu JY, Murphy BD, Tsang BK. 2012. Orphan nuclear receptor NR4A1 is a negative regulator of DHT-induced rat preantral follicular growth. Mol. Endocrinol. 26:2004–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray AA, Gosden RG, Allison V, Spears N. 1998. Effect of androgens on the development of mouse follicles growing in vitro. J. Reprod. Fertil. 113:27–33 [DOI] [PubMed] [Google Scholar]
- 9.Stocco C. 2012. Tissue physiology and pathology of aromatase. Steroids 77:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagnell CA, Mills TM, Costoff A, Mahesh VB. 1982. A model for the study of androgen effects on follicular atresia and ovulation. Biol. Reprod. 27:903–914 [DOI] [PubMed] [Google Scholar]
- 11.Conway BA, Mahesh VB, Mills TM. 1990. Effect of dihydrotestosterone on the growth and function of ovarian follicles in intact immature female rats primed with PMSG. J. Reprod. Fertil. 90:267–277 [DOI] [PubMed] [Google Scholar]
- 12.Kayampilly PP, Menon KM. 2012. AMPK activation by dihydrotestosterone reduces FSH-stimulated cell proliferation in rat granulosa cells by inhibiting ERK signaling pathway. Endocrinology 153:2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pradeep PK, Li X, Peegel H, Menon KM. 2002. Dihydrotestosterone inhibits granulosa cell proliferation by decreasing the cyclin D2 mRNA expression and cell cycle arrest at G1 phase. Endocrinology 143:2930–2935 [DOI] [PubMed] [Google Scholar]
- 14.Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. 2011. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 7:219–231 [DOI] [PubMed] [Google Scholar]
- 15.Daniel SA, Armstrong DT. 1980. Enhancement of follicle-stimulating hormone-induced aromatase activity by androgens in cultured rat granulosa cells. Endocrinology 107:1027–1033 [DOI] [PubMed] [Google Scholar]
- 16.Fitzpatrick SL, Richards JS. 1991. Regulation of cytochrome P450 aromatase messenger ribonucleic acid and activity by steroids and gonadotropins in rat granulosa cells. Endocrinology 129:1452–1462 [DOI] [PubMed] [Google Scholar]
- 17.Jia XC, Ny T, Hsueh AJ. 1990. Synergistic effect of glucocorticoids and androgens on the hormonal induction of tissue plasminogen activator activity and messenger ribonucleic acid levels in granulosa cells. Mol. Cell. Endocrinol. 68:143–151 [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Leung A. 1987. Estrogens, progestogens, and androgens enhance the follicle-stimulating hormone-stimulated plasminogen activator production by cultured rat granulosa cells. Endocrinology 120:2131–2136 [DOI] [PubMed] [Google Scholar]
- 19.Hamel M, Vanselow J, Nicola ES, Price CA. 2005. Androstenedione increases cytochrome P450 aromatase messenger ribonucleic acid transcripts in nonluteinizing bovine granulosa cells. Mol. Reprod. Dev. 70:175–183 [DOI] [PubMed] [Google Scholar]
- 20.Wu YG, Bennett J, Talla D, Stocco C. 2011. Testosterone, not 5α-dihydrotestosterone, stimulates LRH-1 leading to FSH-independent expression of Cyp19 and P450scc in granulosa cells. Mol. Endocrinol. 25:656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeleznik AJ, Little-Ihrig L, Ramasawamy S. 2004. Administration of dihydrotestosterone to rhesus monkeys inhibits gonadotropin-stimulated ovarian steroidogenesis. J. Clin. Endocrinol. Metab. 89:860–866 [DOI] [PubMed] [Google Scholar]
- 22.Jia XC, Kessel B, Welsh TH, Jr, Hsueh AJ. 1985. Androgen inhibition of follicle-stimulating hormone-stimulated luteinizing hormone receptor formation in cultured rat granulosa cells. Endocrinology 117:13–22 [DOI] [PubMed] [Google Scholar]
- 23.Farookhi R. 1985. Effects of aromatizable and nonaromatizable androgen treatments on luteinizing hormone receptors and ovulation induction in immature rats. Biol. Reprod. 33:363–369 [DOI] [PubMed] [Google Scholar]
