

Abstract
Azoles are among the most successful classes of antifungals. They act by inhibiting α-14 lanosterol demethylase in the ergosterol biosynthesis pathway. Oropharyngeal candidiasis (OPC) occurs in about 90% of HIV-infected individuals, and 4 to 5% are refractory to current therapies, including azoles, due to the formation of resistant biofilms produced in the course of OPC. We reasoned that compounds affecting a different target may potentiate azoles to produce increased killing and an antibiofilm therapeutic. 2-Adamantanamine (AC17) was identified in a screen for compounds potentiating the action of miconazole against biofilms of Candida albicans. AC17, a close structural analog to the antiviral amantadine, did not affect the viability of C. albicans but caused the normally fungistatic azoles to become fungicidal. Transcriptome analysis of cells treated with AC17 revealed that the ergosterol and filamentation pathways were affected. Indeed, cells exposed to AC17 had decreased ergosterol contents and were unable to invade agar. In vivo, the combination of AC17 and fluconazole produced a significant reduction in fungal tissue burden in a guinea pig model of cutaneous candidiasis, while each treatment alone did not have a significant effect. The combination of fluconazole and AC17 also showed improved efficacy (P value of 0.018) compared to fluconazole alone when fungal lesions were evaluated. AC17 is a promising lead in the search for more effective antifungal therapeutics.
INTRODUCTION
Fungal infections are on the rise as the at-risk patient population has expanded due mainly to extended life span, increasing numbers of immunosuppressed individuals, and acquisition of resistance by the pathogens. The opportunistic pathogen Candida albicans is currently recognized as the fourth most common cause of nosocomial bloodstream infections (1), and in HIV patients, mucosal candidiasis can be recurrent and debilitating (2).
Oral azole antifungals, such as fluconazole, are effective, are more convenient and better tolerated than topical or intravenous (i.v.) therapies, and therefore have become the drugs of choice. However, HIV patients may not respond well to therapy. Approximately 60% of HIV patients experience a recurrence within 6 months of the initial episode (3). Patients who do not respond to fluconazole may be given amphotericin B or caspofungin, but these drugs are limited by i.v. administration, safety profiles, and drug-drug interactions (2, 4). Ninety percent of HIV-infected individuals will develop oropharyngeal candidiasis (OPC), and refractory oral candidiasis is currently reported in 4 to 5% of HIV-infected individuals (5). Recent studies have found that 25% of patients on highly active antiretroviral therapy still developed oral Candida infections (6). Esophageal candidiasis is an AIDS-defining illness, affecting up to 20% of people with AIDS (7). Esophageal candidiasis causes pain and difficulty swallowing, which can lead to poor patient compliance with medications. Esophageal candidiasis, which must be treated with systemic antifungals, is often the first sign of systemic fungal disease. Prophylactic therapy for recurrent mucosal candidiasis is not recommended due to resistance development that could complicate treatment of systemic disease. Aside from mucosal and systemic disease, biofilms of C. albicans often form on indwelling devices such as catheters and heart valves and represent a common form of infection (8). These infections typically do not respond to antifungal therapy and can develop into a life-threatening disease, with a mortality rate approaching 40% (9, 10). Systemic C. albicans infections are characterized by hyphal growth and invasion into tissues and are a cause of high rates of morbidity and mortality in immunocompromised patients (11).
As the need for antifungal intervention has increased, so too has the prevalence of resistance. Resistance to the newest classes of antifungals, the triazoles and echinocandins, is well documented (12), and there is an unmet need to develop new antifungal agents. One potential strategy to overcome antifungal drug resistance and recurrence is to make currently available antifungals more effective. Standard strategies, such as chemical optimization and improved formulations, have had limited success. An alternative approach is to identify compounds that act in synergy with antifungals. These chemosensitizers may function by simply allowing more drug to enter the cell, or they may act by affecting a pathway that leads to synergy, such as inhibiting a fungal stress response. A comprehensive review of antifungal chemosensitizers has recently been published (13). Some chemosensitizers create fungicidal synergy with the azoles, causing these normally fungistatic agents to become lethal. Such compounds include inhibitors of HSP90 (radicicol and geldanamycin), calcineurin (cyclosporine and FK506), ADP-ribosylation factor cycling (brefeldin A), and calcium signaling (amiodarone) (14–17). While these agents have liabilities, such as immunosuppression and toxicity, they have been shown to improve the efficacy of the azoles in various animal models of fungal infection, validating the approach. Here, we describe the discovery and characterization of a potent new chemosensitizer.
MATERIALS AND METHODS
Strains and drugs.
The C. albicans strains used in this study, SC5314, CAF2-1, ATCC 96901, hip18C, and other clinical isolates, have been previously characterized (18–22). Yeast cells were grown on yeast extract-peptone-dextrose (YPD) agar from frozen (−80°C) glycerol stocks. Single colonies from agar plates that were incubated at 30°C for 24 to 48 h were used to prepare inocula for each experiment. All compounds were obtained from Sigma-Aldrich. Stock solutions were prepared in dimethyl sulfoxide (DMSO) or water and stored at −20°C until use.
Biofilm growth.
C. albicans CAF2-1 cultures were grown and used to inoculate 10 ml of liquid YPD medium in a 125-ml baffled Erlenmeyer flask. These cultures were incubated at 30°C for 24 h. Cells were concentrated by centrifugation and resuspended in RPMI 1640 with l-glutamine and 0.165 M morpholinepropanesulfonic acid (MOPS) (RPMI). The optical density at 600 nm (OD600) of the suspension was adjusted to 0.1, approximately 106 cells/ml. Thirty microliters of this suspension was aliquoted into wells of flat-bottom 384-well microtiter plates (Greiner Bio-One no. 781086). The plates were incubated at 37°C for 48 h to allow biofilm formation.
