

Abstract
NF-κB transcription factors are crucial regulators of inflammation, immunity, stress responses, and cell differentiation. Many studies have demonstrated that ubiquitination of IκB kinase γ (IKKγ), a regulatory subunit of IKK, is instrumental in the activation of IKK and NF-κB. We and others previously identified EGLN3, a member of a family of prolyl hydroxylases, as a negative regulator of the NF-κB pathway. Here we report that EGLN3, but not EGLN1 or -2, interacts with and inhibits K63-linked ubiquitination of IKKγ. The effect appears to be related to inhibition of IKKγ ubiquitination mediated by cIAP1 rather than to stimulation of IKKγ deubiquitination by the deubiquitinases A20 and CYLD (cylindromatosis). EGLN3 does not affect the protein levels of cIAP1 or its E2 ubiquitin-conjugating enzymes UbcH5 and Ubc13. EGLN3 hydroxylase activity is not responsible for its effect on IKKγ ubiquitination and NF-κB signaling. Instead, interaction with IKKγ is required for the ability of EGLN3 to inhibit IKKγ ubiquitination and IKK–NF-κB signaling. EGLN3 competes with cIAP1 for IKKγ binding, leading to inhibition of cIAP1-IKKγ interaction, IKKγ ubiquitination, and IKK–NF-κB signaling. This study provides novel insights into EGLN3 function and sheds new light on the regulation of IKKγ ubiquitination and NF-κB.
INTRODUCTION
EGLN3 (also known as PHD3, HPH1, and SM-20) belongs to the Caenorhabditis elegans gene egl-9 (EGLN) family of prolyl hydroxylases. All three mammalian EGLNs, EGLN1, -2, and -3, catalyze the hydroxylation of the α subunit of hypoxia-inducible factor (HIFα). This results in enhanced HIFα ubiquitination and proteasomal degradation and leads to decreased HIFα activity (1). The rat homolog of EGLN3 is a growth factor-responsive gene originally identified as SM-20 in smooth muscle cells (2). Upregulation of EGLN3 is associated with p53-induced growth arrest and apoptosis in RAS-transformed embryo fibroblasts (3), apoptosis in sympathetic neurons (4), and differentiation of C2C12 myoblasts (5). EGLN3 is also induced in human endothelial cells (6) and in human cancers, such as pancreatic cancer (7) and glioblastoma (8). Although EGLN3 can be induced in many physiologic and pathological settings, the biochemical or biologic function of EGLN3 remains largely to be determined.
We and others have recently reported that EGLN3 is a negative regulator of the NF-κB pathway (9–11). In unstimulated cells, NF-κB is sequestered in the cytoplasm in association with the IκB family of inhibitors. In most cases, degradation of IκB is a prerequisite for NF-κB activation, which is initiated upon phosphorylation by the activated IκB kinase (IKK) complex (12). The IKK complex consists of two catalytic subunits, IKK1 (IKKα) and IKK2 (IKKβ), and a regulatory subunit, IKKγ (also known as NF-κB essential modulator [NEMO]) (12). IKK can be activated by upstream kinases, such as transforming growth factor β-activated kinase 1 (TAK1) (12). Genetic studies have demonstrated that IKKγ is indispensable for the activation of the IKK signalsome and of NF-κB (13–15). Many studies have demonstrated that IKKγ ubiquitination is instrumental in the activation of IKK and NF-κB (16–26). We now report that EGLN3 interacts with and inhibits K63-linked ubiquitination of IKKγ, leading to inhibition of IKK–NF-κB activation. The inhibitory effect on IKKγ ubiquitination is primarily mediated by the carboxyl-terminal region of EGLN3 and does not require EGLN3 prolyl hydroxylase activity. These studies provide novel insights into the regulation of IKKγ and NF-κB and provide evidence for a new biochemical function of EGLN3 that is unrelated to its role as a prolyl hydroxylase.
MATERIALS AND METHODS
Materials.
Dimethyl oxalylglycine (DMOG) was from Frontier Scientific Inc. Smac mimetic (27) was a gift from X. Wang (University of Texas Southwestern Medical Center). Other materials, unless otherwise indicated, were purchased from Sigma.
Cell culture and transfection.
Human embryonic kidney 293 T (HEK293T) cells, HeLa cells, and COS-7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) as described previously (28). Cells were transfected by the use of Lipofectamine 2000 reagent (Invitrogen), following the manufacturer's instructions.
Expression constructs.
The plasmids expressing myc-ubiquitin (myc-Ub), His-Ub, FLAG-EGLN3 and its mutants, FLAG-EGLN1, FLAG-EGLN2, TRAF2 (tumor necrosis factor [TNF] receptor-associated factor 2), IKK1, IKK2, hemagglutinin (HA)-IKKγ, myc-IKKγ, FLAG-IKKγY308S, FLAG-IKKγF312A, myc-IKKγL326P, HA-cIAP1, FLAG-cIAP1 mutant, myc-X-linked inhibitor of apoptosis (myc-XIAP), myc-cIAP1, HA-optineurin, and FLAG-IKKγ truncation mutants (M1 to M5) have been described previously (5, 9, 18, 28–36). FLAG-OTUB1 and its mutant C91S were kindly provided by D. Durocher (37). The LUBAC expression plasmids were described previously (21).
Immunoblotting.
Cells were harvested in lysis buffer 1 (20 mM HEPES [pH 7.4], 120 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride) containing complete protease inhibitor mixture (Roche Applied Science) for 0.5 h at 4°C. The debris was removed by centrifugation at 16,000 × g for 20 min at 4°C. The soluble fractions were recovered. Cellular proteins were resolved on SDS-PAGE and electroblotted onto a polyvinylidene difluoride membrane (Bio-Rad) or a nitrocellulose membrane (Bio-Rad). Following blocking, the membrane was probed with an appropriate primary antibody and then incubated with a corresponding sheep anti-mouse IgG or donkey anti-rabbit IgG conjugated to horseradish peroxidase (Amersham Biosciences). The blots were developed by the ECL or ECL Plus method (Amersham Biosciences). Primary antibodies used in this study included the following: anti-α-tubulin and anti-FLAG (Sigma); anti-myc, anti-IκBα, anti-TRADD, anti-cIAP1, and antiubiquitin (Santa Cruz Biotechnology); anti-phospho-IκBα, anti-IKK2, anti-phospho-IKK1/2, anti-K63-linked ubiquitin, anti-Ubc13, and anti-UbcH5 (Cell Signaling Technology); anti-IKKγ (BD Biosciences and Santa Cruz Biotechnology); anti-COX2 (Cayman Chemical Company and Santa Cruz Biotechnology); anti-HA (Roche Applied Science); anti-HOIP (Abcam); and anti-EGLN3 (Novus Biologicals).
Immunoprecipitation.
Immunoprecipitation was performed as described previously (28). The precleared supernatants were incubated with anti-FLAG M2 affinity gel (Sigma) or anti-myc affinity gel (Sigma) or anti-HA affinity gel (Sigma) at 4°C for 4 h or overnight with constant agitation. The immunoprecipitated materials were analyzed by immunoblotting. For in vivo ubiquitination assays, cells were first lysed in lysis buffer 1 (including 1% SDS) and boiled for 10 min. The denatured lysates were then diluted with lysis buffer 1 (without SDS) followed by immunoprecipitation as described above.
RNA interference.
Smart-pool EGLN3 small interfering RNA (siRNA) and control nontargeting siRNAs were purchased from Dharmacon. CYLD (cylindromatosis) short hairpin RNA (shRNA) and A20 shRNA were described previously (38–40). SiRNAs were transfected into cells by the use of Lipofectamine 2000 reagent or Oligofectamine (Invitrogen) following the manufacturer's protocol.
Preparation of cell extracts and labeling with ubiquitin derivatives.
Deubiquitinase activity assays were conducted as described previously (41, 42). HEK293T cells were transfected with FLAG-tagged EGLN3 or OTUB1 or its catalytically inactive mutant. At 24 h after transfection, cells were lysed in the buffer containing 50 mM Tris (pH 7.6), 150 mM NaCl, 3 mM EDTA, and 0.5% NP-40. Cell extracts and ubiquitin derivatives (Enzo Life Sciences) were incubated at 37°C for 1 h in the buffer containing 50 mM Tris (pH 7.6), 50 mM NaCl, 10% glycerol, and 1 mM EDTA. Reactions were terminated by addition of Laemmli buffer and denatured followed by immunoblotting analysis with anti-FLAG.
In vitro kinase assays.
