

Abstract
Parasites in the genus Plasmodium cause disease throughout the tropic and subtropical regions of the world. P. falciparum, one of the deadliest species of the parasite, relies on glycolysis for the generation of ATP while it inhabits the mammalian red blood cell. The first step in glycolysis is catalyzed by hexokinase (HK). While the 55.3-kDa P. falciparum HK (PfHK) shares several biochemical characteristics with mammalian HKs, including being inhibited by its products, it has limited amino acid identity (∼26%) to the human HKs, suggesting that enzyme-specific therapeutics could be generated. To that end, interrogation of a selected small-molecule library of HK inhibitors has identified a class of PfHK inhibitors, isobenzothiazolinones, some of which have 50% inhibitory concentrations (IC50s) of <1 μM. Inhibition was reversible by dilution but not by treatment with a reducing agent, suggesting that the basis for enzyme inactivation was not covalent association with the inhibitor. Lastly, six of these compounds and the related molecule ebselen inhibited P. falciparum growth in vitro (50% effective concentration [EC50] of ≥0.6 and <6.8 μM). These findings suggest that the chemotypes identified here could represent leads for future development of therapeutics against P. falciparum.
INTRODUCTION
Malaria is a global health concern, infecting more than 200 million people a year. While human malaria is commonly caused by four members of the genus Plasmodium, P. falciparum infection is responsible for the most mortality annually. In addition to disease, the burden of malaria has global economic consequences, with an impact in developing nations that results from reduced worker productivity and markedly increased disability-adjusted life years (DALYs), a measure of disease burden as a consequence of mortality and morbidity (World Health Organization [www.who.int]).
The parasite has a complex life cycle, with the intraerythrocytic stage being primarily responsible for pathology. This stage of Plasmodium lacks a complete tricarboxylic acid (TCA) cycle, and knockout and inhibitor studies of mitochondrial proteins have suggested that the mitochondrion is not a significant contributor to cellular ATP levels (1, 2). Notably, glucose consumption was found to be increased up to 100-fold in infected erythrocytes (3), and lactate levels were ∼20 to 100 times higher than that from uninfected cells (4, 5). These observations suggested that glycolysis was playing a key metabolic role for the parasite during the erythrocytic infection. Supporting this supposition, knockout studies revealed that the hexose transporter responsible for importing glucose was essential to the parasite, and inhibition of glycolysis with glucose analogs rapidly depleted parasite ATP (6, 7).
The first committed step in glycolysis, catalyzed by hexokinase (HK), is the transfer of the γ-phosphoryl group from ATP to glucose. This reaction yields glucose-6-phosphate (G6P), a metabolite with multiple potential fates. First, it can be consumed in glycolysis. Alternatively, if funneled into the pentose phosphate pathway (PPP), the metabolite can serve in the generation of NADPH, which is a key component in the P. falciparum antioxidant defense and de novo nucleotide triphosphate biosynthesis pathways (8).
The importance of glycolysis to the malaria parasite and the observation that the single P. falciparum HK (PfHK) is predicted to share limited (24%) identity with human glucokinases (HsGlk, or HK IV) suggested that this enzyme could serve as a suitable target for therapeutics (Fig. 1). Here, we describe the characterization of recombinant PfHK. Further, we have interrogated a small-molecule library of known HK inhibitors to identify potential lead compounds that inhibit PfHK. Lastly, we have assessed the antiparasitic activity of these molecules against P. falciparum erythrocytic-stage parasites and have found that several are potent antiparasitic compounds.
Fig 1.

Alignments of PfHK, Trypanosoma brucei HK1 (TbHK1; Tb427.10.2010), and human glucokinase (HsGlk; ABS31137.1). Sequences were aligned using CLUSTAL 0 (1.1.0) software. The “*” symbol indicates completely conserved residues, while the colons (:) mark residues that share properties (>0.5 in the Gonnet PAM250 matrix). Periods (.) indicate residues that fall at <0.5 in the Gonnet PAM250 matrix, indicating weak similarity. The boxed residue is the conserved catalytic base (Asp241 in PfHK).
MATERIALS AND METHODS
Chemicals and reagents.
Glucose-6-phosphate dehydrogenase, β-NAD (NAD+), ATP, and glucose were purchased from Sigma (St. Louis, MO). Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (Pittsburgh, PA), while phosphoenol pyruvate (PEP), 2-phenyl-1,2-benzisoselenazol-3(2H)-one (ebselen [Eb]; PubChem substance identifier [SID] 856002) and glucosamine were obtained from VWR International (West Chester, PA). The isobenzothiazolinones and benzamides used (see Table 2) were obtained from the University of Kansas Specialized Chemistry Center (KUSCC).