High-throughput screen.
Biofilm spent medium was discarded and replaced with fresh RPMI containing 100 μg/ml miconazole. Compounds from chemical library plates (http://iccb.med.harvard.edu/libraries/compound-libraries/) were pin transferred by using Epson Robotics (Carson, CA) at the Harvard ICCB-Longwood screening facility. One hundred nanoliters of compound, typically stored in DMSO at 5 mg/ml, was transferred into 30 μl of medium for each well, yielding a final compound concentration of approximately 17 μg/ml. Biofilms were incubated for 48 h at 37°C after compound transfer. Medium containing miconazole and the small molecules was discarded and replaced with phosphate-buffered saline (PBS) containing 0.3 mM resazurin. Biofilms were incubated for an additional 6 h at 37°C, and resazurin reduction was measured by using an EnVision plate reader (PerkinElmer, Waltham, MA) according to the manufacturer's instructions. Hits were determined for each plate by comparing the fluorescence intensity of each experimental well to that of negative-control wells (miconazole alone) and calculating percent inhibition. Only those compounds that resulted in >25% inhibition for both experimental replicates were considered hits.
Cytotoxicity.
Human FaDu epithelial cells (ATCC HTB-43) and HepG2 hepatocytes (ATCC HB-8065) were obtained from the ATCC and grown according to the manufacturer's recommendations. Cells were passaged into 96-well plates (Corning Costar no. 3603) and grown until they reached ∼70% confluence. Dilutions of amantadine, rimantadine, and 2-adamantanamine (AC17) were prepared in fresh growth medium in a separate plate. Cell culture medium was aspirated and replaced with medium containing drugs. The plates were incubated at 37°C and 5% CO2 for 24 h. Cells were washed three times with fresh medium and incubated for an additional 18 h. A total of 0.3 mM resazurin was added, and the plates were incubated for 3 h. Percent inhibition was calculated based on negative-control wells in each plate, and 50% inhibitory concentration (IC50) curves were generated by using XLFit software (IDBS).
Biofilm dose-response and killing assays.
Biofilms were grown as described above, except that 100-μl volumes of cells were dispensed into 96-well plates. After 48 h, fresh RPMI or medium containing 100 μg/ml miconazole was added to wells along with dilutions of AC17. After a further 48 h, biofilms were assayed with 0.3 mM resazurin in a manner similar to the high-throughput screen. IC50 curves were generated by using XLFit software (IDBS). For biofilm killing assays, the biofilms were washed three times with PBS after drug treatment, and the biofilm was resuspended in 100 μl PBS by scraping the bottom of the microtiter plate well with a pipette tip and transferring the solution into an Eppendorf tube. The tube was vortexed vigorously for 30 s, and the suspension was serially diluted and plated onto YPD agar for colony counts.
Agar diffusion assays.
Single colonies of C. albicans grown on YPD agar plates were inoculated into liquid YPD medium and grown overnight at 30°C. The cells were concentrated by centrifugation and washed three times with PBS. Approximately 107 cells/ml were added to prewarmed liquid YPD medium containing 0.75% agar, and 10 ml of this suspension was pipetted onto prepared YPD agar plates. Six-millimeter-diameter BBL filter disks (Becton, Dickinson and Co.) were placed onto the solidified agar surface, and drugs or solvent controls were pipetted onto each filter disk at the desired concentration. Each plate was incubated at 37°C for 24 to 48 h. Etest (bioMérieux) experiments were carried out according to the manufacturer's instructions, and AC17 was added to the RPMI agar medium at 20 μg/ml.
Time-kill assays.
Time-kill experiments were performed in 15-ml sterile polystyrene culture tubes. Cells from cultures grown overnight were washed and resuspended in RPMI medium such that the starting density was 1 × 106 to 5 × 106 CFU/ml. One-milliliter aliquots were dispensed into culture tubes containing voriconazole, AC17, and a combination of voriconazole and AC17 and were placed into a shaking incubator at 37°C. At 0, 24, 48, and 72 h, 100-μl aliquots were washed, diluted, and plated for colony counts on YPD agar.
Transcriptional profiling.
Microarray experiments were performed on cells treated with AC17 and compared to untreated controls. C. albicans cells were diluted to a starting optical density of 0.1 in RPMI medium. Cells were exposed to 100 μg/ml of AC17 for 3 h, while control cultures were exposed to an identical volume of DMSO. Cultures were incubated at 30°C, and cells were harvested by centrifugation and stored at −80°C until RNA was extracted by using a Qiagen RNeasy kit. cDNA labeling, microarray production, and hybridization were performed as previously described (23). Differential gene expression was based on a normalized value of 1.5 and a P value cutoff of 0.05. Gene ontology (GO) groupings were performed by using the Candida Genome Database (http://www.Candidagenome.org/) GoSlim Mapper.
Lipid extraction and GC/MS quantification of sterols.