- 24.El-Hefnawy T, Zeleznik AJ. 2001. Synergism between FSH and activin in the regulation of proliferating cell nuclear antigen (PCNA) and cyclin D2 expression in rat granulosa cells. Endocrinology 142:4357–4362 [DOI] [PubMed] [Google Scholar]
- 25.Falender AE, Lanz R, Malenfant D, Belanger L, Richards JS. 2003. Differential expression of steroidogenic factor-1 and FTF/LRH-1 in the rodent ovary. Endocrinology 144:3598–3610 [DOI] [PubMed] [Google Scholar]
- 26.Sirianni R, Seely JB, Attia G, Stocco DM, Carr BR, Pezzi V, Rainey WE. 2002. Liver receptor homologue-1 is expressed in human steroidogenic tissues and activates transcription of genes encoding steroidogenic enzymes. J. Endocrinol. 174:R13–R17 [DOI] [PubMed] [Google Scholar]
- 27.Askew EB, Gampe RT, Jr, Stanley TB, Faggart JL, Wilson EM. 2007. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 282:25801–25816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC [Google Scholar]
- 29.Wasserman WW, Sandelin A. 2004. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 5:276–287 [DOI] [PubMed] [Google Scholar]
- 30.Zhang CK, Lin W, Cai YN, Xu PL, Dong H, Li M, Kong YY, Fu G, Xie YH, Huang GM, Wang Y. 2001. Characterization of the genomic structure and tissue-specific promoter of the human nuclear receptor NR5A2 (hB1F) gene. Gene 273:239–249 [DOI] [PubMed] [Google Scholar]
- 31.Kollara A, Brown TJ. 2010. Four and a half LIM domain 2 alters the impact of aryl hydrocarbon receptor on androgen receptor transcriptional activity. J. Steroid Biochem. Mol. Biol. 118:51–58 [DOI] [PubMed] [Google Scholar]
- 32.Minsavage GD, Park SK, Gasiewicz TA. 2004. The aryl hydrocarbon receptor (AhR) tyrosine 9, a residue that is essential for AhR DNA binding activity, is not a phosphoresidue but augments AhR phosphorylation. J. Biol. Chem. 279:20582–20593 [DOI] [PubMed] [Google Scholar]
- 33.Tan Z, Huang M, Puga A, Xia Y. 2004. A critical role for MAP kinases in the control of Ah receptor complex activity. Toxicol. Sci. 82:80–87 [DOI] [PubMed] [Google Scholar]
- 34.Mukai R, Shirai Y, Saito N, Fukuda I, Nishiumi S, Yoshida K, Ashida H. 2010. Suppression mechanisms of flavonoids on aryl hydrocarbon receptor-mediated signal transduction. Arch. Biochem. Biophys. 501:134–141 [DOI] [PubMed] [Google Scholar]
- 35.Reiners JJ, Jr, Lee JY, Clift RE, Dudley DT, Myrand SP. 1998. PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen-activated protein kinase kinase. Mol. Pharmacol. 53:438–445 [DOI] [PubMed] [Google Scholar]
- 36.Andrieux L, Langouet S, Fautrel A, Ezan F, Krauser JA, Savouret JF, Guengerich FP, Baffet G, Guillouzo A. 2004. Aryl hydrocarbon receptor activation and cytochrome P450 1A induction by the mitogen-activated protein kinase inhibitor U0126 in hepatocytes. Mol. Pharmacol. 65:934–943 [DOI] [PubMed] [Google Scholar]
- 37.Chen S, Operana T, Bonzo J, Nguyen N, Tukey RH. 2005. ERK kinase inhibition stabilizes the aryl hydrocarbon receptor: implications for transcriptional activation and protein degradation. J. Biol. Chem. 280:4350–4359 [DOI] [PubMed] [Google Scholar]
- 38.Deslypere JP, Young M, Wilson JD, McPhaul MJ. 1992. Testosterone and 5α-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol. Cell. Endocrinol. 88:15–22 [DOI] [PubMed] [Google Scholar]
- 39.Dadras SS, Cai X, Abasolo I, Wang Z. 2001. Inhibition of 5α-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr. 9:183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avila DM, Fuqua SA, George FW, McPhaul MJ. 1998. Identification of genes expressed in the rat prostate that are modulated differently by castration and Finasteride treatment. J. Endocrinol. 159:403–411 [DOI] [PubMed] [Google Scholar]
- 41.Armstrong DT, Dorrington JH. 1976. Androgens augment FSH-induced progesterone secretion by cultured rat granulosa cells. Endocrinology 99:1411–1414 [DOI] [PubMed] [Google Scholar]
