

Abstract
In response to stress, the heart undergoes a remodeling process associated with cardiac hypertrophy that eventually leads to heart failure. A-kinase anchoring proteins (AKAPs) have been shown to coordinate numerous prohypertrophic signaling pathways in cultured cardiomyocytes. However, it remains to be established whether AKAP-based signaling complexes control cardiac hypertrophy and remodeling in vivo. In the current study, we show that AKAP-Lbc assembles a signaling complex composed of the kinases PKN, MLTK, MKK3, and p38α that mediates the activation of p38 in cardiomyocytes in response to stress signals. To address the role of this complex in cardiac remodeling, we generated transgenic mice displaying cardiomyocyte-specific overexpression of a molecular inhibitor of the interaction between AKAP-Lbc and the p38-activating module. Our results indicate that disruption of the AKAP-Lbc/p38 signaling complex inhibits compensatory cardiomyocyte hypertrophy in response to aortic banding-induced pressure overload and promotes early cardiac dysfunction associated with increased myocardial apoptosis, stress gene activation, and ventricular dilation. Attenuation of hypertrophy results from a reduced protein synthesis capacity, as indicated by decreased phosphorylation of 4E-binding protein 1 and ribosomal protein S6. These results indicate that AKAP-Lbc enhances p38-mediated hypertrophic signaling in the heart in response to abrupt increases in the afterload.
INTRODUCTION
In response to increased workload or pathological insults, the heart undergoes a remodeling process associated with cardiomyocyte hypertrophy (1). This response is initially compensatory and causes the ventricular mass to increase as a means of maintaining normal cardiac output. However, concomitant reactivation of a fetal gene program profoundly alters cardiac contractility, calcium handling, and myocardial energetics, which in the long term leads to increased cardiomyocyte death, replacement fibrosis, and heart failure (2).
A-kinase anchoring proteins (AKAPs) constitute a family of scaffolding proteins that tether the cyclic AMP (cAMP)-dependent protein kinase A (PKA), as well as other signaling enzymes, at focal points within cells to ensure the coordination of specific signal transduction events (3, 4). Evidence collected over the last few years indicates that AKAPs coordinate numerous prohypertrophic signaling pathways in cultured cardiomyocytes (neonatal ventricular myocytes [NVMs]) (5–7). However, so far, no study has addressed the implication of these anchoring proteins in cardiac hypertrophy in vivo.
Previous work has identified an anchoring protein expressed in cardiomyocytes, termed AKAP-Lbc, which acts as a RhoA selective guanine nucleotide exchange factor (GEF) (8) and serves as a scaffold for multiple signaling enzymes regulating cardiomyocyte growth (9–12). Silencing of AKAP-Lbc expression in rat NVMs strongly reduces RhoA activation and hypertrophic responses induced by GPCR agonists (9, 10), suggesting a link between AKAP-Lbc-mediated RhoA activation and cardiomyocyte hypertrophy.
Recently, we could show that AKAP-Lbc recruits a signal transduction complex that includes the RhoA effector PKNα, as well as its associated kinases MLTK, MKK3, and p38α, which promotes RhoA-dependent activation of p38α (13). These results, together with recent evidence showing that acute activation of p38 in the adult heart can promote hypertrophy (14, 15), raise the possibility that AKAP-Lbc coordinates p38-regulated hypertrophic responses in the stressed heart.
In the present study, we generated a transgenic (TG)-mouse model displaying cardiomyocyte-specific overexpression of an AKAP-Lbc fragment that competitively inhibits the interaction between AKAP-Lbc and the p38 signaling module. We could show that disruption of the AKAP-Lbc/p38 complex attenuated p38 activation and compensatory cardiomyocyte growth induced by increased workload, which was accompanied by increased apoptosis and the development of early dilated cardiomyopathy. These findings indicate that AKAP-Lbc functions to enhance p38-mediated hypertrophic signaling in the heart in response to abrupt increases in the afterload. They also provide the first in vivo evidence that AKAPs can regulate cardiac hypertrophy and remodeling.
MATERIALS AND METHODS
Animal models.
The Flag-tagged AKAP-Lbc-1570-1764 fragment was subcloned at SalI into the α-myosin heavy chain (α-MHC) clone 22 vector (16) downstream of the 5.8-kb murine α-MHC promoter. Microinjection of the transgene into the pronuclei of fertilized eggs from C57BL6/DBA mice was performed by the Transgenic Animal Facility of the University of Lausanne, Lausanne, Switzerland. Mice were backcrossed onto the C57BL6/N background. PCR genotyping of transgenic mice was performed using the forward primer 5′-GACGATGACGACAAGCTCAA-3′ and the reverse primer 5′-AATGGAGAGGAAATGGCTGA-3′ , resulting in an amplicon of 398 bp.
Transverse aortic constriction.
Twelve-week-old male mice were anesthetized by intraperitoneal (i.p.) injection with a mixture of ketamine-xylazine-acepromazine (65/15/2 mg/kg of body weight). The mice were placed on a warming pad in the supine position, endotracheal intubation was performed, and the mice were connected to a mini-rodent ventilator (tidal volume, 0.2 ml; rate, 120 breaths/min). The thoraxes of the animals were shaved and disinfected with Betadine solution. A small horizontal incision was made at the level of the cranial sternum, allowing visualization of the aortic arch. A suture was placed around the aorta between the origin of the right innominate and left common carotid artery. Transaortic constriction (TAC) was created using a 7.0 silk suture tied twice around the aorta and a 27-gauge needle. The needle was then gently retracted, the incision was closed, and the skin was sutured. For animals undergoing a sham operation, the ligature was placed in an identical location but not tied. Analgesic drug (temgesic [buprenorphine], 0.1 mg/kg) was administered subcutaneously after the surgery. Experiments on the mice were carried out in accordance with the Swiss Animal Protection Ordinance (OPAn). The protocol was approved by the Veterinary Office of the state of Vaud, Switzerland (permit numbers 1547.3 and 2332).
Echocardiography.
Transthoracic echocardiography was performed using a 30-MHz probe and the Vevo 770 Ultrasound machine (VisualSonics, Toronto, ON, Canada). Mice were lightly anesthetized with 1 to 1.5% isoflurane, maintaining the heart rate at 400 to 500 beats per minute. The heart was imaged in two-dimensional (2D) mode in the parasternal long-axis view. From this view, a motion mode (M-mode) cursor was positioned perpendicular to the interventricular septum and the posterior wall of the left ventricle at the level of the papillary muscles. Diastolic (IVS;d) and systolic (IVS;s) internal ventricular septum, diastolic (LVPW;d) and systolic (LVPW;s) left ventricular free posterior wall thickness, and left ventricular internal end-diastolic (LVID;d) and end-systolic (LVID;s) chamber dimensions were measured. The measurements were taken in three separate M-mode images and averaged. Left ventricular fractional shortening (%FS) and ejection fraction (%EF) were also calculated. Fractional shortening is assessed from M mode based on the percent changes of left ventricular end-diastolic and end-systolic diameters. %EF is derived from the following formula: (LV Vol;d − LV Vol;s)/LV Vol;d × 100, where LV Vol;d is end-diastolic and LV Vol;s is end-systolic left ventricular volume.
Histology.
Hearts were collected, washed in ice-cold phosphate-buffered saline (PBS), and fixed in 4% formaldehyde (PBS buffered). After dehydration of the tissue, the hearts were embedded in paraffin blocks. For histological analysis, transversal (short-axis) 3-μm heart sections were obtained. Fibrosis was quantified on deparaffinized sections stained with Masson's Trichrome (Sigma-Aldrich) using Photoshop Elements software.
TUNEL assay.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed on 3-μm deparaffinized transversal sections. Tissue was prepermeabilized with 0.1% Triton X-100 in 0.1% citrate and then treated with 2% proteinase K (Dako). An enzymatic reaction was performed using biotin-16–dUTP and TUNEL enzyme from calf thymus (Roche). To detect biotin-labeled DNA, heart sections were incubated with Alexa Fluor 488-streptavidin (Life Technologies). DAPI (4′,6-diamidino-2-phenylindole) staining was performed to visualize the total number of nuclei. TUNEL-positive nuclei in each heart section were normalized to the number of DAPI-stained nuclei (quantified using ImageJ software). Analysis was performed on two separate heart sections per mouse.
Immunohistochemistry.
Deparaffinized heart sections were immunostained with a rabbit polyclonal antilaminin antibody (Sigma; 1:50 dilution) and an Alexa Fluor 594-labeled goat anti-rabbit secondary antibody (Life Technologies; 1:500 dilution). The cross-sectional areas of cardiomyocytes were quantified using ImageJ software.
RNA preparation and quantitative real-time PCR.