In vitro kinase assays were performed as described previously (9). Cells were transfected as described in the figure legends. Cell extracts were immunoprecipitated with IKKγ antibody. IKK activity was determined in the kinase buffer (9) supplemented with 1 μg of glutathione S-transferase (GST)–IκBα and 0.2 mM ATP. After 30 to 60 min of incubation at 30°C, the reaction was stopped and analyzed by immunoblotting with an antibody specific for phospho-IκBα.
Luciferase reporter assays.
Luciferase reporter assays were conducted as described previously (9). Cells were transfected as described in the figure legends. Luciferase expression was determined using a Dual-Luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer's protocol. The expression of firefly luciferase driven by the NF-κB-responsive element was used as a reporter. pRL-tk (renilla luciferase) was cotransfected to normalize for the transfection efficiency. Luciferase activity was expressed as a ratio of firefly luciferase activity to renilla luciferase activity. Normalized values are reported as the means ± standard deviations (SD) of the results of triplicate transfection. Student's t test for paired samples was used to determine statistical significance.
Expression and purification of GST-IKKγ fusion protein.
Overnight cultures of Escherichia coli BL21(DE3) pLysS (Novagen) transformed with parental or recombinant pGEX4T-2 plasmid containing IKKγ were diluted in LB medium containing ampicillin and incubated at 37°C with shaking to an A600 of 0.6 to 0.8. Isopropyl-d-thiogalactopyranoside (Amersham Biosciences) was then added to reach a final concentration of 1 mM. After an additional 2 h of growth, cells were pelleted at 6,000 × g for 20 min at 4°C and resuspended in GST binding buffer (25 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA) containing 0.1 mg/ml lysozyme (Amersham Biosciences). After sonication, Triton X-100 was added to reach a final concentration of 1% followed by centrifugation at 12,000 × g for 20 min at 4°C. The GST fusion proteins were adsorbed to glutathione-Sepharose 4B beads (Amersham Biosciences) and eluted with 10 mM reduced glutathione (Sigma)–50 mM Tris (pH 8.0).
Purification of FLAG-EGLN3.
HEK293T cells were transfected with FLAG-tagged EGLN3, its fragments, or empty vector. After 24 h, cells were extracted in buffer A (20 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA, and protease inhibitors). Cleared lysates were immunoprecipitated with anti-FLAG. The immunoprecipitates were washed once with buffer B (20 mM HEPES [pH 7.9], 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, and protease inhibitors) and four times with buffer C (20 mM Tris [pH 7.5], 300 mM NaCl, 0.2 mM EDTA, 0.1% NP-40, 20% glycerol, and protease inhibitors). Samples were eluted with 300 to 500 μg/ml FLAG peptide (Sigma).
GST pulldown assay.
GST alone or GST-IKKγ was bound to 20 μl of glutathione resin (Amersham Biosciences) that had been preblocked in phosphate-buffered saline (PBS) containing 0.5% nonfat milk and 0.05% bovine serum albumin and incubated with an equal amount of FLAG-EGLN3 or its fragments at 4°C in the binding buffer (20 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA, and protease inhibitors). After extensive washing, the complexes were eluted with SDS sample buffer and detected by immunoblotting.
RESULTS
EGLN3 inhibits NF-κB activity.
We have previously shown that EGLN3 represses canonical NF-κB signaling in C2C12 skeletal myoblasts (9). To further investigate the mechanism by which EGLN3 regulates NF-κB, we sought to employ HEK293T cells, a more tractable cell line. EGLN3 inhibited NF-κB transcriptional activity induced by the NF-κB activators TRAF2 and IKK2 (9), as demonstrated by luciferase reporter assays (Fig. 1A and B). EGLN3 significantly repressed phosphorylation of IKK2 and that of IκBα (Fig. 1C). These observations indicate that EGLN3 negatively regulates NF-κB in HEK293T cells.
Fig 1.
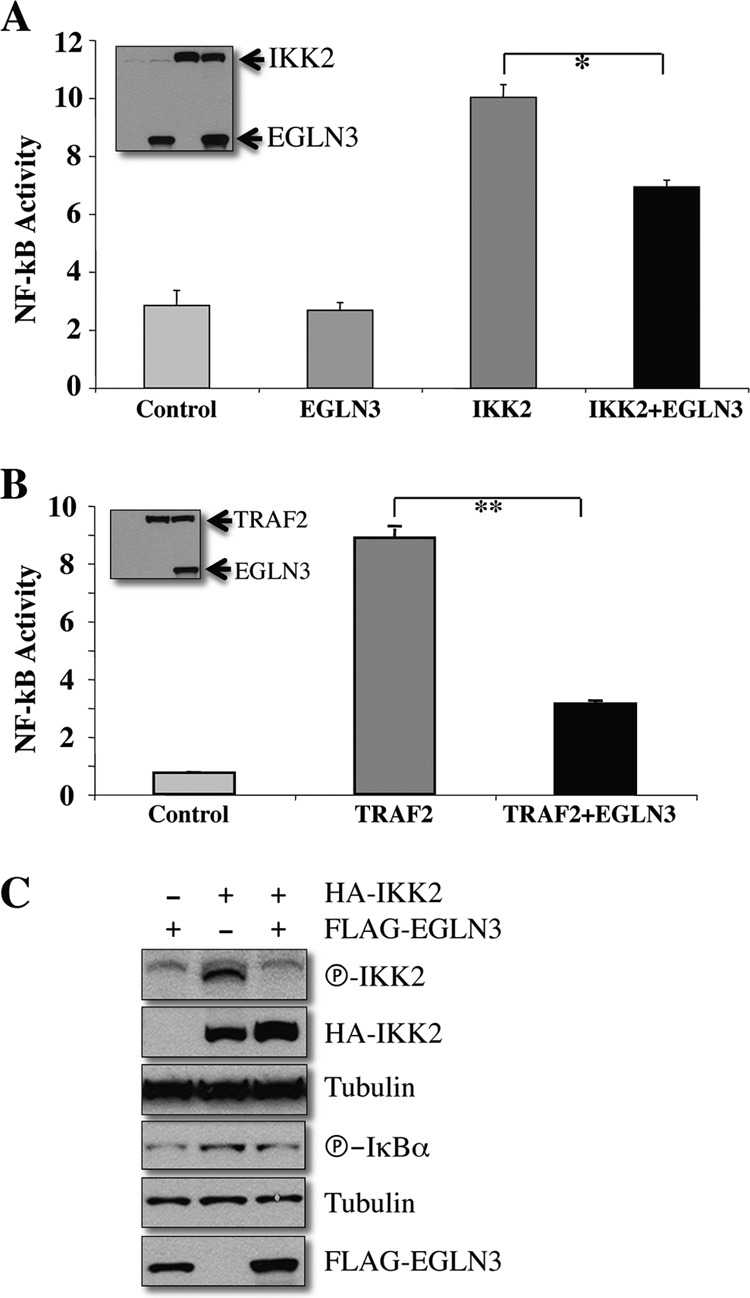
EGLN3 inhibits NF-κB activity in HEK293T cells. (A and B) HEK293T cells were transfected with NF-κB luciferase reporter plasmid and pRL-tk together with the indicated expression plasmids. Luciferase activity was measured and normalized for RL-tk luciferase activity. *, P < 0.05; **, P < 0.01. The expression of transfected genes was examined by immunoblotting. (C) HEK293T cells were transfected with the indicated plasmids. Cell lysates were analyzed by immunoblotting.
EGLN3 interacts with IKKγ.
To explore a potential interaction between EGLN3 and IKKγ, IKKγ was transfected into HEK293T in the presence or absence of EGLN3. EGLN3 was detected in IKKγ immunoprecipitates (Fig. 2A). In reciprocal experiments, IKKγ was detected in EGLN3 immunoprecipitates (Fig. 2B). Similar results were obtained with COS-7 cells (data not shown). Association of EGLN3 with IKKγ appeared far more prominent than with IKK1 and IKK2 (Fig. 2C and D). Binding of IKKγ was selective for EGLN3 in that the other members of the EGLN family, EGLN1 and EGLN2, failed to interact (Fig. 2E). EGLN3 directly interacted with IKKγ in vitro, as demonstrated by GST pulldown assays using purified GST-IKKγ and FLAG-EGLN3 (Fig. 2F). Most importantly, endogenous EGLN3 and IKKγ interacted with each other; deletion of endogenous EGLN3 with its siRNA dramatically diminished the interaction (Fig. 2G). Of note, no interaction occurred between endogenous EGLN3 and IKK2 in unstimulated cells (data not shown). Tumor necrosis factor alpha (TNF-α) had no significant effect on the interaction (Fig. 2H). Iron-binding histidine 196 (H196) and 2-oxoglutarate-coordinating arginine 205 (R205) of EGLN3 are critical for its enzymatic activity; mutation of H196 and R205 leads to loss of EGLN3 hydroxylase activity (9, 31). The catalytically inactive mutants (H196A and R205K) retained the ability to interact with IKKγ (Fig. 2I). Therefore, prolyl hydroxylase activity was not required for the EGLN3-IKKγ interaction.