- 42.Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. 2008. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 18:207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. 2011. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124:1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bussmann UA, Bussmann LE, Baranao JL. 2006. An aryl hydrocarbon receptor agonist amplifies the mitogenic actions of estradiol in granulosa cells: evidence of involvement of the cognate receptors. Biol. Reprod. 74:417–426 [DOI] [PubMed] [Google Scholar]
- 45.Chaffin CL, Trewin AL, Hutz RJ. 2000. Estrous cycle-dependent changes in the expression of aromatic hydrocarbon receptor (AHR) and AHR-nuclear translocator (ARNT) mRNAs in the rat ovary and liver. Chem. Biol. Interact. 124:205–216 [DOI] [PubMed] [Google Scholar]
- 46.Baba T, Mimura J, Nakamura N, Harada N, Yamamoto M, Morohashi K, Fujii-Kuriyama Y. 2005. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol. Cell. Biol. 25:10040–10051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandez-Ochoa I, Barnett-Ringgold KR, Dehlinger SL, Gupta RK, Leslie TC, Roby KF, Flaws JA. 2010. The ability of the aryl hydrocarbon receptor to regulate ovarian follicle growth and estradiol biosynthesis in mice depends on stage of sexual maturity. Biol. Reprod. 83:698–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, Flaws JA. 2007. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol. Reprod. 76:1062–1070 [DOI] [PubMed] [Google Scholar]
- 49.Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. 2010. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 42:813–827 [DOI] [PubMed] [Google Scholar]
- 50.Lee DK, Chang C. 2003. Expression and degradation of androgen receptor: mechanism and clinical implication. J. Clin. Endocrinol. Metab. 88:4043–4054 [DOI] [PubMed] [Google Scholar]
- 51.Ohtake F, Baba A, Fujii-Kuriyama Y, Kato S. 2008. Intrinsic AhR function underlies cross-talk of dioxins with sex hormone signalings. Biochem. Biophys. Res. Commun. 370:541–546 [DOI] [PubMed] [Google Scholar]
- 52.Cheng J, Watkins SC, Walker WH. 2007. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in Sertoli cells. Endocrinology 148:2066–2074 [DOI] [PubMed] [Google Scholar]
- 53.Fassnacht M, Schlenz N, Schneider SB, Wudy SA, Allolio B, Arlt W. 2003. Beyond adrenal and ovarian androgen generation: increased peripheral 5α-reductase activity in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 88:2760–2766 [DOI] [PubMed] [Google Scholar]
- 54.Agarwal SK, Judd HL, Magoffin DA. 1996. A mechanism for the suppression of estrogen production in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 81:3686–3691 [DOI] [PubMed] [Google Scholar]
- 55.Chin D, Shackleton C, Prasad VK, Kohn B, David R, Imperato-McGinley J, Cohen H, McMahon DJ, Oberfield SE. 2000. Increased 5α-reductase and normal 11β-hydroxysteroid dehydrogenase metabolism of C19 and C21 steroids in a young population with polycystic ovarian syndrome. J. Pediatr. Endocrinol. Metabol. 13:253–259 [DOI] [PubMed] [Google Scholar]
- 56.Jakimiuk AJ, Weitsman SR, Magoffin DA. 1999. 5α-Reductase activity in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 84:2414–2418 [DOI] [PubMed] [Google Scholar]
- 57.Goodarzi MO, Shah NA, Antoine HJ, Pall M, Guo X, Azziz R. 2006. Variants in the 5α-reductase type 1 and type 2 genes are associated with polycystic ovary syndrome and the severity of hirsutism in affected women. J. Clin. Endocrinol. Metab. 91:4085–4091 [DOI] [PubMed] [Google Scholar]
- 58.Graupp M, Wehr E, Schweighofer N, Pieber TR, Obermayer-Pietsch B. 2011. Association of genetic variants in the two isoforms of 5α-reductase, SRD5A1 and SRD5A2, in lean patients with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 157:175–179 [DOI] [PubMed] [Google Scholar]