C. albicans SC5314 cells were grown in 20 ml YPD medium in a 125-ml Erlenmeyer flask at 30°C at 180 rpm overnight. Cells were harvested by centrifugation and resuspended in 1 ml of PBS. The OD600 was measured, and cells were diluted to an OD600 of 0.05 in 250 ml of RPMI in a 1-liter Erlenmeyer flask. AC17 and/or voriconazole was added at 5 μg/ml and 1 μg/ml, respectively. An equivalent volume of DMSO was added to control flasks. Cultures were grown for 24 h at 37°C at 180 rpm and harvested by centrifugation at 10,000 × g for 20 min. Pellets were washed twice in PBS and stored at −20°C until use. Total lipids were extracted by chloroform-methanol extraction. Briefly, cell pellets were resuspended in 10 ml of methanol and 20 ml of chloroform. Suspensions were stirred overnight at room temperature and filtered through 3 M Whatman paper. Six milliliters of 0.9% NaCl was added, and the mixture was vigorously shaken. A separation funnel was used to remove the lower layer, which was evaporated under nitrogen. Samples were standardized based on dry weight after extraction to account for growth differences between treatment groups. Samples were analyzed for ergosterol and lanosterol content by gas chromatography-mass spectrometry (GC/MS) at the Kansas Lipidomics Research Center Analytical Laboratory. GC/MS was performed on an Agilent 6890N gas chromatograph coupled to an Agilent 5975N quadrupole mass selective detector. The chromatograph was fitted with a DB-5MS capillary column with a 5% phenyl methyl siloxane stationary phase. The internal standard, cholestanol (Steraloids, Inc.), was included with each sample. Statistical analysis was performed with GraphPad Prism version 6 (GraphPad, La Jolla, CA), using one-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. A P value of <0.05 was considered statistically significant.
Agar invasion assays.
Invasive growth was determined by plating C. albicans cells from a culture grown overnight onto the surface of spider agar (1% nutrient broth [Difco], 1% mannitol, 1.35% agar, 0.2% KH2PO4) and containing a range of AC17 concentrations. Plates were incubated at 37°C for 5 to 7 days. Invasive growth was determined by visual inspection, and colony fringes were examined by using a Zeiss Discovery V12 stereoscope.
Guinea pig model of cutaneous candidiasis.
Prior to infection, each guinea pig was anesthetized with a cocktail of xylazine, ketamine, and acepromazine. The hair was plucked from the left side of each guinea pig's back, and using a stencil, a square of 2.5 by 2.5 cm was marked. The marked skin area was abraded with sterile fine-grit sandpaper. A 100-μl cell suspension containing 107 blastospores was applied onto the abraded area. Guinea pigs received 30 mg/kg of body weight of prednisolone, subcutaneously, 1 day prior to infection and 1 day postinfection in order to induce immunosuppression. Twenty-four hours after inoculation with Candida, animals were treated daily for a period of 6 days. Drugs were dissolved in PBS, and treatments were applied topically. The treatment groups consisted of 0.005% fluconazole (n = 10), 0.005% fluconazole and 2% AC17 (n = 10), 2% AC17 (n = 10), and untreated controls (n = 5). On day 7, a clinical assessment of local changes of the infected skin area was performed. In this assessment, each area was scored as follows: 0 for absent, with no erythema and no evidence of crusting; 1 for mild, with the lesion appearing pink, with minimal inflammation and evidence of light crusting; 2 for moderate, with the lesion appearing red with areas of inflammation and medium crusting present over the infected area; and 3 for severe, with the lesion appearing red over the entire infected area and thick crusting appearing over the infected area. The percent efficacy of each treatment was determined based on the average clinical score for the untreated controls.
After the clinical evaluations, each guinea pig was sacrificed, and the entire skin from the infected square was removed. The samples were homogenized in sterile saline, and serial dilutions were made and plated onto Sabouraud dextrose agar (SDA) supplemented with gentamicin and chloramphenicol. Plates were incubated at 37°C for 48 h, and CFU were then counted. Fungal tissue burden was expressed as log CFU per gram of tissue. The one-way ANOVA with the Bonferroni post hoc test was used to determine significance. Statistical analyses were performed by using SPSS for Windows version 19.0 (SPSS, Chicago, IL), and a P value of <0.05 was considered statistically significant. Animal use was in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals (24), and the Office of Laboratory Animal Welfare. The animal care and use protocol (protocol number 2009-0180) was reviewed and approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
RESULTS
Screen for miconazole potentiators.
Azole antifungals are ineffective against biofilms of C. albicans. We reasoned that it should be possible to identify potentiators of antifungals that will eradicate a biofilm. The rationale of the screen is to add compounds from a chemical library to C. albicans biofilms in the presence of miconazole (which alone has little or no activity against biofilms) and measure their combined effect. In order to make the screen compatible with high-throughput approaches, biofilms were grown in microtiter plates, and reduction of the viability dye resazurin was used as a readout. For the screen, biofilms were grown in 384-well microtiter plates, spent medium was replaced with miconazole (100 μg/ml), and compounds from chemical libraries were pin transferred into wells to give a final concentration of approximately 17 μg/ml. Biofilms were then incubated for 48 h, the drug-containing medium was removed, and biofilms were assayed for activity by using resazurin. More than 60,000 small molecules from various commercial sources were screened, and hits were categorized as having strong, medium, or weak activity based on the percent inhibition of resazurin reduction by miconazole alone (Table 1). The screen was performed in duplicate, producing 244 total hits, for an overall hit rate of 0.39%.
Table 1.
Summary of hits for miconazole potentiatorsa
| Library | No. of compounds screened | No. of hits |
Hit rate (%) | |||
|---|---|---|---|---|---|---|
| S | M | W | Total | |||
| Asinex 1 | 12,378 | 1 | 12 | 13 | 0.11 | |
| ChemBridge 3 | 8,448 | 5 | 42 | 47 | 0.56 | |
| ChemDiv 3 | 11,968 | 1 | 3 | 1 | 5 | 0.04 |
| ChemDiv 4 | 1,056 | 2 | 2 | 0.19 | ||
| Enamine 2 | 26,224 | 2 | 25 | 148 | 175 | 0.67 |
| Maybridge 5 | 3,212 | 2 | 2 | 0.06 | ||
| Commercial total | 63,286 | 3 | 34 | 207 | 244 | 0.39 |
Strong (S) hits had >75% inhibition, while medium (M) hits had >50% inhibition and weak (W) hits had >25% inhibition of resazurin reduction in the presence of miconazole.