Fig 2.

EGLN3 interacts with IKKγ. (A to E) Total cell lysates from HEK293T transfected with the indicated plasmids were immunoprecipitated with anti-HA (A and D), anti-FLAG (B and C), anti-myc (E), or mouse IgG. The immune complex and cell lysates were analyzed by immunoblotting (IB). (F and K) Purified FLAG-EGLN3 or its fragments (F1, F2, or F3) were incubated with GST alone or GST-IKKγ immobilized on glutathione-Sepharose beads. The precipitates or purified FLAG-EGLN3 fragments were analyzed by immunoblotting. Purified FLAG-EGLN3, GST, and GST-IKKγ were visualized by Coomassie blue staining. Con, control; NS, nonspecific band. (G and H) Total cell lysates from HeLa cells treated with the EGLN3 siRNA (or control siRNA) (G) or from HeLa cells treated with TNF-α (10 ng/ml) (H) were immunoprecipitated with anti-IKKγ or control IgG. The immune complex and cell lysates were analyzed by immunoblotting. (I, J, and L) HEK293T cells were transfected with the indicated plasmids. Cell lysates were immunoprecipitated with anti-FLAG (I and L) or anti-HA (J). The immune complex and cell lysates were examined by immunoblotting. The bottom of panel J shows the schematic of wild-type EGLN3 and its fragments used; the bottom of panel L shows a schematic representation of wild-type (WT) IKKγ and its mutants used. FL, full-length EGLN3; CC1, coiled-coil domain 1; CC2, coiled-coil domain 2; LZ, leucine zipper region; ZF, zinc finger region; IP, immunoprecipitation; IgH, heavy chain of IgG; IgL, light chain of IgG; Ig, immunoglobulin; EV, empty vector; CBB, Coomassie blue staining; F-EGLN3, FLAG-tagged EGLN3.
To determine the region of EGLN3 required for IKKγ binding, we cotransfected IKKγ into cells together with three fragments of EGLN3 (F1, F2, and F3), which represent the N-terminal, middle, and C-terminal regions. In coimmunoprecipitation assays, IKKγ exhibited the strongest interaction with the C-terminal region of EGLN3 (Fig. 2J). GST pulldown assays were conducted using purified GST or GST-IKKγ and the EGLN3 fragments. As shown in Fig. 2K, FLAG-tagged F3 (but not F1 or F2) was brought down by GST-IKKγ rather than GST alone. Taking these results together, EGLN3 binds to IKKγ through its C-terminal region. Coimmunoprecipitation experiments were performed to identify the regions on IKKγ required for EGLN3 binding. EGLN3 was barely detected in the immunoprecipitates of two mutants (M3 and M5) that lack the coiled-coil 1 (CC1) domain and leucine zipper (LZ) region, respectively (Fig. 2L). Therefore, the CC1 and LZ domains of IKKγ were required for EGLN3 binding.
EGLN3 inhibits IKKγ ubiquitination.
Because of the importance of IKKγ ubiquitination in regulating NF-κB signaling (16–26, 43), we determined the impact of EGLN3 on IKKγ ubiquitination. As expected, TNF induced the ubiquitination of IKKγ (Fig. 3A). It is worth noting that EGLN3 is expressed at a higher level in HeLa cells than in HEK293T cells. We conducted the loss-of-function experiments by employing HeLa cells. Depletion of endogenous EGLN3 by siRNA potentiated TNF-induced IKKγ ubiquitination (Fig. 3B). In contrast, knockdown of endogenous EGLN3 had only a minimal effect on RIP1 ubiquitination (data not shown). In addition, HeLa cells were transfected with IKKγ and Ub in the presence of another pair of EGLN3 siRNAs. Depletion of EGLN3 increased IKKγ ubiquitination in a concentration-dependent manner (Fig. 3C). Taking these results together, EGLN3 inhibits KKγ ubiquitination. To complement and extend this finding, we performed gain-of-function experiments. HEK293T cells were transfected with plasmid expressing EGLN3 or control vector. Cells were then treated with TNF for different times prior to harvest. IKKγ was immunoprecipitated and then immunoblotted with anti-Ub antibody. Expression of EGLN3 inhibited TNF-induced IKKγ ubiquitination (Fig. 3D). To further verify this observation, we cotransfected HEK293T with IKKγ, Ub, and increasing amounts of EGLN3. IKKγ was immunoprecipitated and then immunoblotted with anti-Ub antibody. EGLN3 inhibited IKKγ ubiquitination in a concentration-dependent manner (Fig. 3E). EGLN3 had no significant effect on IKKγ protein levels (Fig. 3E and data not shown). Similar observations were achieved with COS-7 cells (data not shown). We have shown that EGLN1 and -2 could not interact with IKKγ (Fig. 2E). As expected, EGLN1 and -2 did not affect IKKγ ubiquitination (Fig. 3F). In addition, expression of EGLN2 did not override the effect of EGLN3 on IKKγ ubiquitination (data not shown). Therefore, EGLN3 appears to be unique among the members of the EGLN family in regulating IKKγ ubiquitination. As shown in Fig. 3G, EGLN3 did not affect the ubiquitination of optineurin, a homolog of IKKγ (36), suggesting that EGLN3 did not generally attenuate protein ubiquitination.
Fig 3.

EGLN3 selectively inhibits IKKγ ubiquitination. (A) HeLa cells were treated with 10 ng/ml of TNF-α for the indicated times prior to harvest. Cell lysates were immunoprecipitated with anti-IKKγ. The immunoprecipitates and cell extracts were analyzed by immunoblotting with the indicated antibodies. (B) HeLa cells were transfected with control siRNA (−) or EGLN3 siRNA (+). At 60 h after transfection, cell lysates were prepared after treatment of cells with 10 ng/ml of TNF-α for 10 min and analyzed as described for panel A. (C) HeLa cells were transfected with HA-IKKγ, myc-Ub, and EGLN3 siRNA. Cell lysates were analyzed as described for panel A. (D) HEK293T cells were transfected with a plasmid expressing EGLN3 or an empty vector. At 30 h after transfection, cells were then treated with 10 ng/ml of TNF-α for different times. Cell lysates were analyzed as described for panel A. (E) HEK293T cells were transfected with HA-IKKγ, myc-Ub, and different amounts of FLAG-EGLN3. Cell lysates were immunoprecipitated with anti-HA and then immunoblotted with anti-myc. Cell lysates were examined by immunoblotting. (F to I) HEK293T cells were transfected with the indicated plasmids. Cell lysates were immunoprecipitated with anti-HA (F, G, and H) or anti-FLAG (I). The immune complex was then immunoblotted with anti-myc (F and H) or anti-Ub (G and I). To monitor expression of the transfected genes, cell lysates were examined by immunoblotting with the indicated antibodies. IP, immunoprecipitation; IB, immunoblotting; Ig H, heavy chain of IgG.
To determine the relationship between EGLN3-IKKγ interaction and inhibition of IKKγ ubiquitination, we first cotransfected IKKγ into HEK293T together with three fragments of EGLN3. The C-terminal region of EGLN3, although expressed less abundantly than the full-length EGLN3 or the other two fragments, inhibited IKKγ ubiquitination to a degree similar to that seen with the full-length EGLN3 (Fig. 3H), whereas the N-terminal and middle regions had only minimal effects (Fig. 3H). In sum, the C-terminal region of EGLN3 is important for IKKγ binding and inhibition of IKKγ ubiquitination. We next cotransfected EGLN3 into HEK293T together with different fragments of IKKγ. EGLN3 failed to repress the ubiquitination of two mutants (M3 and M5) that lack the CC1 region and LZ region, respectively (Fig. 3I). Taking these results together, the CC1 and LZ domains of IKKγ are required for EGLN3 binding and for the inhibition of IKKγ ubiquitination by EGLN3. These observations strongly suggest that EGLN3 inhibits IKKγ ubiquitination by physical interaction with IKKγ.
EGLN3 inhibits cIAP1-mediated K63-linked ubiquitination of IKKγ.