Table 2.
Sensitivities of PfHK to TbHK1 inhibitors
Identified in a GSK screen as inhibiting 3D7 P. falciparum growth 97% at 2 μM (http://www.ebi.ac.uk/chemblntd [13]).
ND, not determined.
Recombinant enzyme purification and assay conditions.
The open reading frame PF3D7_0624000 (PlasmoDB) for the P. falciparum hexokinase (UniProt Q02155) was synthesized for codon optimization (Genescript, Piscataway, NJ), sequenced, and cloned into pQE30 (Qiagen, Valencia, CA). Recombinant PfHK, an ∼55.3-kDa protein, was purified, following a protocol developed for heterologous expression and purification of a HK from the African trypanosome. Briefly, a 10-ml bacterial culture of Escherichia coli M15(pREP) harboring pQE30PfHK with PfHK cloned in frame of a 6-His sequence (9) was used to inoculate a 1-liter culture, which was grown to an optical density at 600 nm (OD600) of ∼1 and then induced for 24 h at room temperature (RT) with 500 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and purified as described previously (9).
HK assays were performed in triplicate as described elsewhere, using a coupled reaction to measure enzyme activity (9, 10). In short, the coupled assay uses glucose-6-phosphate dehydrogenase (G6PDH) to convert glucose-6-phosphate (G6P) generated by HK to 6-phosphogluconate with coincident reduction of NADP to NADPH, which is monitored spectrophotometrically at 340 nm. Kinetic analyses were performed using the software program KaleidaGraph 4.1 (Synergy Software, Reading, PA).
Hexokinase inhibitor library.
The collection of small molecules described here as a “selected small-molecule library of HK inhibitors” was originally developed, identified, and characterized as part of a high-throughput screening (HTS) campaign targeting hexokinase 1 from the African trypanosome, Trypanosoma brucei (TbHK1). This screen was completed at the Pittsburgh Molecular Libraries Screening Center (PMLSC) using a 220,233-compound library that was made available as part of the NIH Molecular Libraries Roadmap Initiative. Analog development of lead compound scaffolds, including the isobenzothiazolinones and benzamidobenzoic acid derivatives used here, resulted from a chemical probe development plan described in a probe report from the NIH Molecular Libraries Program (11).
P. falciparum viability assay.
Plasmodium falciparum 3D7 parasites were grown in O+ red blood cells (RBCs) at 1% hematocrit cultured in RPMI (plus l-glutamine, without NaHCO3) supplemented with 0.5% Albumax, 0.37 mM hypoxanthine, 27 mM NaHCO3, 11 mM glucose, and 10 μg/ml gentamicin under 5% CO2, 5% O2, and 90% N2 at 37°C. All assays were performed in triplicate in a total volume of 100 μl with DMSO-only incubations as a negative control. Parasitemia was determined following 72 h of incubation by flow cytometry using acridine orange staining (1.5 μg/ml in phosphate-buffered saline [PBS], 5 min, room temperature [RT], FACSCanto II flow cytometer [BD Biosystems, Franklin Lakes, NJ]).
The dose-response curves for compounds that caused >50% growth inhibition at 10 μM were pursued in a 96-well plate format using an asynchronous parasite culture. The starting parasitemia was 1%. Fifty percent inhibitory concentrations (IC50s) were determined by scoring parasite growth in three separate wells over a 2-fold dilution series starting at 40 μM for each compound. Percent inhibition was calculated by comparison to parasites grown in the DMSO controls from each plate. Chloroquine was screened as a positive control starting at 1 μM. Averages for the triplicates were calculated, and dose-response curves were fitted and IC50s determined using the software program Prism (GraphPad Software).
RESULTS
While the importance of glycolysis to P. falciparum during mammalian infection has been established (3–5), the enzymes involved in the process have not been subjected to in-depth characterization. This is likely due to the difficulty of classically purifying the parasite components from the complex milieu of the red blood cell contents (which includes many related host activities). Additionally, the A-T-rich nature of the Plasmodium genome has historically made production and analysis of recombinant enzymes difficult.