Of the 244 hits, 198 were available for cherry pick retesting, and the screening assay was repeated with each of the hits (see Supplemental Dataset S1 in the supplemental material). Each compound was also counterscreened for the ability to inhibit growth of planktonic Candida cells in YPD medium (see Supplemental Dataset S1 in the supplemental material). Among the strong hits that showed no activity on their own but potentiated miconazole was 2-adamantanamine (AC17), a member of the Enamine 2 library and a close structural analog of the antivirals amantadine and rimantadine (Fig. 1).
Fig 1.
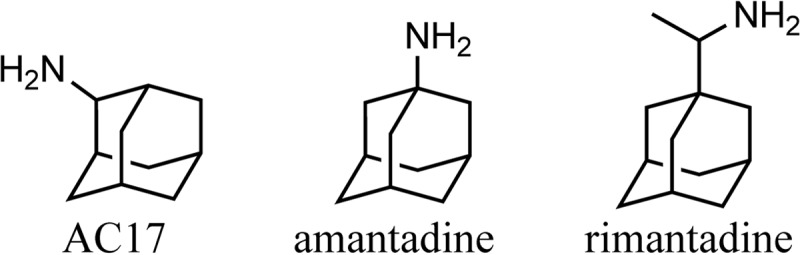
Structures of AC17, amantadine, and rimantadine.
Validation of AC17.
AC17 demonstrated a sigmoidal dose-response curve against C. albicans biofilms when tested in combination with miconazole (Fig. 2A). AC17 had no activity against biofilms when it was present alone. Similarly, AC17 did not show killing activity against biofilm cells (Fig. 2B), and neither did miconazole. However, the combination of AC17 and miconazole had a strong killing activity, diminishing the number of live biofilm cells by over 100-fold (Fig. 2B). Amantadine (IC50 = 40 μM) was 8-fold less potent than AC17 (IC50 = 5 μM), and rimantadine (IC50 = 185 μM) had over 35-fold-lower activity. AC17 did not exhibit cytotoxicity when tested against human epithelial cells and hepatocytes (IC50 = >200 μg/ml).
Fig 2.

Potentiation of miconazole action against C. albicans biofilms by AC17. (A) Dose response of AC17 in combination with a subinhibitory miconazole concentration (50 μg/ml). Percent inhibition of resazurin reduction was calculated based on miconazole-alone controls, and the IC50 (3.15 ± 1.62 μg/ml) is an average determined from 25 clinical isolates by using XLFit software. (B) Survival of C. albicans CAF2-1 biofilms exposed to miconazole (100 μg/ml) and AC17 (40 μg/ml) alone or in combination. The limit of detection was 10 cells per biofilm.
Since miconazole is primarily a topical treatment and is known to have multiple mechanisms of action at high concentrations, we needed to know whether synergy with AC17 was specific to miconazole or extended to other azole antifungals, including the systemically administered triazoles. In agar diffusion assays, AC17 at 10 μg/ml had no effect alone but increased clearing within the zone of inhibition for voriconazole, fluconazole, and miconazole (Fig. 3).
Fig 3.

Agar diffusion growth inhibition assays with AC17 and azole antifungals. For disk diffusion experiments on YPD agar, 20 μg miconazole (mic) or fluconazole (flu) and AC17 (10 μg) were added to filter disks, while 2 μg voriconazole (vori) was used. For voriconazole and fluconazole Etest strips, 10 μg/ml of AC17 was added to RPMI medium. C. albicans was added to each plate prior to the addition of filter disks or Etest strips.
Increased clearing within the zone of inhibition was confirmed in a similar experiment using Etest strips, which contained gradients of fluconazole or voriconazole. Etest strips were placed onto RPMI agar or agar containing AC17, which was previously streaked with C. albicans (Fig. 3). In the Etest assays, potentiation was detected at very low, clinically achievable concentrations (below 1 and 0.1 μg/ml for fluconazole and voriconazole, respectively). It was apparent in the disk diffusion assays that the addition of AC17 did not cause the zone of inhibition to increase; however, the increased clearing that was detected could be indicative of cidal activity. To test this hypothesis, time-kill experiments were performed. Cells were exposed to AC17, voriconazole, or a combination of both agents. The concentration of each agent was based on the agar diffusion experiments, and higher concentrations of voriconazole and AC17 (1 and 10 μg/ml, respectively) were used for fluconazole-resistant strain ATCC 96901 than for wild-type strain SC5314 (0.1 and 5 μg/ml, respectively). At 24, 48, and 72 h, samples from each treatment were removed, washed, and plated for colony counts. As expected, exposure to voriconazole or AC17 alone did not reduce colony counts, but the combination of the two was lethal (Fig. 4A and B).
Fig 4.

Effects of voriconazole and AC17 on C. albicans in time-kill assays. Wild-type SC3514 (A) or fluconazole-resistant ATCC 96901 (B) cells were diluted in RMPI medium containing voriconazole (0.1 or 1 μg/ml), AC17 (5 or 10 μg/ml), or combinations of each agent. After 24, 48, and 72 h, samples from each culture tube were washed, diluted, and plated for colony counts.
Mechanism of action.