Two deubiquitinases, A20 and CYLD (cylindromatosis), have been documented to remove polyubiquitin chains from IKKγ and thereby attenuate IKKγ ubiquitination (44, 45). Although A20 shRNA and CYLD shRNA efficiently reduced A20 expression and CYLD expression, respectively (Fig. 4A), and increased IKKγ ubiquitination (Fig. 4B), depletion of CYLD or A20 did not abrogate the effect of EGLN3 on IKKγ ubiquitination (Fig. 4B). To exclude the possibility that CYLD and A20 work coordinately in deubiquitinating IKKγ, we simultaneously depleted both deubiquitinases using their shRNAs. Deletion of both CYLD and A20 markedly enhanced IKKγ ubiquitination but did not abolish the inhibitory effect of EGLN3 on IKKγ ubiquitination (Fig. 4C), demonstrating that neither CYLD nor A20 is essential for inhibition of IKKγ ubiquitination by EGLN3.
Fig 4.

The deubiquitinating enzymes A20 and CYLD are not involved in EGLN3-mediated inhibition of IKKγ ubiquitination. (A) HEK293T cells were transfected with FLAG-CYLD, or FLAG-A20, in the presence of pSuper, pSuper-CYLD shRNA, or pSuper A20 shRNA. At 48 h posttransfection, cell lysates were analyzed by immunoblotting. (B) HEK293T cells were transfected with the indicated plasmids. Cell lysates were analyzed with anti-HA, anti-FLAG, or anti-tubulin antibodies. (C) HEK293T cells were transfected with the indicated plasmids. Cell lysates were subjected to denatured immunoprecipitation with anti-HA. The immmunoprecipitates and lysates were evaluated by immunoblotting. (D and E) HEK293T cells were transfected with FLAG-tagged EGLN3 or OTUB1 or its catalytically inactive mutant C91S (OTUB1C/S). Cell extracts were prepared and labeled with ubiquitin derivatives as detailed in Materials and Methods. F, FLAG; Ub-Vs, ubiquitin vinyl sulfone; Ub-VME, ubiquitin vinyl methyl ester; Ub-Cl, chloroethyl ubiquitin; Ub-Br, bromoethyl ubiquitin; IP, immunoprecipitation; IB, immunoblotting; IgH, heavy chain of IgG; TBS, Tris-buffered saline.
We next determined whether EGLN3 has an intrinsic deubiquitinase activity. It has been documented that a deubiquitinase can react with a chemically modified ubiquitin probe (41, 42). However, EGLN3 did not show an activity to react with any of four widely used ubiquitin probes (Fig. 4D and E). In contrast, the deubiquitinase OTUB1, but not its catalytically inactive mutant, reacted with bromoethyl Ub (Ub-Br) (Fig. 4E). Therefore, these experiments suggested that EGLN3 does not possess deubiquitinating activity and has no significant effect on IKKγ deubiquitination.
It has been suggested that cellular inhibitor of apoptosis 1 (cIAP1) is responsible for IKKγ ubiquitination (16). We therefore examined whether EGLN3-sensitive IKKγ ubiquitination was catalyzed by cIAP1. In agreement with the previous observation (16), cIAP1 targeted IKKγ for ubiquitination (Fig. 5A); however, cIAP1 did not ubiquitinate IKK1 or IKK2 (Fig. 5B). Smac mimetic has been shown to trigger autodegradation of cIAP1 and cIAP2 (27, 46, 47). Treatment of cells with Smac mimetic dramatically depleted cIAP1 protein levels and robustly blocked IKKγ ubiquitination (Fig. 5C). EGLN3-mediated inhibition of IKKγ ubiquitination was not observed (Fig. 5C). Unlike wild-type cIAP1, a cIAP1 mutant defective for enzymatic activity failed to target IKKγ for ubiquitination (Fig. 5D). In contrast, X-linked inhibitor of apoptosis (XIAP), which is a homolog of cIAP1 and a Ub ligase (28), did not affect IKKγ ubiquitination (Fig. 5E). As expected, cIAP1-induced IKKγ ubiquitination was substantially reduced by EGLN3 (Fig. 5D and E), suggesting that EGLN3 could block cIAP1-mediated ubiquitination of IKKγ.
Fig 5.
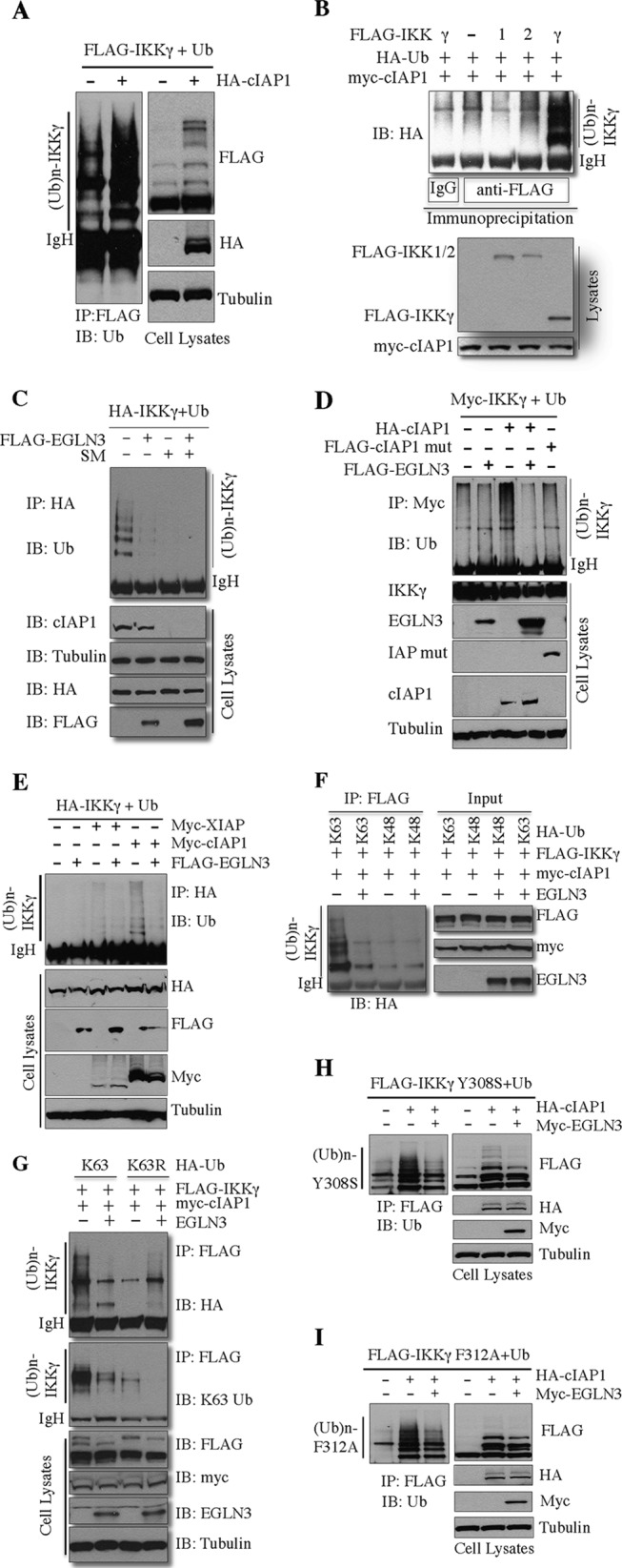
EGLN3 suppresses cIAP1-mediated IKKγ ubiquitination. (A to I) HEK293T cells were transfected with the indicated plasmids. (C) At 6 h or so posttransfection, cells were treated with PBS or Smac mimetic (100 nM). Cell lysates were applied to in vivo ubiquitination assays as described in Materials and Methods. Cell lysates were analyzed by immunoblotting. IP, immunoprecipitation; IB, immunoblotting; IgH, heavy chain of IgG; mut, mutant; SM, Smac mimetic.
To determine whether cIAP1 targets IKKγ for K63-linked polyubiquitination, we employed several Ub mutants (40). K48 and K63 Ub mutants, in which all Ub lysine (K) residues except K48 or K63 were mutated to arginine (R), promote the assembly of K48-linked and K63-linked Ub chains, respectively. K63R, in which only K63 was mutated to R, disrupts the assembly of the K63-linked Ub chain. The in vivo ubiquitination assays demonstrated that EGLN3 inhibited K63-linked ubiquitination of IKKγ (Fig. 5F and G).
Given that IKKγ is a Ub-binding protein (32, 33), we sought to further determine whether cIAP1 could ubiquitinate IKKγ mutants defective for Ub-binding ability (32, 33) and whether EGLN3 could block this effect. Four different IKKγ mutants were transfected into HEK293T, in combination with cIAP1 and EGLN3. All mutants were ubiquitinated by cIAP1, and their ubiquitination was markedly inhibited by EGLN3 (Fig. 5H and I and data not shown). Collectively, our studies provided strong evidence that EGLN3 inhibits cIAP1-mediated K63-linked ubiquitination of IKKγ.