Determination of the mRNA levels of α- and β-MHC, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), skeletal α-actin, collagen I, and collagen III in the ventricles of wild-type (WT) and TG mice was carried out by real-time reverse transcription (RT)-PCR analysis using a 7500 fast real-time PCR system (Applied Biosystems). Total RNA was extracted from ventricular tissues using the RNeasy fibrous tissue minikit (Qiagen), and single-stranded cDNA was synthesized from 2 μg of total RNA by using random hexamers (Applied Biosystems) and SuperScript II reverse transcriptase (Invitrogen). RT-PCR mixtures were prepared in 1× PCR universal master mix (Applied Biosystems) using 100 ng of the reverse-transcribed RNA and selected TaqMan gene expression assays (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as an invariant internal control.
Quantification of the transgene copy number and mRNA expression by real-time PCR.
Genomic DNA was extracted from tail biopsy specimens (4 mm) taken from WT and TG mice after sacrifice. The biopsy specimens were incubated overnight at 56°C in 750 μl of 50 mM Tris-HCl, pH 8.0, 100 mM EDTA, 100 mM NaCl, 1% SDS, and 0.5 mg/ml proteinase K (Merck). Samples were mixed for 5 min and combined with 250 μl of 6 M NaCl. After centrifugation for 15 min at 14,000 rpm, 750 μl of the supernatant was mixed with 500 μl of isopropanol to precipitate the DNA. Finally, the DNA pellet was washed with 70% ethanol, dried, and resuspended in Tris-EDTA (TE) buffer.
The transgene copy number integrated into the genome was quantified on a 7500 fast real-time PCR system using 10 and 20 ng of genomic DNA from transgenic mice, Power SYBR green 2× PCR master mix (Applied Biosystems), and specific primers to amplify the transgene (forward, 5′-GACGATGACGACAAGCTCAA-3′ , and reverse, 5′-GCCAGCAGAGTACTGCACAC-3′ ). To calculate the number of copies integrated in the genome, a standard curve was generated using a plasmid containing the transgene.
Determination of the mRNA levels of the AKAP-Lbc-1570-1764 fragment in the ventricles of WT and TG mice was carried out by real-time RT-PCR analysis using a 7500 fast real-time PCR system (Applied Biosystems). Extraction of the total RNA and reverse transcription were performed as described above. RT-PCR mixtures were prepared in Power SYBR green 2× PCR master mix (Applied Biosystems) using 100 ng of the reverse-transcribed RNA and specific primers to amplify the transgene (forward, 5′-AACCACCGGAGTGAGCTG-3′ , and reverse, 5′-GATGAGGATGGCTTGTTTCC-3′ ). GAPDH mRNA was used as an invariant internal control.
Immunoprecipitation experiments.
(i) From rat NVMs. For immunoprecipitation experiments, noninfected or infected rat NVMs were lysed in 1 ml of buffer A (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% Na deoxycholate, 5 mg/ml aprotinin, 10 mg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). The cell lysates were incubated for 4 h at 4°C on a rotating wheel. The solubilized material was centrifuged at 100,000 × g for 30 min at 4°C, and the supernatants were dialyzed twice in 5 liters of buffer B (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% [wt/vol] Triton X-100) at 4°C. The dialyzed supernatants were then incubated for 2 h at 4°C with 4 μg of polyclonal anti-AKAP-Lbc or with 4 μg of rabbit IgG (negative control) on a rotating wheel. Next, all the samples were incubated with 20 μl of protein A-Sepharose beads for 2 h at 4°C on a rotating wheel. Following a brief centrifugation on a benchtop centrifuge, the pelleted beads were washed five times with buffer B and twice with PBS, and proteins were eluted in SDS-PAGE sample buffer (65 mM Tris-HCl, pH 6.8, 2% SDS, 5% glycerol, 5% 2-mercaptoethanol) by boiling samples for 3 min at 95°C. The eluted proteins were analyzed by SDS-PAGE and Western blotting.
(ii) From heart lysates.
Heart ventricular tissues were disrupted with a Polytron homogenizer in 1 ml of buffer C (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% [wt/vol] Triton X-100, 2 μg/ml aprotinin, 4 μg/ml leupeptin, 4 μg/ml pepstatin, and 1 mM PMSF). The cell lysates were incubated for 1 h at 4°C on a rotating wheel, followed by centrifugation at 14,000 rpm for 30 min at 4°C. The protein concentration of the supernatants was determined by Lowry assay, and 1 mg of total homogenates was incubated for 2 h at 4°C with 15 μl of anti-FLAG M2 agarose beads (Sigma). Following a brief centrifugation, the pelleted beads were washed three times with 1 ml of buffer C, and proteins were eluted in SDS-PAGE sample buffer (65 mM Tris, pH 6.8, 2% SDS, 5% glycerol, 5% 2-mercaptoethanol) by boiling the samples for 3 min at 95°C.
SDS-PAGE and Western blotting.
Samples denatured in SDS-PAGE sample buffer were separated on acrylamide gels and electroblotted onto nitrocellulose membranes. The blots were incubated with primary antibodies and horseradish-conjugated secondary antibodies (Amersham). The following primary antibodies were used for immunoblotting: affinity-purified rabbit polyclonal anti-AKAP-Lbc (Covance; 0.1 mg/ml; 1:1,000 dilution), mouse monoclonal anti-Flag (Sigma; 4.9 mg/ml; 1:2,000 dilution), rabbit polyclonal anti-phospho-p38 (threonine 180 and tyrosine 182; 1:1,000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-p38α (Cell Signaling Technologies; 1:1,000 dilution), mouse monoclonal anti-MKK3 (Assay Designs; 1:1,000 dilution), rabbit polyclonal anti-MKK3 (Cell Signaling Technologies; 1:1,000 dilution), mouse monoclonal anti-MLTK (Abnova; 1:500 dilution), mouse monoclonal anti-PKNα (BD Biosciences Pharmingen; 1:1,000 dilution), rabbit polyclonal anti-phospho-extracellular signal-regulated kinase 1/2 (anti-phospho-ERK1/2) (threonine 202 and tyrosine 204; 1:500 dilution; Santa Cruz Biotechnology), rabbit polyclonal anti-ERK1/2 (Santa Cruz Biotechnology; 1:500 dilution), rabbit polyclonal anti-phospho-c-Jun N-terminal kinase (anti-phospho-JNK) (threonine 183 and tyrosine 185; 1:500 dilution; Cell Signaling Technologies), rabbit polyclonal anti-JNK (Cell Signaling Technologies; 1:500 dilution), rabbit polyclonal anti-phospho-PKD (serines 744 and 748; 1:1,000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-PKD (Cell Signaling Technologies; 1:1,000 dilution), mouse monoclonal anti-phospho-Akt (serine 473; 1:1,000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-Akt (Cell Signaling Technologies; 1:1,000 dilution), rabbit polyclonal anti-MYH7 (Santa Cruz Biotechnology; 1:500 dilution), mouse monoclonal antiactin (Sigma; 1:1,000 dilution), rabbit anti-phospho-4E-BP1 (threonine 70), rabbit polyclonal anti-phospho-S6 ribosomal protein (serine 240 and serine 244; 1:1,000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-S6 ribosomal protein (Cell Signaling Technologies; 1:1,000 dilution), rabbit polyclonal anti-P70 S6 kinase 1 (Cell Signaling Technologies; 1:1,000 dilution), and rabbit polyclonal anti-mTOR (Cell Signaling Technologies; 1:1,000 dilution).
Rat neonatal ventricular myocyte preparation.
Rat NVMs were prepared from 1- to 2-day-old Sprague-Dawley rats. Excised hearts were digested by three cycles of enzymatic digestion at 37°C for 15 min, using a mixture of 0.45 mg/ml collagenase type II (Worthington) and 1 mg/ml pancreatin (Sigma), followed by centrifugation (800 rpm; 10 min). The cells contained in the final pellet were suspended in maintenance medium (80% Dulbecco's modified Eagle's medium [DMEM], 20% M199 medium [Invitrogen], 1% penicillin-streptomycin solution [Invitrogen], and 1% HEPES) supplemented with 10% fetal calf serum and 5% horse serum (Invitrogen, Gibco) and seeded on T75 cell culture flasks to deplete fibroblasts. After two sequential steps of 1 h of differential plating, nonadherent neonatal myocytes were seeded in cell culture dishes precoated with 0.2% gelatin. After 24 h, the medium was changed, and the cells were cultured in maintenance medium supplemented with 5% horse serum. Cardiomyocyte culture purity was >95%, as assessed by immunocytochemistry using an anti-α-actinin monoclonal antibody.
Production of lentiviruses.