Indeed, our initial forays into expression of PfHK in E. coli were unsuccessful despite repeated efforts using a battery of expression vectors and bacterial cell lines. Expression was finally achieved using a synthetic PfHK gene optimized with a codon bias that reflected those codons employed by E. coli. The recombinant enzyme, which was purified to ∼95% homogeneity (as determined by Coomassie staining of an SDS-PAGE gel), displayed Michaelis-Menten kinetics when substrate levels were increased (Fig. 2A) and had apparent Km values for glucose and ATP (0.62 ± 0.06 and 0.66 ± 0.08 mM, respectively) similar to those from yeast HK and T. brucei TbHK1 (Table 1) (9). The PfHK kcat value was 1.2 × 104 min−1, similar to that reported for a recently characterized T. brucei HK1 (2.9 × 104 min−1) (9). PfHK had an affinity for MgCl2 (apparent Km = 2.7 ± 0.4 mM) similar to that of TbHK1 (apparent Km = 0.92 ± 0.21 mM). PfHK lacked detectable activity when CaCl2 or MnCl2 was included in place of MgCl2 in the standard assay. The optimal pH for enzyme activity was found to be about pH 7.4, with enzyme activity reduced ∼50% by a 1.2 pH unit change in either the basic or acidic direction (Fig. 2B).
Fig 2.
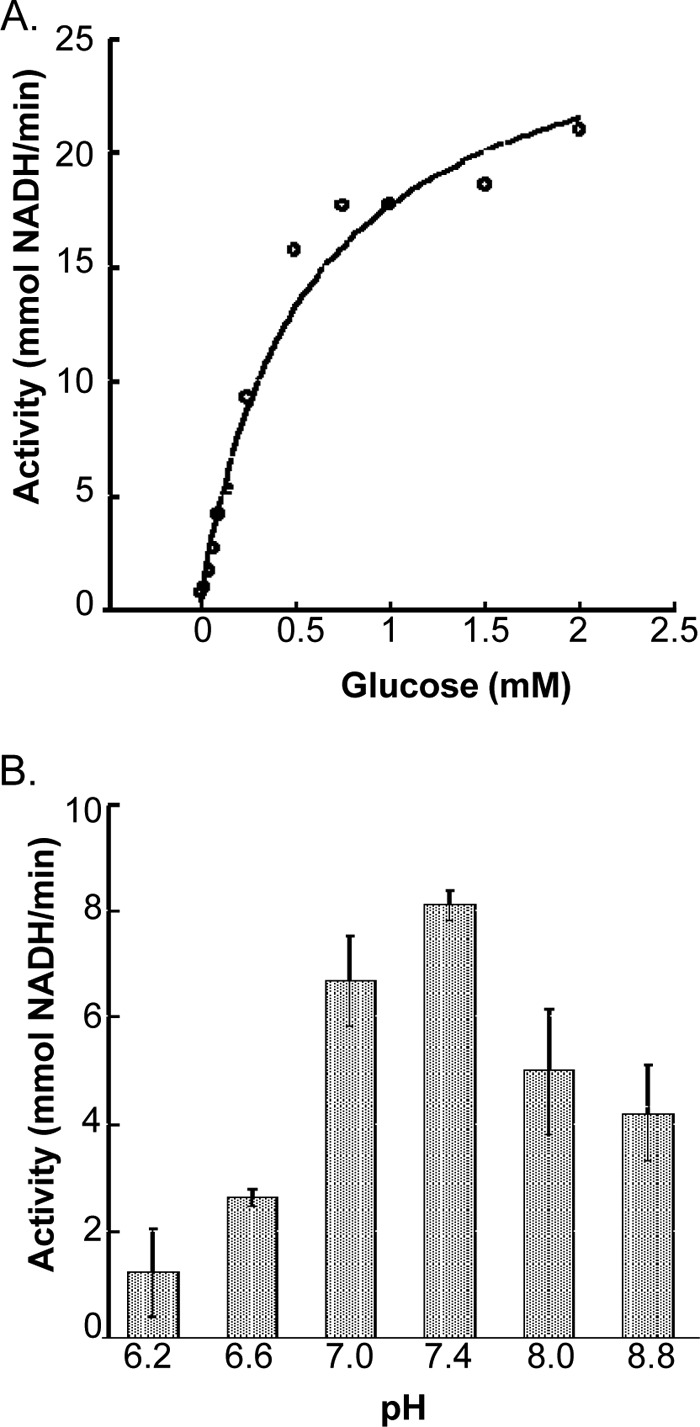
Effects of glucose concentration and pH on PfHK activity. (A) PfHK (200 ng) activity in response to increasing glucose concentration. (B) The pH profile of PfHK (200 ng) activity. Three different buffers with buffering capacity in the range of pH tested were used for the assay: piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.2, 6.5, and 7.0), triethanolamine (pH 7.4), and Tris (pH 8.0 and 8.8). Reactions were carried out in triplicate, and standard deviations are indicated.