The effect of EGLN3 on IKKγ ubiquitination does not depend upon prolyl hydroxylase activity.
EGLN3 is a prolyl hydroxylase which requires O2, Fe2+, and 2-oxoglutrate for its catalytic activity. Dimethyl oxalylglycine (DMOG) is a widely used pharmacologic inhibitor of hydroxylases, including EGLN3 (9). To determine whether EGLN3 hydroxylase activity is required for inhibition of IKKγ ubiquitination, we transfected IKKγ and Ub into HEK293T in the presence or absence of EGLN3. At 24 h following transfection, cells were left untreated or treated with DMOG for an additional 24 h. DMOG did not block the inhibitory effect of EGLN3 on IKKγ ubiquitination (Fig. 6A). However, DMOG increased HIF1α expression (data not shown), indicating that DMOG was effective at the concentration used to examine IKKγ ubiquitination. Importantly, catalytically inactive mutants (H196R, H196A, and R205K) blocked IKKγ ubiquitination to an extent similar to that seen with wild-type EGLN3 (Fig. 6B and C). Moreover, the H196A and R205K mutants significantly inhibited cIAP1-mediated ubiquitination of IKKγ mutants (Y308S and F312A) that are deficient in Ub-binding ability (Fig. 6D and E). Therefore, EGLN3 hydroxylase activity is not essential for its effect on IKKγ ubiquitination.
Fig 6.

Hydroxylase activity of EGLN3 is not required for inhibition of IKKγ ubiquitination. (A) HEK293T cells were transfected with the indicated plasmids. At 24 h posttransfection, cells were treated or not treated with DMOG (1 mM) for 24 h. Cell lysates were immunoprecipitated with anti-HA. The immunoprecipitates and cell lysates were assayed by immunoblotting. (B to E) HEK293T cells were transfected with the indicated plasmids. Cell lysates were subjected to in vivo ubiquitination assays as described in Materials and Methods. Cell lysates were analyzed by immunoblotting. IP, immunoprecipitation; IB, immunoblotting; IgH, heavy chain of IgG; EV, empty vector.
EGLN3 inhibits cIAP1-mediated IKKγ ubiquitination by interfering with the interaction between IKKγ and cIAP1.
To explore the mechanism by which EGLN3 decreases cIAP1-mediated IKKγ ubiquitination, we first examined whether EGLN3 promoted degradation of cIAP1 or its E2 ubiquitin-conjugating enzymes Ubc13 and UbcH5C. Ectopic expression of EGLN3 (Fig. 7A and B) or depletion of endogenous EGLN3 (Fig. 7C and D) with siRNA had no significant effect on the abundance of cIAP1, Ubc13, or UbcH5C proteins. In addition, coimmunoprecipitation studies failed to demonstrate an interaction between EGLN3 and Ubc 13 or UbcH5C (data not shown).
Fig 7.
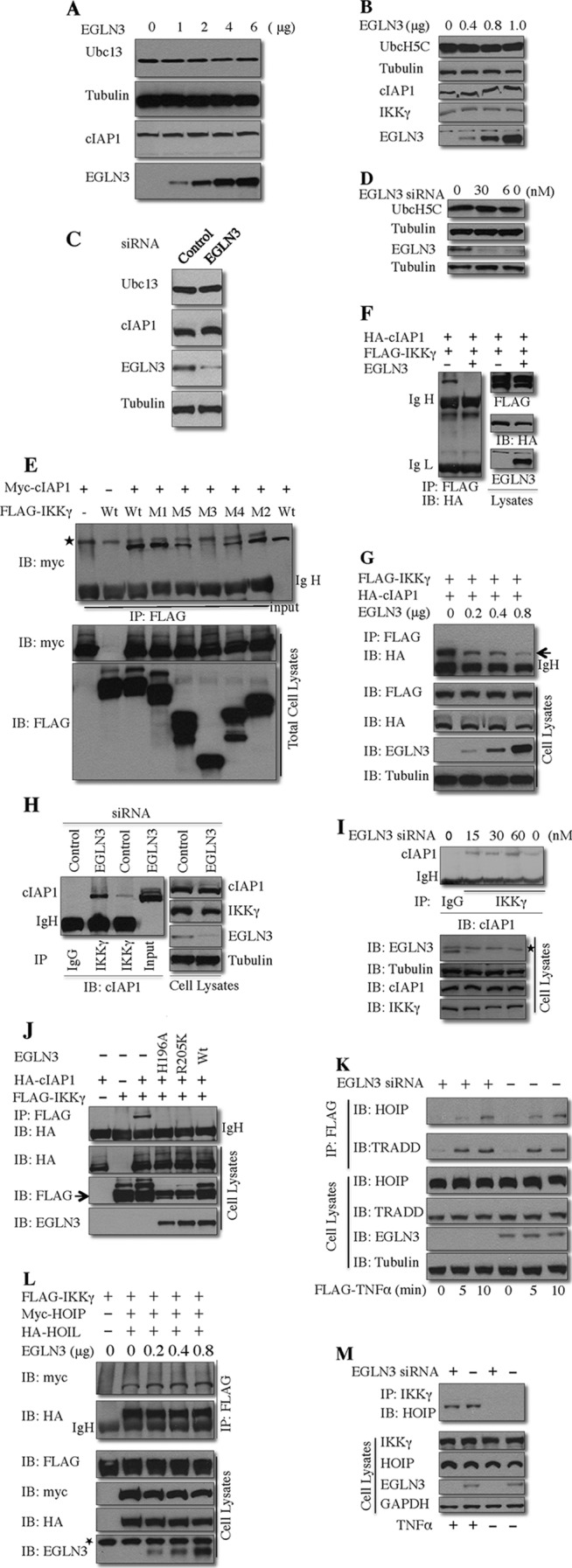
EGLN3 competes with cIAP1 for IKKγ binding. (A and B) HEK293T cells were transfected with various amounts of EGLN3. Cell lysates were analyzed by immunoblotting. (C and D) HeLa cells were transfected with EGLN3 or control siRNA for 48 h. Prior to harvest, cells were treated with TNF-α (10 ng/ml) for 10 min. Cell lysates were analyzed by immunoblotting. (E, F, G, and J) The cell lysates from HEK293T cells transfected with the indicated plasmids were immunoprecipitated with anti-FLAG. The immunoprecipitates and cell lysates were immunoblotted with the indicated antibodies. (H and I) HeLa cells were transfected with siRNA. At 48 h posttransfection, cells were treated with TNF-α (10 ng/ml) for 5 to 10 min. Cell lysates were immunoprecipitated with anti-IKKγ or control IgG. The immunoprecipitates and cell lysates were analyzed by immunoblotting with the indicated antibodies. (K) HeLa cells were transfected with EGLN3 siRNA or control siRNA. At 48 h posttransfection, cells were treated with FLAG–TNF-α for different times. The anti-FLAG immunoprecipitates and lysates were evaluated by immunoblotting. (L) HEK293T cells were transfected with the indicated plasmids. The anti-FLAG immune complex and lysates were analyzed by immunoblotting. (M) HeLa cells were transfected with EGLN3 siRNA or control siRNA. At 48 h posttransfection, cell lysates were prepared and subjected to immunoprecipitation with anti-IKKγ. The immunoprecipitates and lysates were examined by immunoblotting. IP, immunoprecipitation; IB, immunoblotting; IgH, heavy chain of immunoglobulin; IgL, light chain of immunoglobulin; *, nonspecific band.
We next tested the hypothesis that EGLN3 represses cIAP1-mediated IKKγ ubiquitination by interfering with the interaction between cIAP1 and IKKγ. Consistent with the finding that IKKγ is a substrate for cIAP1, coimmunoprecipitation assays demonstrated that cIAP1 formed a complex with IKKγ (Fig. 7E to J). Furthermore, the interaction of endogenous cIAP1 and IKKγ was observed (Fig. 7H and I). To define the domains of IKKγ responsible for cIAP1 binding, cIAP1 was cotransfected into HEK293T cells with a panel of IKKγ mutants (see Fig. 2L). The M3 and M5 mutants, which lack the CC1 region and LZ region, respectively, were greatly impaired in their ability to bind cIAP1, as determined by coimmunoprecipitation assays (Fig. 7E). Our results suggested that the CC1 and, to a lesser extent, LZ domains contributed to cIAP1 binding. Therefore, cIAP1 and EGLN3 both physically interact with IKKγ through the same domains. These observations suggest that EGLN3 may compete with cIAP1 for IKKγ binding, thereby leading to attenuating IKKγ ubiquitination. In support of this hypothesis, expression of EGLN3 blocked the interaction between IKKγ and cIAP1 in a concentration-dependent manner (Fig. 7F and G). Expression of EGLN3 attenuated the interaction of endogenous IKKγ and cIAP1 (data not shown), and depletion of EGLN3 facilitated the interaction between IKKγ and cIAP1 (Fig. 7H and I) in concentration-dependent fashions.