Vesicular stomatitis virus G protein (VSV-G)-pseudotyped lentiviruses were produced by cotransfecting 293-T cells with 20 μg of the empty polyclonal antibody (PAb) vector, the PAb vector containing the cDNA encoding AKAP-Lbc short hairpin RNA (shRNA) (5–7), or the Flag-tagged AKAP-Lbc-1570-1764 fragment (13), 15 μg of pCMVDR8.91, and 5 μg of pMD2.VSVG, using the calcium phosphate method. The culture medium (DMEM and 100 mg/ml gentamicin, 5% fetal calf serum [FCS]) was replaced by serum-free DMEM 8 h after transfection. Cell supernatants were collected 48 h later and filtered through a 0.22-μm filter unit. Lentiviruses were concentrated using Centricon Plus-70 (nominal molecular weight limit, 100,000) columns (Millipore) and resuspended in PBS. Virus titers were determined by infecting 293-T cells using serial dilutions of the viral stocks and by scoring puromycin-resistant clones (at 6 days after infection). Titers determined using this method were between 2 × 108 and 7 × 108 transducing units (TU)/ml.
Lentiviral infection.
Rat NVMs were infected at 90% confluence using PAb-based lentiviruses at a multiplicity of infection (MOI) of 50 in maintenance medium containing 5% horse serum and 8 μg/ml Polybrene; 48 h after infection, the rat NVMs were incubated with fresh maintenance medium for an additional 24 h. The rat NVMs were subsequently starved for 24 h in serum-free maintenance medium before stimulation for an additional 24 h with 10−4 M phenylephrine (PE).
Adult mouse cardiomyocyte isolation.
Mice were sacrificed by cervical dislocation, and their hearts were excised by cutting the aorta at the level of the aortic arch. Ventricular myocytes were isolated by enzymatic gravimetric perfusion of the heart. In summary, the heart was cannulated by the aorta and mounted on a Langendorff system, where it underwent retrograde perfusion with control solution (135 mM NaCl, 4 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 10 mM HEPES, pH 7.4, 11 mM glucose) at 37°C to wash out the blood. Digestion of the tissue was performed by perfusion of the heart with 25 ml of control solution complemented with 1 mg/ml type II collagenase (Worthington, Switzerland) and 50 μM CaCl2 at 37°C. The digested ventricles were placed on a petri dish containing dissociation solution (control solution plus 100 μM CaCl2 at 37°C) and gently dissociated with the help of forceps to liberate individual cardiomyocytes. The suspension of cells and ventricular tissues was filtered through a 100-μm nylon mesh to recover the isolated cardiomyocytes, which were subsequently washed in 10 ml of new dissociation solution. Phase-contrast photographs of the sedimented cardiomyocytes were taken using an inverted microscope (40× objective). The maximal cardiomyocyte width and length were measured using ImageJ software.
Statistical analysis.
All values in the figures and text are presented as means and standard errors of the mean (SEM). Paired data were analyzed by Student's t test. One-way analysis of variance (ANOVA) followed by Tukey post hoc tests with Bonferroni's corrections was used for multiple comparisons. P values of <0.05 were considered statistically significant.
RESULTS
AKAP-Lbc assembles a PKNα-based signaling complex controlling p38 activation in cardiomyocytes.
Several lines of evidence indicate that activated p38 is strongly implicated in pathological cardiac remodeling (14, 15, 17–19). However, it is currently poorly understood how signals controlling the activity of this key kinase are integrated and coordinated within cardiac cells to generate specific stress responses.
Previous experiments performed in HEK-293 cells indicate that AKAP-Lbc can coordinate a p38-activating complex. To initially assess whether such a complex is also assembled in cardiomyocytes, lysates from rat NVMs were submitted to immunoprecipitation using antibodies against AKAP-Lbc or control rabbit IgGs. Western blot analysis of the coimmunoprecipitated proteins revealed the presence of PKNα, MLTK, MKK3, and p38α, suggesting that p38α and its upstream activating kinases form a complex with AKAP-Lbc in cardiomyocytes (Fig. 1A and B).
Fig 1.

AKAP-Lbc assembles a PKNα-based p38 activation complex in cardiomyocytes. (A) Rat NVM extracts were subjected to immunoprecipitation with either nonimmune IgGs or affinity-purified anti-AKAP-Lbc antibodies. Proteins in the lysates and immunoprecipitates were identified by immunoblotting using antibodies against PKNα, MLTK, MKK3, and p38α and AKAP-Lbc, as indicated. (B) Rat NVMs were infected with control lentiviruses or lentiviruses encoding the Flag-AKAP-Lbc-1570-1764 competitor fragment (Comp.). AKAP-Lbc was immunoprecipitated from infected NVM lysates as indicated in panel A. The presence of associated kinases was detected by immunoblotting using specific antibodies, as indicated. (C) Rat NVMs infected as indicated in panel B were serum starved and subsequently treated or not for 24 h with 10−4 M PE. The amounts of total and phosphorylated p38, ERK1/2, and JNK, as well as the expression of Flag-AKAP-Lbc-1570-1764, in the cell lysates were determined by immunoblotting using specific antibodies, as indicated. (D) Quantitative analysis of phosphorylated p38 was performed by densitometry. The amount of phospho-p38 was normalized to the total amount of p38. The results are presented as means and SEM of four independent experiments. Statistical differences were analyzed using the Student t test. *, P < 0.05 versus control cardiomyocytes treated with PE.
AKAP-Lbc recruits PKNα and its associated kinases through a binding site located in its N-terminal regulatory region (13). To determine whether this AKAP-Lbc domain can be used as a competitive inhibitor of the interaction between the endogenous anchoring protein and the PKNα-based p38-activating complex, rat NVMs were infected with control lentiviruses or lentiviruses encoding the Flag-tagged PKNα binding fragment of AKAP-Lbc (residues 1570 to 1764), and the interaction between AKAP-Lbc and the various kinases was assessed by coimmunoprecipitation. Our results indicate that the overexpressed AKAP-Lbc competitor fragment (Fig. 1B, Comp.) strongly reduces the ability of endogenous PKNα, MLTK, MKK3, and p38α to interact with the anchoring protein, suggesting that it functions as an efficient competitive inhibitor (Fig. 1B). In line with this notion, we could show that disruption of the AKAP-Lbc/p38 activation complex in cardiomyocytes selectively inhibits p38 activation induced by stress signals, as shown by the fact that overexpression of the competitor in rat NVMs inhibits PE-induced p38 phosphorylation by 75% (Fig. 1C, upper gel, and D) without affecting PE-mediated phosphorylation of ERK1/2 and JKN (Fig. 1C and D). The implication of AKAP-Lbc in PE-mediated p38 activation was confirmed by gene-silencing experiments showing that knockdown of AKAP-Lbc expression in rat NVMs reduces the ability of PE to promote p38 phosphorylation by about 80% (Fig. 2A, upper gel, and B). Altogether, these findings suggest that the AKAP-Lbc competitor acts as a selective blocker of AKAP-Lbc-dependent activation of p38 in cardiomyocytes and could represent a valuable tool to investigate the role of the AKAP-Lbc/p38 activation complex in the heart.
Fig 2.