In order to determine how AC17 functions, we performed microarray studies on cells treated with AC17 and compared the transcriptional response to that of DSMO controls. Since AC17 lacks direct growth-inhibitory effects at high concentrations, cells were exposed to high concentrations of AC17 (100 μg/ml) for 3 h. A total of 104 genes were differentially expressed in response to AC17 (see Supplemental Dataset S2 in the supplemental material). Functional characterization of these genes using GO annotations revealed that 22 of them were involved in lipid metabolism (see Supplemental Dataset S2 in the supplemental material). Among the upregulated genes were members of the ergosterol biosynthetic pathway, including ERG1, ERG7, ERG10, ERG11, ERG13, ERG24, ERG27, and ERG251 and the transcription factor UPC2, which is induced upon ergosterol depletion (25). The upregulation of the ergosterol pathway was surprising, since AC17 does not inhibit growth. We decided to test whether AC17 inhibits ergosterol production in order to validate the results of the microarray experiments. Sterols were extracted from cells exposed to AC17 or voriconazole and compared to untreated samples by using GC/MS. AC17 (5 μg/ml) caused a 68% decrease in ergosterol content compared to DMSO controls, while voriconazole (1 μg/ml) caused a 95% decrease (Fig. 5A). While AC17 did not cause a further reduction in ergosterol content for voriconazole-treated cells, this was probably due to the fact that voriconazole caused an almost complete, 95% reduction in ergosterol under the conditions tested. Voriconazole alone caused an accumulation of the sterol intermediate lanosterol, and this effect was abolished in the presence of the combination (Fig. 5B). This finding suggests that AC17 inhibits a point in the ergosterol pathway that is upstream of α-14 lanosterol demethylase, the target of voriconazole. Thereby, AC17 prevented lanosterol accumulation in voriconazole-treated cells. The microarray studies also revealed that a number of other pathways were affected by AC17. Interestingly, 15.4% of the genes differentially regulated by AC17 were involved in filamentation, even though the cells for the microarray experiment were grown at 30°C. The ability of C. albicans to form hyphae is an important virulence factor required for pathogenesis and invasion into tissues (26). We tested the ability of AC17 to prevent invasion of C. albicans into spider agar. AC17 completely abolished invasion of wild-type strain SC5314 at a fairly low concentration of 3 μg/ml (Fig. 6). AC17 also prevented invasion for a larger panel of 17 clinical isolates of C. albicans. AC17 was effective in all cases, and 1 μg/ml, which was the lowest concentration tested, was sufficient to prevent invasion for 15 of the isolates.
Fig 5.

Ergosterol (A) and lanosterol (B) contents of cells exposed to AC17, voriconazole, or a combination of the two agents. Lipids were extracted from cells exposed to AC17 (5 μg/ml), voriconazole (1 μg/ml), or a combination of the two agents and compared to DMSO controls by using GC/MS quantification. Asterisks indicate statistical significance with a P value of <0.05.
Fig 6.
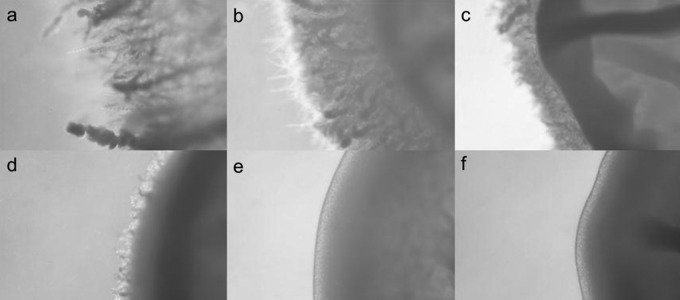
Effect of AC17 on invasion by C. albicans into agar. Strain SC5314 was plated onto spider agar containing various concentrations of AC17. Plates were incubated for 5 days at 37°C, and invasion was determined by examining colonies using a stereomicroscope. Shown are representative colonies from plates containing 0 μg/ml (a), 0.5 μg/ml (b), 1 μg/ml (c), 2 μg/ml (d), 3 μg/ml (e), and 4 μg/ml (f) of AC17.
In vivo efficacy.
A guinea pig model of cutaneous candidiasis (27, 28) was used to evaluate the efficacy of AC17 in vivo. Since fluconazole alone is effective in this model, a subinhibitory dose of fluconazole was established, which did not decrease the pathogen burden or lesion size. A fluconazole concentration of 0.005% was determined to have a suboptimal clinical effect that would be appropriate for combination studies with AC17. Next, a similar study was conducted to test the effects of AC17 (2%) alone (n = 10) or in combination with 0.005% fluconazole (n = 10). An infected, untreated control group (n = 5) was also included for comparison. The combination of AC17 and fluconazole caused a statistically significant reduction in fungal tissue burden (P value of <0.001) and demonstrated statistically significant efficacy (P value of 0.001) compared to untreated controls (Table 2). Fluconazole or AC17 treatment alone did not produce a statistically significant effect compared to untreated controls. Importantly, the combination of fluconazole and AC17 showed significant efficacy (P value of 0.018) compared to fluconazole given alone (Table 2).
Table 2.
Effect of AC17 with fluconazole in a guinea pig model of cutaneous candidiasis
| Test compound(s) | Dose(s) (%)c | Efficacy (%) | Mean tissue burden (log CFU/g tissue) ± SD |
|---|---|---|---|
| Fluconazole | 0.005 | 26.5 | 2.24 ± 1.11 |
| AC17 | 2 | 22.6 | 3.26 ± 0.52 |
| Fluconazole + AC17 | 0.005 + 2 | 60.8a,b | 1.49 ± 0.90a |
| Untreated control | NA | 3.58 ± 0.73 |
P value of <0.05 compared to the untreated control.
P value of <0.05 compared to fluconazole alone.
NA, not applicable.