Because EGLN3 inhibits cIAP1-mediated IKKγ ubiquitination via a noncatalytic mechanism, we determined whether EGLN3 hydroxylase activity is essential for the effect of EGLN3 on the cIAP1-IKKγ interaction. As shown in Fig. 7J, H196A and R205K, two enzymatically inactive mutants of EGLN3, inhibited cIAP1-IKKγ interaction to an extent similar to that seen with wild-type EGLN3. Collectively, the data presented above suggest that EGLN3 inhibits cIAP1-mediated IKKγ ubiquitination through a hydroxylase-independent process involving interference with the interaction between IKKγ and cIAP1.
It has been previously reported that cIAP1 is required for the recruitment of the linear ubiquitin chain assembly complex (LUBAC) to the TNF-R1 signaling complex (TNF-RSC), which plays an important role in TNF-induced NF-κB signaling (48). LUBAC is an E3 ubiquitin ligase and is composed of a catalytic subunit (HOIP) and two regulatory subunits, HOIL and Sharpin; IKKγ interacts with HOIP (24–26). LUBAC facilitates the recruitment of IKKγ into the TNF-RSC (48). To test whether LUBAC is responsible for EGLN3-sensitive ubiquitination of IKKγ, cells were transfected with IKKγ and either LUBAC or cIAP1 (as a control). Whereas cIAP1 efficiently ubiquitinated IKKγ, LUBAC had a minimal effect (data not shown).
Like TRADD, a well-known component of the TNF-RSC, HOIP was recruited to the TNF-RSC in response to TNF stimulation; depletion of EGLN3 had no significant effect on the recruitment of HOIP to TNF-RSC (Fig. 7K). This observation suggests that regulation of NF-κB signaling by EGLN3 is unlikely to occur through the recruitment of LUBAC to TNF-RSC. This observation also suggests that EGLN3 does not impair the catalytic activity of cIAP1, because the catalytic activity of cIAP1 is required for LUBAC recruitment to the TNF-RSC (48). To determine whether EGLN3 affects the interaction between LUBAC and IKKγ, cells were transfected with IKKγ and LUBAC (HOIP and HOIL) in the presence or absence of EGLN3. Coimmunoprecipitation assays indicated that IKKγ interacted with both HOIP and HOIL; this interaction was not impaired by coexpressed EGLN3 (Fig. 7L). Furthermore, EGLN3 knockdown did not affect TNF-induced interaction of HOIP and KKγ (Fig. 7M). These results suggest that EGLN3 does not interfere with the interaction between LUBAC and IKKγ.
EGLN3 inhibition of IKK–NF-κB signaling requires its interaction with IKKγ.
To explore the functional consequence of the mechanistic observations described above, we tested the effect of wild-type EGLN3 and several of its mutants (Fig. 8A) on the activity of IKK and NF-κB. As shown in Fig. 8B, enzymatically inactive mutants (H196A and R205K) inhibited the IKK phosphorylation and activity (as demonstrated by an in vitro kinase assay) to an extent similar to that seen with wild-type EGLN3. Cox2 is a well-known NF-κB target (49). TNF dramatically induced COX2 expression in HeLa cells (Fig. 8D). Depletion of EGLN3 increased TNF-induced COX2 expression (Fig. 8E), whereas overexpression of EGLN3 decreased it (Fig. 8F). Moreover, two mutants, H196A and R205K, impeded TNF-induced COX2 expression as efficiently as wild-type EGLN3 (Fig. 8F). As shown in Fig. 3H, EGLN3 inhibits ubiquitination of IKKγ primarily via its C-terminal region (F3). Interestingly, the F3 fragment but not the F1 or F2 fragment efficiently blocked IKK phosphorylation and activity (Fig. 8C). Furthermore, F3, but not F1, attenuated TNF-induced COX2 expression (Fig. 8G). Taken together, our results indicated that EGLN3 inhibited IKKγ ubiquitination and thereby the activity of the IKK–NF-κB signaling pathway. The physical interaction between EGLN3 and IKKγ is required for the inhibitory role of EGLN3 in IKKγ ubiquitination and IKK–NF-κB signaling.
Fig 8.
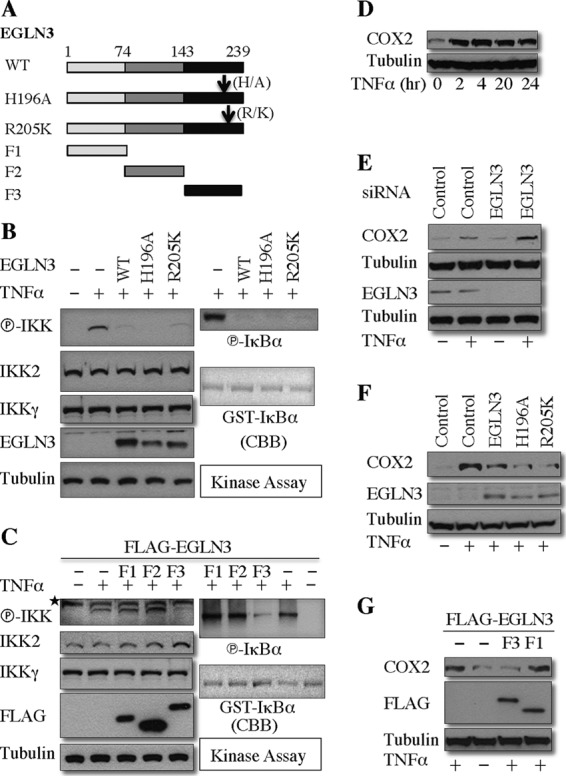
Interaction with IKKγ is required for the ability of EGLN3 to inhibit the activity of IKK and NF-κB. (A) Schematic of the wild type and the mutants of EGLN3 used. (B and C) HEK293T cells were transfected with the indicated plasmids and treated or not treated with TNF-α. Cell lysates were subjected to in vitro kinase assays with GST-IκBα as the substrate. The cell lysates were examined by immunoblotting. GST-IκBα was visualized by Coomassie blue staining. (D) HeLa cells were treated with TNF-α for various times. The cell lysates were evaluated by immunoblotting. (E to G) HeLa cells were transfected with siRNAs (E), or the indicated expression plasmids (F and G), and then treated or not treated with TNF-α for 16 h. The cell lysates were analyzed by immunoblotting. CBB, Coomassie blue staining; *, nonspecific band; A, alanine; H, histidine; K, lysine; R, arginine.
DISCUSSION
In this report, we have identified and characterized a physical and functional interaction between EGLN3 and IKKγ. IKKγ was initially identified as a core component of the IKK signalsome composed of IKK1, IKK2, and IKKγ (50). IKKγ is essential for the activation of the IKK signalsome and NF-κB (13–15). The central position of IKKγ in the canonical NF-κB pathway makes it an attractive target for identifying novel regulators of this pathway. Numerous IKKγ binding proteins have been reported, as exemplified by p47, an essential molecule for Golgi membrane fusion (51), annexin-1 (52), ZNF216 (53), and CARDINAL (54). They are involved in the regulation of the IKK2/NF-κB pathway. Interestingly, they have all been reported to interact with IKKγ but not IKK2. These studies suggested that IKKγ regulates IKK2/NF-κB activation through multiple mechanisms. In the current study, immunoprecipitation assays indicated that in unstimulated cells, endogenous EGLN3 interacted with IKKγ (but not IKK2). This raises the possibility that EGLN3 sequesters IKKγ from IKK2, cIAP1, or another molecule(s), thereby altering the threshold for IKK2/NF-κB activation.
We have demonstrated that EGLN3 interacts with IKKγ via its C-terminal region. We have shown that the C-terminal region of EGLN3 plays a key role in inhibiting K63-linked IKKγ ubiquitination and NF-κB signaling. Meanwhile, we have presented evidence showing that deletion of either the coiled-coil domain 1 (CC1) or leucine zipper domain (LZ) of IKKγ abrogates their ability to interact with EGLN3 and confers resistance to EGLN3-mediated inhibition of IKKγ ubiquitination. Therefore, the CC1 and LZ domains of IKKγ are required for EGLN3 binding and for the inhibition of IKKγ ubiquitination by EGLN3. These findings strongly suggest EGLN3 inhibits IKKγ ubiquitination by physical interacting with IKKγ.