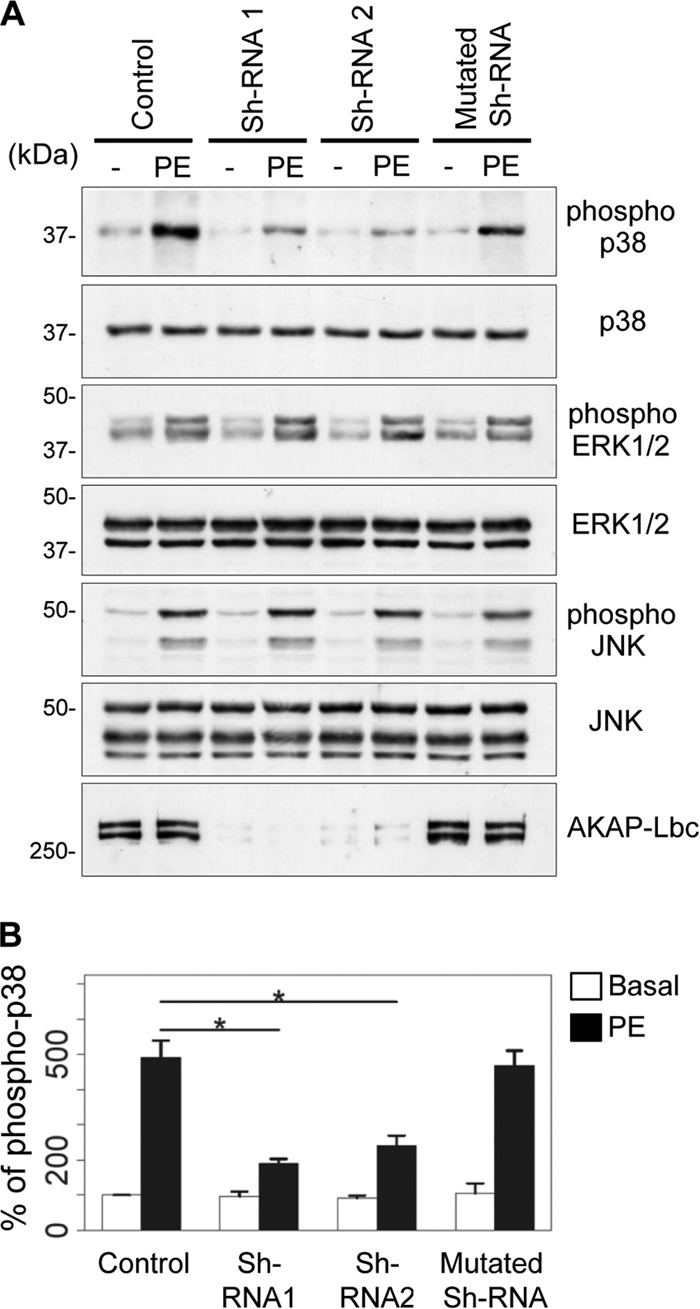
Silencing of AKAP-Lbc impairs p38 phosphorylation in response to α1-adrenergic receptor (α1-AR) stimulation. (A) Rat NVMs were infected with empty lentiviruses (Control), lentiviruses carrying two independent AKAP-Lbc-specific shRNAs (shRNA1 and shRNA2), or lentiviruses carrying a mutated shRNA. Seventy-two hours after infection, cardiomyocytes were serum starved for an additional 24 h and subsequently incubated for 15 min with or without 10−4 M PE. The amounts of total and phosphorylated p38, ERK1/2, and JNK in cell lysates were determined by immunoblotting using specific antibodies, as indicated. Silencing of AKAP-Lbc in rat NVMs was confirmed by Western blotting using an AKAP-Lbc-specific antibody. (B) Quantitative analysis of phosphorylated p38 was performed by densitometry. The amount of phospho-p38 was normalized to the total amount of p38. The results are presented as the means and SEM of four independent experiments. Statistical significance was analyzed using an ANOVA test, followed by Tukey posttests with Bonferroni corrections. *, P < 0.05 compared with the amount of phospho-p38 measured in nonstimulated control cardiomyocytes (Control).
Generation of a mouse model expressing the AKAP-Lbc competitor in cardiomyocytes.
To investigate the role of the AKAP-Lbc/p38 signaling complex in the heart, we generated a transgenic-mouse model overexpressing the competitor fragment of AKAP-Lbc in cardiomyocytes. The transgene was composed of the cDNA encoding the Flag-tagged AKAP-Lbc-1570-1764 fragment expressed under the control of the cardiac tissue-specific mouse α-MHC gene promoter (16). Rabbit β-globin IVS2 intronic sequences, encoding a polyadenine tail, were added at the end of the transgene to stabilize the mRNA (Fig. 3A). The purified transgene was microinjected into the pronuclei of fertilized eggs from C57BL6/DBA mice, which were subsequently backcrossed to a C57BL6/N background. Real-time PCR analysis revealed that approximately 20 copies of the transgene integrated in the genome of the transgenic mice (Fig. 3B), resulting in a 4,000-fold overexpression of the AKAP-Lbc-1570-1764 fragment mRNA in heart tissues compared to wild-type mice (Fig. 3C). Expression of the competitor fragment (Comp.) in cardiac lysates was also visualized by Western blotting using anti-Flag antibodies (Fig. 3D). The transgenic mice, were born in normal Mendelian ratios, developed to adulthood, were fertile, exhibited normal life spans, and displayed normal global cardiac structure and function (Table 1). This suggest that cardiac overexpression of the competitor does not affect heart function at baseline.
Fig 3.

Cardiac tissue-specific overexpression of the AKAP-Lbc competitor fragment inhibits p38 activation induced by pressure overload. (A) Diagram of the transgene used to drive cardiac tissue-specific overexpression of the AKAP-Lbc-1570-1764 fragment in mice. (B) Quantitative real-time PCR analysis of the transgene copy numbers integrated in the genomes of two transgenic mice. (C) Quantitative real-time PCR analysis of the AKAP-Lbc-1570-1764 fragment mRNA expression in WT and transgenic (Comp.) mouse hearts. (D) Western blot analysis of the expression of the Flag-AKAP-Lbc-1570-1764 fragment (Comp.) in heart lysates from WT and transgenic mice using anti-Flag antibodies (upper gel). Actin was used as a loading control (bottom gel). (E) The Flag-AKAP-Lbc-1570-1764 fragment (Comp.) was immunoprecipitated (IP) from transgenic mouse heart lysates using anti-Flag antibodies. Immunoprecipitations from wild-type heart lysates were performed in parallel as a control. Proteins in the lysates and immunoprecipitates were identified by immunoblotting using antibodies against PKNα, MLTK, MKK3, and p38α and the Flag tag. (F) Lysates from WT and transgenic (Comp.) mice were subjected to immunoprecipitation with either nonimmune IgGs or affinity-purified anti-AKAP-Lbc antibodies. Proteins in the lysates and immunoprecipitates were identified by immunoblotting using specific antibodies, as indicated. (G and H) Lysates from wild-type hearts were subjected to immunoprecipitation with either nonimmune IgGs or affinity-purified anti-AKAP-Lbc antibodies. p38β and p38γ in the lysates and immunoprecipitates were identified by immunoblotting using specific antibodies, as indicated. (I) Wild-type mice were subjected to 0 min, 30 min, 2 h, or 6 h of TAC. Heart lysates from the different time points were loaded in SDS-PAGE gels and analyzed by Western blotting. The amounts of total and phosphorylated p38 in cell lysates were determined by immunoblotting using specific antibodies, as indicated. (J) Transgenic mice (Comp.) and their WT littermates were subjected to TAC or a sham operation for a period of 6 h. The amounts of total and phosphorylated p38, as well as the expression of the Flag-AKAP-Lbc-1570-1764 fragment in heart lysates, were determined by immunoblotting using specific antibodies, as indicated. (K) Quantitative analysis of phosphorylated p38 was performed by densitometry. The amount of phospho-p38 was normalized to the total amount of p38. Values are presented as means and SEM. Four mice were analyzed for each group. Statistical differences were analyzed using the Student t test. *, P < 0.05 versus WT-TAC.
Table 1.
Physiological and echocardiographic parameters of WT and AKAP-Lbc competitor-overexpressing mice at baseline
| Parametera | Valueb |
|
|---|---|---|
| WT (n = 56) | AKAP-Lbc competitor overexpressing (n = 45) | |
| Genotype (%) | 52.43 | 47.57 |
| Body wt (g) | 28.19 ± 0.30 | 28.12 ± 0.28 |
| Heart rate (beats/min) | 485 ± 5.06 | 484.1 ± 7.30 |
| Cardiac wall thickness (mm) | ||
| Intraventricular septum; diastole | 0.699 ± 0.005 | 0.696 ± 0.006 |
| LV free wall; diastole | 0.684 ± 0.006 | 0.673 ± 0.007 |
| LV internal diameter; diastole | 4.17 ± 0.05 | 4.29 ± 0.06 |
| LV Vol (μl); diastole | 78.11 ± 2.36 | 83.46 ± 2.73 |
| Cardiac function (%) | ||
| Fractional shortening | 27.96 ± 1.11 | 26.34 ± 1.12 |
| Ejection fraction | 53.58 ± 1.64 | 51.13 ± 1.73 |
LV, left ventricular; HW/BW, heart weight to body weight ratio measured by transthoracic echocardiography.
Values are presented as mean ± SEM.
To determine whether the AKAP-Lbc competitor fragment can sequester PKNα, as well as its interacting kinases, in the heart, the Flag-tagged competitor was immunoprecipitated from heart lysates of transgenic mice using anti-Flag antibodies, and associated kinases were detected by Western blotting. Our results indicate that PKNα, MLTK, MKK3, and p38 are recruited by the competitor (Fig. 3E). Importantly, the overexpressed AKAP-Lbc competitor fragment (Comp.) impaired the ability of p38α and its upstream kinases to coimmunoprecipitate with the anchoring protein from heart tissues, thus confirming the notion that it can efficiently disrupt the cardiac AKAP-Lbc/p38 complex (Fig. 3F). We could not detect the presence of p38β or p38γ in AKAP-Lbc immunoprecipitates, suggesting that cardiac AKAP-Lbc preferentially interacts with p38α (Fig. 3G and H).
The AKAP-Lbc/p38 activation complex mediates pressure overload-induced p38 activation in the heart.