DISCUSSION
New approaches are needed to overcome antifungal drug resistance and tolerance. Miconazole is used as a standard treatment of vaginal and oropharyngeal candidiasis and is applied at very high concentrations. At these levels, miconazole has additional modes of action, apart from inhibiting ergosterol biosynthesis. This explains the effectiveness of topical miconazole in the killing of planktonic cells of C. albicans. However, even miconazole at high concentrations is ineffective against biofilms, which probably explains the recurrent nature of these infections. In this study, we screened for potentiators of miconazole acting against C. albicans biofilms formed in wells of a microtiter plate. The screen was based on inhibition of resazurin reduction by cells in a biofilm.
The screen produced a number of hits, including 2-adamantanamine (AC17), which strongly potentiated miconazole. AC17 did not prevent growth of planktonic cells or affect biofilms on its own. This distinguishes AC17 from many of the other known potentiators of azoles, such as cyclosporine, amiodarone, HSP90, or calcineurin inhibitors. AC17 appears to be mechanistically distinct from other azole potentiators. Synergy between cyclosporine or radicicol and the azoles occurs through inhibition of calcineurin, but AC17 did not inhibit calcineurin according to a UTR2-lacZ reporter construct (data not shown) (22). Synergy between amiodarone, an antiarrhythmic agent, and the azoles occurs through disruption of calcium homeostasis. However, amiodarone also has fungicidal activity on its own and causes pulmonary fibrosis and thyroid and liver toxicity. Very recently, doxycycline was found to exhibit fungicidal synergy with the azoles. Synergy with doxycycline occurred at high concentrations, which are unlikely to be clinically achievable, and was due to iron chelation (29). Indeed, the link between iron chelation, membrane fluidity, and increased azole susceptibility was previously established for C. albicans (30).
Unlike miconazole, which can be fungicidal against planktonic cells, other azoles are only fungistatic. Therefore, it was interesting that fluconazole and voriconazole showed fungicidal activity in the presence of AC17. Unexpectedly, the combination of voriconazole and AC17 was also synergistic against fluconazole-resistant strain ATCC 96901. Fluconazole resistance is a major problem, and it is unlikely that the combination of fluconazole and AC17 would be effective if a strain completely extrudes fluconazole through efflux. Although the mechanism of fluconazole resistance for ATCC 96901 is unknown, the combination of AC17 and voriconazole was lethal. Additional research is needed to characterize the effectiveness of AC17 in combination with azoles against strains with different mechanisms of azole resistance.
Although AC17 was a primary screening hit, this molecule had been studied since it is a close structural analog of the antiviral amantadine. In fact, the only difference between amantadine and AC17 is a shift of the amino group from the tertiary carbon to the secondary carbon on the adamantane ring. The availability of additional amantadine analogs, such as rimantadine, allowed us to perform a preliminary structure-activity relationship analysis for antifungal activity on this class of compounds. As mentioned above, amantadine did synergize with miconazole, although it had an 8-fold-decreased potency compared to AC17. This indicates that there is some degree of flexibility for changing the position of the amine on the adamantyl core. Additional analysis indicated that the amino group itself was essential for activity, and any additional nitrogen substitution was not well tolerated and resulted in a dramatic reduction in activity. Given that rather minor expansions around the adamantane core were not tolerated, and substitutions of the core itself were not optimal, we conclude that there is a highly specific interaction between AC17 and its fungal target.
Amantadine inhibits the M2 proton channel of the influenza A virus, preventing viral uncoating and replication (31). AC17 was previously evaluated for antiviral activity, and it was found to be 8-fold less potent than amantadine (32). AC17 had low toxicity, with a 50% lethal dose (LD50) of 250 mg/kg in mice, which is actually better than amantadine (198 mg/kg) according to the NLM Toxicology database. Amantadine is also used to treat some of the symptoms of Parkinson's disease. For Parkinson's disease, daily doses of up to 400 mg are tolerated in humans, and the therapeutic effect may be due to increases in dopamine levels, although the exact mechanism is unknown. Apparently, the antiviral and neurological mechanisms of amantadine are unrelated. It is currently unknown whether AC17 also has neurological effects similar to those of amantadine, and we are investigating this possibility. To our knowledge, this is the first report of antifungal properties of AC17, and the increased antifungal potency of AC17 compared to amantadine suggests that there is an opportunity to repurpose analogs of other FDA-approved medications.
AC17 was found to inhibit ergosterol biosynthesis, providing insight into the mechanism of synergy with the azoles. Inhibitors that block different steps of the ergosterol biosynthesis pathway are known to act synergistically. For example, synergy between terbinafine, which targets squalene epoxidation (Erg1p), and the azoles, which target α-14 lanosterol demethylase (Erg11p), is well documented (33–36). The antifungal amorolfine, which inhibits the Δ14 reduction and Δ7-8 isomerization steps in the ergosterol pathway (37), also synergized with azoles in a murine model of dermatophytosis (38). While the specific target of AC17 is currently unknown, AC17 probably inhibits a point in the ergosterol pathway distinct from that of α-14 lanosterol demethylase, which results in fungicidal synergy. Since the addition of AC17 prevented the accumulation of lanosterol caused by voriconazole, it is likely that AC17 inhibits a target in the ergosterol pathway that is upstream of α-14 lanosterol demethylase. Furthermore, AC17 likely targets a nonessential gene, since it does not inhibit growth. However, we cannot rule out the possibility that AC17 partially inhibits an essential target. Additional experiments to further characterize the precise target of AC17 are under way.