Importantly, the depletion of EGLN3 by RNA interference increases IKKγ ubiquitination, whereas overexpression of EGLN3 decreases IKKγ ubiquitination. Dimethyl oxalylglycine (DMOG) is a widely used pharmacologic inhibitor of hydroxylases, including EGLN3 (9). Using the pharmacologic inhibitor DMOG, we have provided evidence that EGLN3 hydroxylase activity is not required to inhibit the ubiquitination of IKKγ. In addition, the enzymatically inactive mutants of EGLN3 interact and inhibit the ubiquitination of IKKγ as efficiently as wild-type EGLN3 and inhibit IKK2 phosphorylation and activity to an extent similar to that seen with wild-type EGLN3. These studies thus define a new role for EGLN3 that is independent of its hydroxylase activity.
EGLN3 has been shown to catalyze hydroxylation of several proteins such as the α subunits of HIF, the large subunit of RNA polymerase II, β2-adrenergic receptor, and pyruvate kinase M2 (44, 55–59). Although most of the biologic roles of EGLNs have been tied to its prolyl hydroxylase activity, several recent studies have also suggested that EGLNs may have functions independent of prolyl hydroxylase activity. For example, a recent study reported that EGLN1 prolyl hydroxylase negatively regulated NF-κB signaling in a hydroxylase-dependent manner (60). However, a subsequent study indicated that EGLN1 inhibition of NF-κB pathway was independent of its hydroxylase activity (61). Our previous study suggested that the inhibition of NF-κB by EGLN3 during the differentiation of C2C12 skeletal myoblasts was largely dependent on its hydroxylase activity (9). In contrast, a recently published study suggested that the inhibition of NF-κB by EGLN3 in several cell lines was not dependent upon its enzymatic activity (10). Taken together, the evidence suggests that the EGLN family of prolyl hydroxylases regulates the NF-κB pathway through hydroxylase-dependent and -independent mechanisms in a cell type- and context-specific manner.
In spite of great efforts in the last decade, the molecular mechanism underlying IKK2 activation remains to be completely elucidated. It has been established that activation of the NF-κB pathway critically requires ubiquitin (Ub) signaling (23). One of the key issues is exactly how ubiquitination induces IKK2 activation. Recruitment of LUBAC to the TNF-R1 signaling complex (TNF-RSC) is required for efficient TNF-induced NF-κB signaling; cIAP1 plays an important role in the recruitment of LUBAC to the TNF-RSC, which is dependent on its catalytic activity (48). EGLN3 had no significant effect on the interaction between LUBAC and IKKγ or the recruitment of LUBAC to the TNF-RSC. This suggests that LUBAC recruitment is not responsible for the regulation of NF-κB signaling by EGLN3 and that the effect of EGLN3 on cIAP1-mediated IKKγ ubiquitination is not related to its ability to compromise the catalytic activity of cIAP1. Although emerging evidence highlights the significance of the LUBAC in the NF-κB pathway, more direct evidence for a role of the M1-linked ubiquitination in NF-κB activation is still lacking.
RIP1 ubiquitination is essential for TNF-α-induced NF-κB activation (62). In contrast to this study, a recent report suggests that ubiquitinated RIP1 does not contribute to the recruitment of the TAK1 and IKK complexes to the TNF-RSC (63). cIAP1 is an E3 ligase for RIP1 (64). In HeLa cells, we found that EGLN3 deletion had minimal effect on RIP1 ubiquitination (data not shown), further suggesting that EGLN3 has no significant effect on the cIAP1 enzymatic activity.
Accumulating evidence supports the notion that IKKγ is the key factor for transducing the Ub signal to the TAK1 and IKK complexes. IKKγ is able to interact with the M1- and K63-linked poly-Ub chains (22, 33, 65). However, binding of IKKγ to distinct types of poly-Ub chains may not be sufficient for the full activation of IKK2. IKKγ has been shown to regulate activation of IKK and NF-κB by a mechanism that is dependent upon IKKγ ubiquitination (16–26). IKKγ ubiquitination may trigger the conformational change in the IKK complex, which is required for full activation of IKK2 (22, 66). Alternatively, the poly-Ub chains attached on IKKγ can function as a platform to nucleate the TAK1-TAB2-TAB3 complex (67). The nuclear protein localization 4 zinc finger (NZF) domain of TAB2 and TAB3 interacts specifically with the K63 poly-Ub chains (68). Interaction of TAB2 and TAB3 with the K63 poly-Ub chains provokes oligomerization-dependent autophosphorylation and activation of TAK1, which in turn induces phosphorylation and activation of IKK2 as well as subsequent activation of NF-κB (12, 50). In the current study, we showed that cIAP1 targets IKKγ for K63-linked polyubiquitination. In support of the previous observation that IKKγ is not necessary for the recruitment of the LUBAC to the TNF-RSC (48), our current data suggest that cIAP1-mediated IKKγ ubiquitination may not be important for the LUBAC recruitment. We therefore favor a model whereby ubiquitinated IKKγ may potentiate IKK2 activation by inducing the conformational change of the IKK complex and/or by serving as a platform to recruit and activate the TAK1 complex. Further studies are required to explore the mechanisms for activation of IKK2 by the ubiquitinated IKKγ.
The inhibition of IKKγ ubiquitination by EGLN3 was independent of CYLD or A20. In addition, we found no evidence that EGLN3 acted as a deubiquitinase. Importantly, we have shown that cIAP1 and EGLN3 both physically interact with IKKγ through the same domains (CC1 and LZ domains). Consistent with these observations, we have demonstrated that EGLN3 interrupts the interaction between IKKγ and cIAP1. Taking these findings together, we propose that EGLN3 competes with cIAP1 for the interaction with IKKγ, leading to interruption of cIAP1-IKKγ interaction and inhibition of IKKγ ubiquitination and thereby of IKK2/NF-κB activation.
Our data also suggest that the regulation of IKKγ by EGLN3 affects NF-κB activity. For example, depletion of EGLN3 increased TNF-induced expression of COX2, a well-known downstream target of NF-κB, whereas overexpression of EGLN3 decreased it. Moreover, the enzymatically inactive mutants of EGLN3 impeded TNF-induced COX2 expression as efficiently as the wild-type EGLN3. Consistent with our finding that EGLN3 interacts with and inhibits ubiquitination of IKKγ primarily via its C-terminal region (F3), the F3 fragment (but not the F1 or F2 fragment) blocked IKK activity and TNF-induced COX2 expression. We therefore conclude that the interaction between EGLN3 and IKKγ is required for the ability of EGLN3 to inhibit IKKγ ubiquitination and thereby alter IKK–NF-κB signaling.
NF-κB transcription factors are crucial regulators of inflammation, immunity, stress responses, cell apoptosis, and differentiation. Aberrant activation of NF-κB has been linked to human diseases, including chronic inflammatory diseases, autoimmune and metabolic disorders, and cancers (50). The activation of NF-κB by many stimuli is typically transient, limited by the action of negative regulatory mechanisms (50). Termination of an NF-κB response by these negative regulatory mechanisms prevents persistent and pathological activation of NF-κB signaling, which may be linked to the diseases described above. Our report provides novel insights into a negative regulator of NF-κB activity. Further study of the molecular mechanisms underlying EGLN3 regulation of NF-κB signaling may therefore contribute to novel therapeutic strategies for the treatment of inflammatory diseases and cancers.
ACKNOWLEDGMENTS
We are grateful to D. Abbott, L. Arend, K. Arimoto, C. Duckett, D. Durocher, E. Harhaj, A. Israel, Y. Jin, J. Li, K. Iwai, A. MacDonald, K. Nakayama, Z. Ronai, P. Storz, X. Wang, Z. Wu, and X. Yang for providing us with valuable reagents.