It is currently poorly understood how stress signals controlling the activity of p38 are integrated and coordinated within cardiac cells to generate specific responses. To determine whether the AKAP-Lbc/p38 complex is involved in the coordination of stress-induced p38 activation in the heart, we subjected 12-week-old male mice to TAC. Time course analysis revealed that maximal p38 phosphorylation is already reached at 6 h of TAC, suggesting that p38-mediated signaling is rapidly activated after constriction (Fig. 3I, upper gel). Interestingly, overexpression of the AKAP-Lbc competitor strongly reduces TAC-induced p38 phosphorylation, suggesting that the AKAP-Lbc/p38 complex mediates the activation of the kinase in response to pressure overload (Fig. 3J, upper gel, and K). Control experiments revealed that overexpression of the AKAP-Lbc competitor did not affect TAC-induced phosphorylation of additional prohypertrophic kinases, including ERK, JNK, PKD, and Akt, suggesting that the competitor fragment might specifically inhibit AKAP-Lbc-mediated p38 activation (Fig. 4A). To determine whether the AKAP-Lbc/p38 activation complex also participates in the activation of p38 induced by stimuli that do not regulate hypertrophy, we determined the impact of overexpressing the competitor fragment on the ability of doxorubicin, a cardiotoxic drug, to promote cardiac p38 activation. Transgenic and wild-type mice were intraperitoneally injected with PBS or 20 mg/kg doxorubicin (Fig. 4B, DOX) and sacrificed 5 days later. Our results indicate that overexpression of the AKAP-Lbc competitor does not affect doxorubicin-induced p38 phosphorylation, suggesting that the AKAP-Lbc/p38 complex does not participate in this pathway (Fig. 4B, upper gel, and C).
Fig 4.

Selectivity of the inhibitory effect of the AKAP-Lbc competitor fragment. (A) Transgenic mice overexpressing the competitor fragment of AKAP-Lbc (Comp.) and their WT littermates were subjected to TAC or a sham operation. The amounts of total and phosphorylated ERK1/2 and JNK, PKD, and Akt in heart lysates were determined by immunoblotting using specific antibodies, as indicated. (B) Cardiac overexpression of the AKAP-Lbc competitor fragment does not affect doxorubicin-induced p38 activation in the heart. Transgenic (Comp.) and WT mice were injected intraperitoneally with PBS or 20 mg/kg doxorubicin (DOX). The mice were sacrificed 5 days later, and the hearts were harvested. The amounts of total and phosphorylated p38, as well as the expression of the Flag-AKAP-Lbc-1570-1764 fragment in heart lysates, were determined by immunoblotting using specific antibodies, as indicated. (C) Quantitative analysis of phosphorylated p38 was performed by densitometry. The amount of phospho-p38 was normalized to the total amount of p38. Four mice were analyzed for each group. Values are presented as means and SEM. Statistical differences were analyzed using the Student t test. No statistically significant differences were found.
Disruption of the AKAP-Lbc/p38 signaling complex leads to early dilated cardiomyopathy in response to pressure overload.
To investigate whether the inhibition of the AKAP-Lbc/p38 signaling pathway in cardiomyocytes affects the cardiac adaptive response to pressure overload, we subjected transgenic mice overexpressing the AKAP-Lbc competitor and their wild-type littermates to 2 weeks of transverse aortic constriction or a sham operation. Histological heart sections and transthoracic M-mode echocardiography of WT mice that had undergone a TAC operation showed the expected adaptive response of the myocardium to pressure overload, characterized by an LVPW and IVS significantly thicker than values measured at baseline and in the group that had undergone a sham operation (Fig. 5 and 6A, B, and E and Table 1). At the same time, end-systolic and end-diastolic LVID and LV Vol were slightly reduced in WT mice after TAC (Fig. 6C to E). In contrast, mice overexpressing the competitor fragment of AKAP-Lbc that were subjected to 2 weeks of pressure overload had thinner cardiac walls (IVS and LVPW) and larger left ventricular internal diameter and volume than WT animals after TAC (Fig. 5 and 6A to E). In addition, transgenic mice that had undergone a TAC operation exhibited compromised cardiac activity, as indicated by significantly reduced FS and EF compared with WT mice (Fig. 6E to G). Altogether, these findings suggest that transgenic mice develop early dilated cardiomyopathy in response to TAC.
Fig 5.

Mice overexpressing the AKAP-Lbc competitor fragment in cardiomyocytes develop dilated cardiomyopathy in response to pressure overload. Shown are Masson's Trichrome-stained transversal histological sections of WT and transgenic (Competitor) mouse hearts. For each group, two sequential heart sections (A and B) from two mouse hearts (1 and 2) are shown.
Fig 6.

Disruption of the AKAP-Lbc/p38 complex in cardiomyocytes promotes cardiac dysfunction. (A to D) Echocardiographic parameters in transgenic mice overexpressing the AKAP-Lbc-1570-1764 fragment (Comp.) and their WT littermates after 2 weeks of TAC or a sham operation. Shown are end-diastolic (IVS;d) and end-systolic (IVS;s) intraventricular septum thickness (A), end-diastolic (LVPW;d) and end-systolic (LVPW;s) left ventricular posterior wall thickness (B), end-diastolic (LVID;d) and end-systolic (LVID;s) left ventricular internal diameter (C), and end-diastolic (LV Vol;d) and end-systolic (LV Vol;s) left ventricular volume (D). (E) Transthoracic M-mode echocardiography tracings from wild-type and transgenic mice 2 weeks after surgery (arrows indicate LIVD;s and LIVD;d). (F and G) FS and EF measured by transthoracic echocardiography. WT-sham, n = 14; WT-TAC, n = 20; competitor-sham, n = 10; and competitor-TAC, n = 15. Values are presented as means and SEM. Statistical differences were analyzed using the Student t test. *, P < 0.05 versus WT-TAC; **, P < 0.01 versus WT-TAC; ***, P < 0.001 versus WT-TAC.
Disruption of the AKAP-Lbc/p38 complex in cardiomyocytes enhances pressure overload-induced apoptosis.
To determine the cause of early cardiac dysfunction induced by pressure overload in mice overexpressing the AKAP-Lbc competitor fragment, we evaluated apoptosis and interstitial fibrosis on heart sections from WT and transgenic mice subjected to 2 weeks of TAC or a sham operation. Our results indicate that TAC-induced cellular apoptosis, assessed by the number of TUNEL-positive nuclei per heart section, was enhanced 2-fold in transgenic mice compared to WT animals (WT-TAC, 0.16% ± 0.014%; competitor-TAC, 0.22% ± 0.026%) (Fig. 7A and B). In contrast, quantification of the fibrotic area in heart sections stained with Masson's Trichrome revealed no significant differences in the fibrotic response of the heart to pressure overload between transgenic and wild-type animals (WT-TAC, 3.04% ± 0.77%; competitor-TAC, 3.49% ± 0.61%; P = 0.6) (Fig. 7C). In line with this observation, the stimulatory effect of TAC on myocardial collagen I and III mRNA expression was enhanced only marginally in transgenic mice compared to their WT littermates, as assessed by real-time PCR analysis (Fig. 7D). Altogether, these findings indicate that the disruption of the AKAP-Lbc/p38 complex in cardiomyocytes enhances cardiac apoptosis but has a modest or no effect on cardiac fibrosis.
Fig 7.

Disruption of the AKAP-Lbc/p38 complex in cardiomyocytes enhances pressure overload-induced apoptosis. Transgenic (Comp.) and WT mice were subjected to 2 weeks of TAC or a sham operation. Histological analyses were done on transversal heart sections. (A) Representative TUNEL and DAPI stainings of transversal heart sections. (B) Quantification of apoptosis. The number of TUNEL-positive nuclei was normalized to the total number of nuclei per heart section. The data from two different heart sections were averaged for each mouse. WT-sham, n = 5; WT-TAC, n = 6; competitor-sham, n = 5; competitor-TAC, n = 8; P < 0.05 versus WT-TAC. (C) Quantitative assessment of fibrosis. Heart sections from the indicated groups of animals were stained with Masson's Trichrome. WT-sham, n = 6; WT-TAC, n = 7; competitor-sham, n = 5; competitor-TAC, n = 9. (D) The relative mRNA expression of collagen I and III in heart lysates from the indicated mouse groups was assessed by quantitative real-time PCR. Values were normalized to the expression of GAPDH and are presented as means and SEM. Statistical differences were analyzed using the Student t test. No statistically significant differences were found.
Disruption of the AKAP-Lbc/p38 signaling complex inhibits pressure overload-induced cardiomyocyte hypertrophy.
Cardiomyocyte hypertrophy is an early compensatory response to cardiac stress or insult aimed at normalizing wall tension and at maintaining normal cardiac output. An impaired ability to undergo hypertrophy could therefore result in increased cardiac stress that might negatively impact heart function. We therefore determined whether the early appearance of functional alterations in stressed transgenic hearts could be related to an inability of ventricular cardiomyocytes to sustain an increase in cell size, as assessed by measuring the cardiomyocyte cross-sectional area on heart sections immunostained against laminin or stained with hematoxylin and eosin (Fig. 8A and B). TAC increased the cardiomyocyte cross-sectional area in wild-type mice by 61% (Fig. 8C). In contrast, this hypertrophic effect was reduced by 2-fold in transgenic mice overexpressing the AKAP-Lbc competitor (Fig. 8C).