The ability of AC17 to inhibit invasion by C. albicans into agar is one of its most striking antifungal phenotypes. Indeed, AC17 was effective at concentrations below 5 μM, which is an excellent value in a whole-cell assay. Invasion is mediated by the ability of C. albicans to form hyphae by undergoing a morphological transition called filamentation. The ability to transition between yeast and hyphae and invade tissue is a key virulence factor. It was surprising that AC17 was able to abolish invasion at very low concentrations, while growth was not inhibited at much higher levels (>250 μM). However, AC17-treated cells were found to have diminished ergosterol levels, and decreased ergosterol levels could account for prevention of invasion. A number of ergosterol mutants also have defects in filamentation (39, 40), and invasion is prevented in cells exposed to sub-MICs of azoles (41–43). Hyphal cells have been reported to contain 10 times more ergosterol than yeast cells (44), suggesting that a partial block of ergosterol biosynthesis may have greater effects on filamentation. Alternatively, activation of a cell membrane or cell wall compensatory pathway may allow for yeast cell growth in the presence of AC17 and not invasion. Taken together, the agar invasion and azole synergy phenotypes can both be explained, since AC17 functions through inhibition of ergosterol biosynthesis. However, the unique phenotypic properties of AC17, such as the disconnect between growth inhibition and invasion, suggest that AC17 may target a component of the ergosterol pathway for which a chemical inhibitor is unknown.
AC17 was found to improve clinical efficacy compared to fluconazole in a guinea pig model of cutaneous candidiasis. In this model, the combination of AC17 and fluconazole caused significantly reduced fungal burden compared to that in untreated controls, while each agent alone was ineffective. Interestingly, fluconazole is not typically used as a topical agent, and these data raise the intriguing possibility of exploring topical formulations of fluconazole in combination with AC17. Identification of an effective potentiator of the azoles opens the path toward further developing AC17 into a therapeutic and for the search for additional synergistically acting compounds for eradication of fungal biofilms.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Institute of Chemistry and Cell Biology Screening Facility of Harvard University for allowing us to screen their small-molecule collection. We also thank Thilani Samarakoon for performing the GC/MS analysis.
The sterol analysis described in this work was performed at the Kansas Lipidomics Research Center Analytical Laboratory. The Kansas Lipidomics Research Center was supported by the National Science Foundation (EPS 0236913, MCB 0455318, and DBI 0521587), the Kansas Technology Enterprise Corporation, K-IDeA Networks of Biomedical Research Excellence (INBRE) of the National Institutes of Health (P20RR16475), and Kansas State University. Research reported in this publication was supported by the National Institutes of Health under award numbers R43AI074258, R44AI074258, and R43DE020880.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 20 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00294-13.
REFERENCES
- 1. Edmond MB, Wallace SE, McClish DK, Pfaller MA, Jones RN, Wenzel RP. 1999. Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin. Infect. Dis. 29:239–244 [DOI] [PubMed] [Google Scholar]
- 2. Skiest DJ, Vazquez JA, Anstead GM, Graybill JR, Reynes J, Ward D, Hare R, Boparai N, Isaacs R. 2007. Posaconazole for the treatment of azole-refractory oropharyngeal and esophageal candidiasis in subjects with HIV infection. Clin. Infect. Dis. 44:607–614 [DOI] [PubMed] [Google Scholar]
- 3. Vazquez JA. 2000. Therapeutic options for the management of oropharyngeal and esophageal candidiasis in HIV/AIDS patients. HIV Clin. Trials 1:47–59 [DOI] [PubMed] [Google Scholar]
- 4. Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H. 2009. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recommend. Rep.58(RR4):1–207 [PubMed] [Google Scholar]
- 5. Vazquez JA. 2010. Optimal management of oropharyngeal and esophageal candidiasis in patients living with HIV infection. HIV AIDS (Auckl.) 2:89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pomarico L, Cerqueira DF, de Araujo Soares RM, de Souza IP, de Araujo Castro GF, Socransky S, Haffajee A, Teles RP. 2009. Associations among the use of highly active antiretroviral therapy, oral candidiasis, oral Candida species and salivary immunoglobulin A in HIV-infected children. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 108:203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Darouiche RO. 1998. Oropharyngeal and esophageal candidiasis in immunocompromised patients: treatment issues. Clin. Infect. Dis. 26:259–272 [DOI] [PubMed] [Google Scholar]
- 8. Chandra J, Mukherjee PK, Ghannoum MA. 2012. Candida biofilms associated with CVC and medical devices. Mycoses 55:46–57 [Google Scholar]
- 9. Zaoutis TE, Heydon K, Localio R, Walsh TJ, Feudtner C. 2007. Outcomes attributable to neonatal candidiasis. Clin. Infect. Dis. 44:1187–1193 [DOI] [PubMed] [Google Scholar]
- 10. Leleu G, Aegerter P, Guidet B. 2002. Systemic candidiasis in intensive care units: a multicenter, matched-cohort study. J. Crit. Care 17:168–175 [DOI] [PubMed] [Google Scholar]
- 11. Ruhnke M. 2006. Epidemiology of Candida albicans infections and role of non-Candida-albicans yeasts. Curr. Drug Targets 7:495–504 [DOI] [PubMed] [Google Scholar]