Footnotes
Published ahead of print 3 June 2013
REFERENCES
- 1.Kaelin WG, Jr, Ratcliffe PJ. 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30:393–402 [DOI] [PubMed] [Google Scholar]
- 2.Wax SD, Rosenfield CL, Taubman MB. 1994. Identification of a novel growth factor-responsive gene in vascular smooth muscle cells. J. Biol. Chem. 269:13041–13047 [PubMed] [Google Scholar]
- 3.Madden SL, Galella EA, Riley D, Bertelsen AH, Beaudry GA. 1996. Induction of cell growth regulatory genes by p53. Cancer Res. 56:5384–5390 [PubMed] [Google Scholar]
- 4.Lipscomb EA, Sarmiere PD, Crowder RJ, Freeman RS. 1999. Expression of the SM-20 gene promotes death in nerve growth factor-dependent sympathetic neurons. J. Neurochem. 73:429–432 [DOI] [PubMed] [Google Scholar]
- 5.Fu J, Menzies K, Freeman RS, Taubman MB. 2007. EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J. Biol. Chem. 282:12410–12418 [DOI] [PubMed] [Google Scholar]
- 6.Gerber SA, Yatsula B, Maier CL, Sadler TJ, Whittaker LW, Pober JS. 2009. Interferon-γ induces prolyl hydroxylase PHD3 through a STAT1-dependent mechanism in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 29:1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Y, Loos M, Giese N, Hines OJ, Diebold I, Gorlach A, Metzen E, Pastorekova S, Friess H, Buchler P. 2010. PHD3 regulates differentiation, tumour growth and angiogenesis in pancreatic cancer. Br. J. Cancer 103:1571–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henze AT, Riedel J, Diem T, Wenner J, Flamme I, Pouyseggur J, Plate KH, Acker T. 2010. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 70:357–366 [DOI] [PubMed] [Google Scholar]
- 9.Fu J, Taubman MB. 2010. Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-κB-dependent pathway. J. Biol. Chem. 285:8927–8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. 2010. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKβ independent of hydroxylase activity. Gastroenterology 138:606–615 [DOI] [PubMed] [Google Scholar]
- 11.Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. 2011. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479:122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheidereit C. 2006. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene 25:6685–6705 [DOI] [PubMed] [Google Scholar]
- 13.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. 2000. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 14:854–862 [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. 2000. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 5:981–992 [DOI] [PubMed] [Google Scholar]
- 15.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. 2000. Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 5:969–979 [DOI] [PubMed] [Google Scholar]
- 16.Tang ED, Wang CY, Xiong Y, Guan KL. 2003. A role for NF-κB essential modifier/IκB kinase-γ (NEMO/IKKγ) ubiquitination in the activation of the IκB kinase complex by tumor necrosis factor-α. J. Biol. Chem. 278:37297–37305 [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. 2004. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 427:167–171 [DOI] [PubMed] [Google Scholar]
- 18.Abbott DW, Wilkins A, Asara JM, Cantley LC. 2004. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 14:2217–2227 [DOI] [PubMed] [Google Scholar]
- 19.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14:289–301 [DOI] [PubMed] [Google Scholar]
- 20.Sebban-Benin H, Pescatore A, Fusco F, Pascuale V, Gautheron J, Yamaoka S, Moncla A, Ursini MV, Courtois G. 2007. Identification of TRAF6-dependent NEMO polyubiquitination sites through analysis of a new NEMO mutation causing incontinentia pigmenti. Hum. Mol. Genet. 16:2805–2815 [DOI] [PubMed] [Google Scholar]
- 21.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. 2009. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 11:123–132 [DOI] [PubMed] [Google Scholar]
- 22.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I. 2009. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136:1098–1109 [DOI] [PubMed] [Google Scholar]
- 23.Grabbe C, Husnjak K, Dikic I. 2011. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12:295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. 2011. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471:591–596 [DOI] [PubMed] [Google Scholar]
- 25.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, Iwai K. 2011. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471:633–636 [DOI] [PubMed] [Google Scholar]
- 26.Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J, Tokunaga F, Androulidaki A, Nakagawa T, Pasparakis M, Iwai K, Sundberg JP, Schaefer L, Rittinger K, Macek B, Dikic I. 2011. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471:637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Du F, Wang X. 2008. TNF-α induces two distinct caspase-8 activation pathways. Cell 133:693–703 [DOI] [PubMed] [Google Scholar]
- 28.Fu J, Jin Y, Arend LJ. 2003. Smac3, a novel Smac/DIABLO splicing variant, attenuates the stability and apoptosis-inhibiting activity of X-linked inhibitor of apoptosis protein. J. Biol. Chem. 278:52660–52672 [DOI] [PubMed] [Google Scholar]
- 29.Nakayama K, Gazdoiu S, Abraham R, Pan ZQ, Ronai Z. 2007. Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem. J. 401:217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. 2010. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U. S. A. 107:15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato M, Sakota M, Nakayama K. 2010. Human PRP19 interacts with prolyl-hydroxylase PHD3 and inhibits cell death in hypoxia. Exp. Cell Res. 316:2871–2882 [DOI] [PubMed] [Google Scholar]
- 32.Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. 2010. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell 40:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A. 2009. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 28:2885–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu S, Yang X. 2003. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J. Biol. Chem. 278:10055–10060 [DOI] [PubMed] [Google Scholar]
- 35.Csomos RA, Wright CW, Galban S, Oetjen KA, Duckett CS. 2009. Two distinct signalling cascades target the NF-κB regulatory factor c-IAP1 for degradation. Biochem. J. 420:83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mankouri J, Fragkoudis R, Richards KH, Wetherill LF, Harris M, Kohl A, Elliott RM, Macdonald A. 2010. Optineurin negatively regulates the induction of IFNβ in response to RNA virus infection. PLoS Pathog. 6:e1000778. 10.1371/journal.ppat.1000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC, O'Donnell L, Kumakubo A, Munro M, Sicheri F, Gingras AC, Natsume T, Suda T, Durocher D. 2010. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466:941–946 [DOI] [PubMed] [Google Scholar]
- 38.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. 2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 424:797–801 [DOI] [PubMed] [Google Scholar]
- 39.Storz P, Doppler H, Ferran C, Grey ST, Toker A. 2005. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem. J. 387:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, Harhaj EW. 2008. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 9:254–262 [DOI] [PubMed] [Google Scholar]
- 41.Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. 2002. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 9:1149–1159 [DOI] [PubMed] [Google Scholar]
- 42.Cotto-Rios XM, Jones MJ, Busino L, Pagano M, Huang TT. 2011. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. J. Cell Biol. 194:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niu J, Shi Y, Iwai K, Wu ZH. 2011. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 30:3741–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. 2003. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 424:801–805 [DOI] [PubMed] [Google Scholar]
- 45.Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. 2007. IκB kinase β phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Mol. Cell. Biol. 27:7451–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. 2007. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131:669–681 [DOI] [PubMed] [Google Scholar]
- 47.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. 2007. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131:682–693 [DOI] [PubMed] [Google Scholar]
- 48.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. 2009. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36:831–844 [DOI] [PubMed] [Google Scholar]
- 49.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. 2000. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403:103–108 [DOI] [PubMed] [Google Scholar]
- 50.Hayden MS, Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 51.Shibata Y, Oyama M, Kozuka-Hata H, Han X, Tanaka Y, Gohda J, Inoue J. 2012. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat. Commun. 3:1061. 10.1038/ncomms2068 [DOI] [PubMed] [Google Scholar]
- 52.Bist P, Leow SC, Phua QH, Shu S, Zhuang Q, Loh WT, Nguyen TH, Zhou JB, Hooi SC, Lim LH. 2011. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-κB: implication in breast cancer metastasis. Oncogene 30:3174–3185 [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Teng L, Li L, Liu T, Chen D, Xu LG, Zhai Z, Shu HB. 2004. ZNF216 is an A20-like and IκB kinase γ-interacting inhibitor of NFκB activation. J. Biol. Chem. 279:16847–16853 [DOI] [PubMed] [Google Scholar]
- 54.Bouchier-Hayes L, Conroy H, Egan H, Adrain C, Creagh EM, MacFarlane M, Martin SJ. 2001. CARDINAL, a novel caspase recruitment domain protein, is an inhibitor of multiple NF-κB activation pathways. J. Biol. Chem. 276:44069–44077 [DOI] [PubMed] [Google Scholar]
- 55.Bruick RK, McKnight SL. 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294:1337–1340 [DOI] [PubMed] [Google Scholar]
- 56.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468 [DOI] [PubMed] [Google Scholar]
- 57.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. 2001. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472 [DOI] [PubMed] [Google Scholar]
- 58.Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS. 2009. Oxygen-regulated β2-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci. Signal. 2:ra33. 10.1126/scisignal.2000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. 2011. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145:732–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. 2006. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. U. S. A. 103:18154–18159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ. 2009. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 15:527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. 2006. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22:245–257 [DOI] [PubMed] [Google Scholar]
- 63.Ramakrishnan P, Baltimore D. 2011. Sam68 is required for both NF-κB activation and apoptosis signaling by the TNF receptor. Mol. Cell 43:167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30:689–700 [DOI] [PubMed] [Google Scholar]
- 65.Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M, Beisker W, Schmidt-Supprian M, Krappmann D. 2011. NF-κB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-κB activation. J. Biol. Chem. 286:26107–26117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulathu Y, Komander D. 2012. Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13:508–523 [DOI] [PubMed] [Google Scholar]
- 67.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. 2004. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 15:535–548 [DOI] [PubMed] [Google Scholar]
- 68.Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D. 2009. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10:466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]