Fig 8.

Disruption of the AKAP-Lbc/p38 signaling complex inhibits cardiomyocyte hypertrophy in response to pressure overload. Transgenic mice overexpressing the competitor fragment of AKAP-Lbc (Comp.) and their WT littermates were subjected to 2 weeks of TAC or a sham operation. (A and B) Antilaminin (A) and hematoxylin and eosin (B) stainings of transversal heart sections from the different groups of mice. (C) Quantification of cardiomyocyte cross-sectional areas from the indicated groups (WT-sham, n = 6; WT-TAC, n = 7; competitor-sham, n = 5; competitor-TAC, n = 9; an average of 350 cells were analyzed per mouse). (D) Representative cardiomyocytes (phase-contrast images) isolated from WT and transgenic (Comp.) animals subjected to 2 weeks of aortic constriction. (E) Quantitative analysis of the width, length, and area (width times length) of cardiomyocytes from hearts of the indicated groups of mice (WT-sham, n = 3; WT-TAC, n = 6; competitor-sham, n = 3; competitor-TAC, n = 3; an average of 500 cells were analyzed per mouse). Values are presented as means and SEM. Statistical differences were analyzed using the Student t test. **, P < 0.01 versus WT-TAC; ***, P < 0.0001 versus WT-TAC.
To confirm these findings, we isolated adult cardiomyocytes from hearts of WT and TG mice 2 weeks after TAC or a sham operation (Fig. 8D). Remarkably, the increase in cell width and cell surface area induced by TAC was strongly reduced in cardiomyocytes from transgenic mice compared to cardiomyocytes from WT mice (Fig. 8E, left and right). Cardiomyocytes overexpressing the competitor fragment of AKAP-Lbc were slightly longer than control cardiomyocytes at baseline (WT-sham, 124.65 ± 1.41 μm; competitor-sham, 131.79 ± 1.70 μm; P = 0.0013) but did not show any further increase in cell length after TAC (Fig. 8E, middle).
In line with these findings, transfection of a green fluorescent protein (GFP)-tagged competitor fragment of AKAP-Lbc in rat NVMs significantly decreased the growth response of cardiomyocytes to PE treatment compared with control cardiomyocytes expressing only GFP (Fig. 9). Altogether, these findings suggest that the AKAP-Lbc/p38 complex is crucially involved in the transduction of signals controlling adaptive concentric cardiomyocyte growth in response to cardiac stress.
Fig 9.
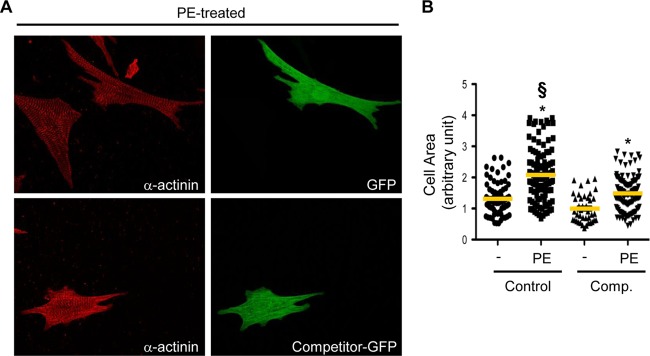
The AKAP-Lbc competitor fragment impairs the hypertrophic response of rat NVMs induced by α1-AR stimulation. (A) Rat NVMs were transfected with GFP (control) or with the GFP-tagged AKAP-Lbc competitor fragment (Competitor-GFP). Seventy-two hours after transfection, cardiomyocytes were serum starved for 24 h and incubated for an additional 24 h in the absence or presence of 10−4 M PE. The cells were then fixed, permeabilized, and incubated with anti-α-actinin (red) monoclonal antibodies, as well as rhodamine-conjugated anti-mouse secondary antibodies. GFP expression was visualized directly by fluorescent excitation at 490 nm. (B) Mean cell surface areas of cardiomyocytes transfected and treated or not with PE. The cell surface area was determined on a total of 120 GFP-positive cardiomyocytes derived from four independent experiments by using Image J software. Statistical significance was analyzed using an ANOVA test followed by Tukey posttests with Bonferroni corrections. *, P < 0.05 compared with the cell surface area measured in nonstimulated cardiomyocytes transfected with GFP (Control). §, P < 0.05 compared with the cell surface area measured in PE-stimulated cardiomyocytes transfected with GFP (Control).
The AKAP-Lbc/p38 signaling complex regulates mTOR signaling.
Hypertrophic cardiomyocyte growth induced by stress stimuli is mainly driven by an increase in protein synthesis controlled by the mammalian target of rapamycin (mTOR) (20, 21). Based on this evidence, we raised the hypothesis that the AKAP-Lbc/p38 signaling complex might mediate cardiomyocyte hypertrophy through the regulation of the mTOR signaling pathway. We therefore assessed the phosphorylation of the translation repressor protein 4E-BP1 and of the S6 ribosomal protein (S6rp), two direct downstream targets of mTOR (20), in cardiac protein extracts from WT and transgenic mice that had undergone a sham or TAC operation.
Interestingly, the increase in 4E-BP1 and S6rp phosphorylation induced by 2 weeks of TAC was significantly reduced in transgenic mice expressing the competitor fragment of AKAP-Lbc compared to their WT littermates, as assessed by Western blotting using antibodies recognizing phosphothreonine 70 of 4E-BP1 and phosphoserines 240 and 244 of S6rp, respectively (Fig. 10A, D, and G). While the total protein levels of 4E-BP1 were increased after TAC in both wild-type and transgenic mice (Fig. 10F), the intensities of the two high-molecular-weight β- and γ-bands were reduced (Fig. 10E) and that of the lower-molecular-weight α-band was increased in transgenic mice subjected to TAC, indicating reduced 4E-BP1 phosphorylation. The total protein levels of mTOR were slightly increased after TAC in both WT and transgenic mice (Fig. 10A and B), whereas expression of S6 kinase (P70-S6K1) was constant in all the groups (Fig. 10A and C). Collectively, these findings suggest that the AKAP-Lbc/p38 activation complex controls pressure overload-induced activation of the mTOR-regulated growth pathway in the heart.
Fig 10.

The AKAP-Lbc/p38 complex mediates activation of mTOR signaling in response to TAC. (A) Transgenic mice overexpressing the competitor fragment of AKAP-Lbc (Comp.) and their WT littermates were subjected to 2 weeks of TAC or a sham operation. Heart lysates from the different groups of mice were loaded on SDS-PAGE gels and analyzed by Western blotting. The paired lanes represent samples from two different animals. Phospho-4E-BP1 and phospho-S6rp were detected using antibodies recognizing phospho-threonine 70 of 4E-BP1 and phospho-serine 240 and 244 of S6rp, respectively. The amounts of mTOR, P70-S6K1, 4EBP1, S6rp, and Flag-AKAP-Lbc-1570-1764 were detected using specific antibodies, as indicated. (B to G) Quantitative analysis of mTOR (B), P70-S6K1 (C), phosphorylated 4E-BP1 (D), 4EBP1-β (E), total 4EBP1 (F), and S6rp (G) was performed by densitometry. *, P < 0.05 versus WT-TAC. (H) Wild-type mice administered PBS or SB203580 (10 mg/kg) were subsequently subjected to TAC or a sham operation. Heart lysates from the different groups of mice were loaded in SDS-PAGE gels and analyzed by Western blotting. Phospho-S6rp and S6rp were detected using specific antibodies, as indicated. (I) Quantitative analysis of S6rp was performed by densitometry. Four mice were analyzed for each group. The results are presented as means and SEM. Statistical differences were analyzed using the Student t test. *, P < 0.05 versus PBS-TAC.
To provide evidence for a direct link between TAC-induced p38 activation and increased mTOR signaling, wild-type mice were administered PBS or the p38 inhibitor SB203580 (10 mg/kg) and subsequently subjected to a sham operation or TAC. We could show that p38 inhibition significantly reduced S6rp phosphorylation induced by TAC (Fig. 10H and I), suggesting that cardiac p38 activation can contribute at least to downstream mTOR signaling.
Fetal genes are induced after aortic constriction in transgenic mice overexpressing the AKAP-Lbc competitor fragment.