- 12. Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 125:S3–S13. 10.1016/j.amjmed.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 13. Campbell BC, Chan KL, Kim JH. 2012. Chemosensitization as a means to augment commercial antifungal agents. Front. Microbiol. 3:79. 10.3389/fmicb.2012.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cowen LE, Carpenter AE, Matangkasombut O, Fink GR, Lindquist S. 2006. Genetic architecture of Hsp90-dependent drug resistance. Eukaryot. Cell 5:2184–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Onyewu C, Blankenship JR, Del Poeta M, Heitman J. 2003. Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 47:956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Epp E, Vanier G, Harcus D, Lee AY, Jansen G, Hallett M, Sheppard DC, Thomas DY, Munro CA, Mullick A, Whiteway M. 2010. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLoS Pathog. 6:e1000753. 10.1371/journal.ppat.1000753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta SS, Ton VK, Beaudry V, Rulli S, Cunningham K, Rao R. 2003. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J. Biol. Chem. 278:28831–28839 [DOI] [PubMed] [Google Scholar]
- 18. Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179–182 [DOI] [PubMed] [Google Scholar]
- 19. Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Espinel-Ingroff A, Rodriguez-Tudela JL, Martinez-Suarez JV. 1995. Comparison of two alternative microdilution procedures with the National Committee for Clinical Laboratory Standards reference macrodilution method M27-P for in vitro testing of fluconazole-resistant and -susceptible isolates of Candida albicans. J. Clin. Microbiol. 33:3154–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. LaFleur MD, Qi Q, Lewis K. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, Cowen LE. 2009. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 5:e1000532. 10.1371/journal.ppat.1000532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sellam A, Askew C, Epp E, Tebbji F, Mullick A, Whiteway M, Nantel A. 2010. Role of transcription factor CaNdt80p in cell separation, hyphal growth, and virulence in Candida albicans. Eukaryot. Cell 9:634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 25. Hoot SJ, Oliver BG, White TC. 2008. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 154:2748–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sudbery PE. 2011. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9:737–748 [DOI] [PubMed] [Google Scholar]
- 27. Ghannoum MA, Long L, Kim HG, Cirino AJ, Miller AR, Mallefet P. 2010. Efficacy of terbinafine compared to lanoconazole and luliconazole in the topical treatment of dermatophytosis in a guinea pig model. Med. Mycol. 48:491–497 [DOI] [PubMed] [Google Scholar]
- 28. Ghannoum MA, Long L, Pfister WR. 2009. Determination of the efficacy of terbinafine hydrochloride nail solution in the topical treatment of dermatophytosis in a guinea pig model. Mycoses 52:35–43 [DOI] [PubMed] [Google Scholar]
- 29. Fiori A, Van Dijck P. 2012. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob. Agents Chemother. 56:3785–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prasad T, Chandra A, Mukhopadhyay CK, Prasad R. 2006. Unexpected link between iron and drug resistance of Candida spp.: iron depletion enhances membrane fluidity and drug diffusion, leading to drug-susceptible cells. Antimicrob. Agents Chemother. 50:3597–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pinto LH, Lamb RA. 2007. Controlling influenza virus replication by inhibiting its proton channel. Mol. Biosyst. 3:18–23 [DOI] [PubMed] [Google Scholar]
- 32. Zoidis G, Kolocouris N, Foscolos GB, Kolocouris A, Fytas G, Karayannis P, Padalko E, Neyts J, De Clercq E. 2003. Are the 2-isomers of the drug rimantadine active anti-influenza A agents? Antivir. Chem. Chemother. 14:153–164 [DOI] [PubMed] [Google Scholar]
- 33. Weig M, Muller FM. 2001. Synergism of voriconazole and terbinafine against Candida albicans isolates from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. Antimicrob. Agents Chemother. 45:966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barchiesi F, Falconi Di Francesco L, Scalise G. 1997. In vitro activities of terbinafine in combination with fluconazole and itraconazole against isolates of Candida albicans with reduced susceptibility to azoles. Antimicrob. Agents Chemother. 41:1812–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghannoum MA, Elewski B. 1999. Successful treatment of fluconazole-resistant oropharyngeal candidiasis by a combination of fluconazole and terbinafine. Clin. Diagn. Lab. Immunol. 6:921–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barrett-Bee K, Dixon G. 1995. Ergosterol biosynthesis inhibition: a target for antifungal agents. Acta Biochim. Pol. 42:465–479 [PubMed] [Google Scholar]
- 37. Polak AM. 1992. Preclinical data and mode of action of amorolfine. Clin. Exp. Dermatol. 17(Suppl 1):8–12 [DOI] [PubMed] [Google Scholar]
- 38. Polak A. 1993. Combination of amorolfine with various antifungal drugs in dermatophytosis. Mycoses 36:43–49 [DOI] [PubMed] [Google Scholar]
- 39. Jia N, Arthington-Skaggs B, Lee W, Pierson CA, Lees ND, Eckstein J, Barbuch R, Bard M. 2002. Candida albicans sterol C-14 reductase, encoded by the ERG24 gene, as a potential antifungal target site. Antimicrob. Agents Chemother. 46:947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pasrija R, Krishnamurthy S, Prasad T, Ernst JF, Prasad R. 2005. Squalene epoxidase encoded by ERG1 affects morphogenesis and drug susceptibilities of Candida albicans. J. Antimicrob. Chemother. 55:905–91315845783 [Google Scholar]
- 41. Ha KC, White TC. 1999. Effects of azole antifungal drugs on the transition from yeast cells to hyphae in susceptible and resistant isolates of the pathogenic yeast Candida albicans. Antimicrob. Agents Chemother. 43:763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Odds FC, Cockayne A, Hayward J, Abbott AB. 1985. Effects of imidazole- and triazole-derivative antifungal compounds on the growth and morphological development of Candida albicans hyphae. J. Gen. Microbiol. 131:2581–2589 [DOI] [PubMed] [Google Scholar]
- 43. Kontoyiannis DP, Tarrand J, Prince R, Samonis G, Rolston KV. 2001. Effect of fluconazole on agar invasion by Candida albicans. J. Med. Microbiol. 50:78–82 [DOI] [PubMed] [Google Scholar]
- 44. Hitchcock CA, Barrett-Bee KJ, Russell NJ. 1989. The lipid composition and permeability to the triazole antifungal antibiotic ICI 153066 of serum-grown mycelial cultures of Candida albicans. J. Gen. Microbiol. 135:1949–1955 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.