Cardiomyocyte hypertrophy is usually paralleled by a concomitant reactivation of a fetal gene program in which the expression of α-MHC is reduced and that of β-MHC, α-skeletal actin, and ANP is increased (22). To address the impact of inhibiting the formation of the AKAP-Lbc/p38 activation complex on TAC-induced fetal gene expression, α-MHC, β-MHC, α-skeletal actin, ANP, and BNP mRNA levels were assessed by real-time PCR in ventricular tissues of WT and transgenic mice that were subjected to TAC or sham operations (Fig. 11A to D). The increase in β-MHC expression induced by TAC was significantly greater in transgenic mice than in WT mice (Fig. 11A, bottom). This caused the β-MHC/α-MHC ratio to increase by 2-fold (Fig. 11B). A similar trend was observed for ANP and BNP (Fig. 11C). Western blot analysis confirmed the higher expression of β-MHC and ANP in heart lysates from transgenic mice subjected to TAC (Fig. 11E to G). In contrast, α-skeletal actin was induced to similar extents in WT and transgenic mice after TAC (Fig. 11D). The observation that TAC induces a stronger upregulation of some fetal genes in transgenic mice than in WT mice indicates that disruption of the AKAP-Lbc/p38 signaling complex in cardiomyocytes exacerbates cardiac stress induced by pressure overload.
Fig 11.

Disruption of the AKAP-Lbc/p38 signaling complex enhances the α- to β-MHC isoform switch induced by pressure overload. (A to D) Quantitative real-time PCR analysis of the expression of α-MHC and β-MHC (A), ANP and BNP (C), and α-skeletal actin (D) 2 weeks after TAC or sham surgery. The β-MHC/α-MHC ratio is shown in panel B. WT-sham, n = 14; WT-TAC, n = 14; competitor-sham. n = 6; competitor-TAC, n = 9. (E) Heart lysates from the different groups of mice were loaded on SDS-PAGE gels and analyzed by Western blotting. β-MHC and ANP were detected using specific antibodies, as indicated. (F and G) Quantitative analysis of β-MHC (F) and ANP (G) was performed by densitometry. Values are presented as means and SEM. Statistical differences were analyzed using the Student t test. *, P < 0.05 versus WT-TAC; **, P < 0.01 versus WT-TAC.
DISCUSSION
During the last few years, several studies using primary cultures of rat NVMs as a model system have shown that AKAPs organize signaling complexes that regulate hypertrophic signaling pathways (9–12, 23–27). While these findings contributed importantly to our understanding of how this family of scaffolding proteins coordinates hypertrophic signals at the cellular level, it remains to be established whether AKAP-based signaling complexes can control cardiac hypertrophy in vivo.
In the present study, we show that the AKAP-Lbc/p38 activation complex contributes to the compensatory hypertrophic response induced by pressure overload (Fig. 12). Our findings indicate that AKAP-Lbc assembles a signaling module containing the RhoA effector PKNα and the kinases MLTK, MKK3, and p38, which controls cardiomyocyte growth through the activation of the mTOR signaling pathway (Fig. 12). Activated mTOR promotes the phosphorylation of 4E-BP1 and the activation of the p70S6K-S6 pathway, which in turn positively regulate protein synthesis (Fig. 12). Importantly, impairment of the adaptive hypertrophic response to pressure overload induced by disrupting the AKAP-Lbc/p38 activation complex is associated with cardiomyocyte apoptosis and early ventricular dilation and dysfunction. Overall, these findings provide the first evidence for the implication of A-kinases anchoring proteins in cardiac hypertrophy in vivo, and in particular, they highlight the importance of individual protein-protein interactions within AKAP-based signaling complexes in mediating transduction events associated with cardiac remodeling.
Fig 12.
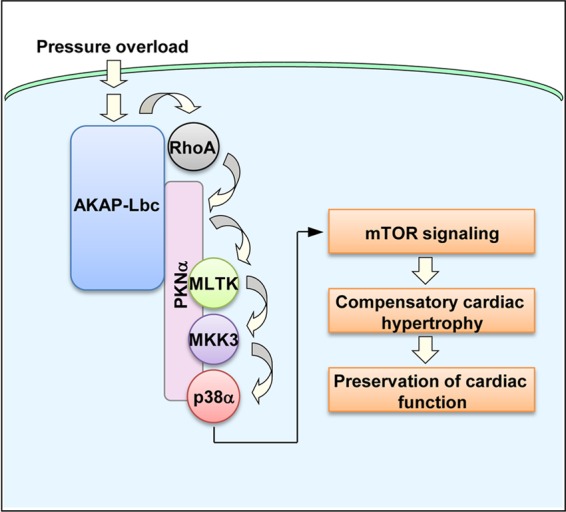
Model illustrating the role of the AKAP-Lbc/p38 activation complex in the regulation of compensatory cardiac hypertrophy. AKAP-Lbc assembles a signaling complex, which includes the scaffolding protein PKNα, as well as MLTK, MKK3, and p38α. In response to pressure overload, AKAP-Lbc promotes the formation of RhoA-GTP, which in turn induces the sequential activation of PKNα, MLTK, MKK3, and p38α within the AKAP-Lbc complex. This results in the activation of the mTOR signaling pathway that controls protein synthesis and promotes compensatory cardiac hypertrophy, which temporarily preserves the function of the stressed heart.
Recent studies indicate that AKAP-Lbc can recruit the scaffolding protein kinase suppressor of Ras (KSR-1) to form a signaling complex that efficiently relays signals from Raf, through MEK, and on to ERK1/2 (28). These findings suggest that AKAP-Lbc act as a scaffold that can coordinate the activation of multiple mitogen-activated protein kinase (MAPK) pathways. Our current results showing that ERK1/2 activation is not affected upon knockdown of AKAP-Lbc in cardiomyocytes is consistent with the fact that there is little or no KSR-1 in heart cells (28).
While previous findings indicate that PKNα exerts prohypertrophic effects in cardiomyocytes, the molecular pathways controlling this response were not identified (29). We now provide evidence that PKNα assembles a macromolecular signaling complex that controls p38 activation and cardiomyocyte growth in response to pressure overload. Our findings showing that disruption of the interaction between AKAP-Lbc and the PKNα-associated complex strongly compromises the ability of the heart to sustain compensatory hypertrophy provide the first demonstration of the functional importance of PKNα anchoring in vivo.
The conclusion that the AKAP-Lbc/p38 activation complex participates in the hypertrophic response induced by pressure overload is supported by cardiac wall thickness measured by echocardiography, cardiomyocyte cross-sectional area determined in histological sections, and cardiomyocyte dimensions assessed on isolated adult cardiomyocytes. These findings are in accordance with earlier in vitro data suggesting that silencing of AKAP-Lbc expression in rat NVMs inhibits the hypertrophic response induced by different stress stimuli (9, 10).
Our current findings indicating that cardiac AKAP-Lbc associates preferentially with p38α but not with p38β and p38γ (Fig. 3G and H) are in line with our previous results obtained in HEK-293 cells (13). The role of p38α in cardiac hypertrophy has been controversial over the last 10 years. While several initial studies concluded that this stress kinase positively regulates cell growth in primary cultures of rat NVMs (17, 30, 31), subsequent studies using in vivo models in which myocardial p38α was chronically activated or inhibited concluded that the kinase does not induce hypertrophy (18, 19, 32–34). However, this paradigm has been challenged by recent studies showing that acute (inducible) activation of p38α in the adult heart promotes cardiomyocyte growth (14, 15). Our current results showing that disruption of the AKAP-Lbc/p38 activation complex inhibits pressure overload-induced hypertrophy support this view. The discrepancy between studies based on chronic versus acute manipulation of p38α signaling could arise from the fact that chronic alteration of p38 expression and/or activation could alter p38-mediated regulation of cardiomyocyte proliferation and/or differentiation at earlier developmental stages (35) and cause secondary compensatory changes.
Our observation that the AKAP-Lbc competitor fragment inhibits pressure overload-induced p38 activation (Fig. 3) without affecting basal p38 activity suggests that the AKAP-Lbc/p38 activation complex is mobilized only in response to cardiac stress and does not contribute to p38 signaling at baseline. These findings are in line with the observation that cardiac AKAP-Lbc expression is strongly increased in response to aortic banding (Fig. 13) and are consistent with a role of AKAP-Lbc in mediating p38 activation and cardiomyocyte hypertrophy in response to abrupt rises in afterload.
Fig 13.

Cardiac AKAP-Lbc mRNA is upregulated in response to TAC. (A and B) Real-time PCR (A) and Western blot (B) analyses of AKAP-Lbc expression were performed on total RNA samples extracted from C57BL6 mouse hearts that were subjected to a sham operation or to 2 weeks of TAC. Five mice were analyzed for each group. (C) Quantitative analysis of AKAP-Lbc protein expression was performed by densitometry. Statistical differences were analyzed using the Student t test. Values are presented as means and SEM. *, P < 0.05 versus sham.
Our current findings indicate that disruption of AKAP-Lbc-mediated p38 signaling in cardiomyocytes reduces the activation of the mTOR pathway induced by aortic banding, as shown by an inhibition of 4E-BP1 and S6 phosphorylation (Fig. 10). Knowing that mTOR-mediated regulation of protein synthesis controls the hypertrophic response induced by pressure overload (20, 21), we conclude that the AKAP-Lbc-RhoA-p38 transduction pathway mediates cardiomyocyte growth through the modulation of mTOR signaling. In line with these results, recent findings indicate that RhoA and p38 can both mediate mTOR activation and promote cardiomyocyte hypertrophy (36). While the cardiac signaling pathway linking the AKAP-Lbc/p38 complex to mTOR activation remains to be identified, recent findings suggest the existence of a cardioprotective p38-mTOR pathway in the heart (37).
It is formally possible that, in addition to the mTOR signaling pathway identified in this study, other transduction cascades mediate cardiac hypertrophy downstream of the AKAP-Lbc/p38 complex. In this respect, recent data suggest that MAPK-activated protein kinase 2 (MK2), a direct effector of p38, can control hypertrophy and remodeling in the adult mouse heart, as shown by the fact that cardiac MK2 inactivation reduces the increase in left ventricular wall thickness and cardiomyocyte cross-sectional area induced by p38 (14).
Dissociation of the AKAP-Lbc/p38 activation complex results in a blunted hypertrophic response to pressure overload, which is associated with unfavorable cardiac remodeling characterized by increased apoptosis, upregulation of stress genes, left ventricular dilation, and reduced left ventricular contractile capacity (Fig. 5 and 6) but not with significantly increased cardiac fibrosis (Fig. 7). Increased myocardial apoptosis and alterations in the relative abundances of α-MHC and β-MHC in the sarcomere might both represent leading causes of the cardiac dysfunction observed in transgenic mice subjected to TAC (Fig. 6), since they can reduce the myocardial contractile mass and affect the cross-bridge cycling kinetics within sarcomeres, respectively (38).
Remarkably, a similar association between impaired compensatory hypertrophy and exacerbated maladaptive remodeling has been recently described in mice with cardiac ablation of raptor (20, 21). It appears therefore that inhibiting mTOR signaling in cardiomyocytes significantly affects adaptive hypertrophy in response to rapid rises in afterload. This can significantly increase cardiac stress and cause early ventricular dysfunctions that eventually compromise the ability of the heart to sustain normal cardiac output.
In conclusion, our current study provides the first in vivo evidence for the role of AKAP signaling complexes in cardiac remodeling. In particular, it identifies the AKAP-Lbc/p38 activation complex as a crucial mediator of the cardiac hypertrophic response to hemodynamic overload. It also indicates that cardiac delivery of molecular competitors targeting individual protein-protein interactions represents a valid strategy to dissect the functions of unique transduction complexes in the heart.
ACKNOWLEDGMENTS
We acknowledge Monique Nenniger-Tosato for excellent technical assistance in preparing primary cultures of rat NVMs, Susanna Cotecchia for helpful discussions, and Philippe Julien for helpful assistance in preparing figures.
This work was supported by grant 3100A0-138289 from the Swiss National Science Foundation (to D.D) and by grants from the Muschamp and Novartis Foundations (to D.D.). H.A. was supported by grant 310030B-135693 from the Swiss National Science Foundation.
Footnotes
Published ahead of print 28 May 2013
REFERENCES
- 1. Hill JA, Olson EN. 2008. Cardiac plasticity. N. Engl. J. Med. 358: 1370– 1380 [DOI] [PubMed] [Google Scholar]
- 2. Towbin JA, Bowles NE. 2002. The failing heart. Nature 415: 227– 233 [DOI] [PubMed] [Google Scholar]
- 3. Pawson CT, Scott JD. 2010. Signal integration through blending, bolstering and bifurcating of intracellular information. Nat. Struct. Mol. Biol. 17: 653– 658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scott JD, Dessauer CW, Tasken K. 2013. Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53: 187– 210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diviani D. 2008. Modulation of cardiac function by A-kinase anchoring proteins. Curr. Opin. Pharmacol. 8: 166– 173 [DOI] [PubMed] [Google Scholar]
- 6. Diviani D, Dodge-Kafka KL, Li J, Kapiloff MS. 2011. A-kinase anchoring proteins: scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol. 301: H1742– H1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carnegie GK, Burmeister BT. 2011. A-kinase anchoring proteins that regulate cardiac remodeling. J. Cardiovasc. Pharmacol. 58: 451– 458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diviani D, Soderling J, Scott JD. 2001. AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 276: 44247– 44257 [DOI] [PubMed] [Google Scholar]
- 9. Appert-Collin A, Cotecchia S, Nenniger-Tosato M, Pedrazzini T, Diviani D. 2007. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 104: 10140– 10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. 2008. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32: 169– 179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edwards HV, Scott JD, Baillie GS. 2012. The A-kinase-anchoring protein AKAP-Lbc facilitates cardioprotective PKA phosphorylation of HSP20 on Ser(16). Biochem. J. 446: 437– 443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. del Vescovo CD, Cotecchia S, Diviani D. 2013. AKAP-Lbc anchors IKKbeta to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol. Cell Biol. 33: 14– 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cariolato L, Cavin S, Diviani D. 2011. A-kinase anchoring protein (AKAP)-Lbc anchors a PKN-based signaling complex involved in alpha1-adrenergic receptor-induced p38 activation. J. Biol. Chem. 286: 7925– 7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Streicher JM, Ren S, Herschman H, Wang Y. 2010. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ. Res. 106: 1434– 1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marber MS, Rose B, Wang Y. 2011. The p38 mitogen-activated protein kinase pathway—a potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 51: 485– 490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palermo J, Gulick J, Colbert M, Fewell J, Robbins J. 1996. Transgenic remodeling of the contractile apparatus in the mammalian heart. Circ. Res. 78: 504– 509 [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. 1998. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 273: 2161– 2168 [DOI] [PubMed] [Google Scholar]
- 18. Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. 2001. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 98: 12283– 12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, Otsu K. 2004. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24: 10611– 10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Ruegg MA, Pedrazzini T, Brink M. 2011. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123: 1073– 1082 [DOI] [PubMed] [Google Scholar]
- 21. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G. 2010. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120: 2805– 2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frey N, Katus HA, Olson EN, Hill JA. 2004. Hypertrophy of the heart: a new therapeutic target? Circulation 109: 1580– 1589 [DOI] [PubMed] [Google Scholar]
- 23. Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. 2005. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J. Cell Sci. 118: 5637– 5646 [DOI] [PubMed] [Google Scholar]
- 24. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. 2005. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437: 574– 578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li J, Negro A, Lopez J, Bauman AL, Henson E, Dodge-Kafka K, Kapiloff MS. 2010. The mAKAPbeta scaffold regulates cardiac myocyte hypertrophy via recruitment of activated calcineurin. J. Mol. Cell. Cardiol. 48: 387– 394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. 2011. Phospholipase C epsilon scaffolds to mAKAP and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 286: 23012– 23021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abrenica B, AlShaaban M, Czubryt MP. 2009. The A-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 46: 674– 681 [DOI] [PubMed] [Google Scholar]
- 28. Smith FD, Langeberg LK, Cellurale C, Pawson T, Morrison DK, Davis RJ, Scott JD. 2010. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12: 1242– 1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takagi H, Hsu CP, Kajimoto K, Shao D, Yang Y, Maejima Y, Zhai P, Yehia G, Yamada C, Zablocki D, Sadoshima J. 2010. Activation of PKN mediates survival of cardiac myocytes in the heart during ischemia/reperfusion. Circ. Res. 107: 642– 649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. 1997. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J. Cell Biol. 139: 115– 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charron F, Tsimiklis G, Arcand M, Robitaille L, Liang Q, Molkentin JD, Meloche S, Nemer M. 2001. Tissue-specific GATA factors are transcriptional effectors of the small GTPase RhoA. Genes Dev. 15: 2702– 2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. 2003. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Invest. 111: 1475– 1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, Wang Y, Muslin AJ. 2003. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Invest. 111: 833– 841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. 2005. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J. Biol. Chem. 280: 669– 676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Engel FB. 2005. Cardiomyocyte proliferation: a platform for mammalian cardiac repair. Cell Cycle 4: 1360– 1363 [DOI] [PubMed] [Google Scholar]
- 36. Zeidan A, Hunter JC, Javadov S, Karmazyn M. 2011. mTOR mediates RhoA-dependent leptin-induced cardiomyocyte hypertrophy. Mol. Cell. Biochem. 352: 99– 108 [DOI] [PubMed] [Google Scholar]
- 37. Hernandez G, Lal H, Fidalgo M, Guerrero A, Zavilde J, Force T, Pombo CM. 2011. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 317: 2938– 2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frey N, Olson EN. 2003. Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65: 45– 79 [DOI] [PubMed] [Google Scholar]