Table 1.
Kinetic characterization of rPFHKa
| HK | kcat (min−1) | Apparent Km (mM) |
|
|---|---|---|---|
| Glucose | ATP | ||
| rPfHK | 1.2 × 104 | 0.62 ± 0.06 | 0.66 ± 0.08 |
| rTbHK1b | 2.9 × 104 | 0.09 ± 0.02 | 0.28 ± 0.1 |
rPfHK and rTbHK1, recombinant PfHK and TbHK1.
Data from reference 9.
The enzyme lacked detectable activity when alternative phosphoryl donors, including UTP, GTP, CTP, TTP, ITP, and PPi, were tested. Notably, ATP at concentrations greater than 3.5 mM was inhibitory to the reaction (data not shown). Products also inhibited the enzyme, with inclusion of ADP (10 mM) in the reaction inhibiting the enzyme ∼94% (Fig. 3). PPi was also inhibitory, while AMP only modestly (∼22%) inhibited PfHK at 10 mM. G6P, another product of PfHK, inhibited the enzyme ∼80% at 10 mM.
Fig 3.
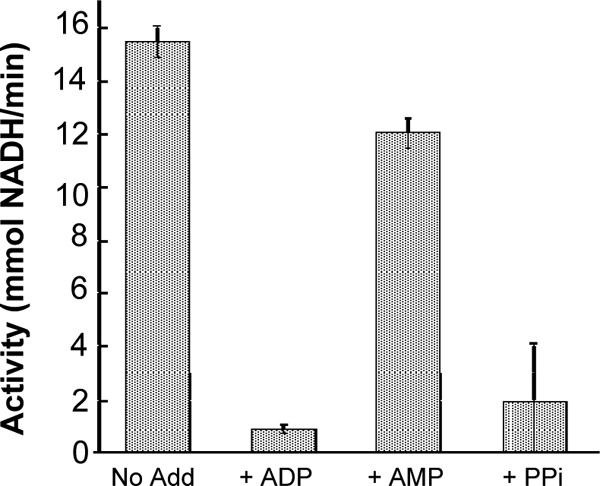
Effects of phosphoryl-bearing compounds on PfHK activity. PfHK (200 ng) activity was assessed in the presence of 5 mM ATP supplemented with ADP, PPi, or AMP (all at 5 mM). Assays were performed in triplicate as described in Materials and Methods, and standard deviations are indicated.
Identification of small-molecule inhibitors of PfHK.
The completion of an HTS for inhibitors of T. brucei hexokinase 1 (TbHK1) yielded several scaffolds that have since been further elaborated to resolve structure-activity relationships against that enzyme (12). We have taken advantage of the reagents and assays developed therein to interrogate this selected small-molecule library of HK inhibitors for PfHK inhibitors. These compounds have been rigorously characterized. For example, they do not inhibit the coupled reporter enzyme, are typically poor inhibitors of human glucokinase, have variable activity against TbHK1, and are minimally toxic to a number of mammalian cell lines (including A549, IMR-90, HeLa, and MDA-MB-231 (12).
Of the 16 hits obtained from the HTS effort against TbHK1, two groups were pursued as scaffolds for analog development against TbHK1. Our interest in the first analog group, which was based on five representative isobenzothiazolinones found in the TbHK1 HTS, was particularly piqued because one analog, SID 92126115, had been previously identified in a screen performed by GlaxoSmithKline of more than 2 million compounds for molecules with anti-Plasmodium activity (http://www.ebi.ac.uk/chemblntd) (13). SID 92126115, which inhibited parasite growth 97% at 2 μM, was a potent PfHK inhibitor (IC50 = 0.16 ± 0.04 μM) (Table 2). Other isobenzothiazolinones lacking the halogen substituents (SID 85747718 and SID 17387000) had slightly increased IC50s, though substitution of the sulfur atom with a selenium atom (to yield SID 856002, ebselen [Eb]) increased potency of inhibition, a trend similar to that seen when Eb inhibition of TbHK1 was compared to that seen with SID 17387000. Interestingly, Eb and SID 17387000 inhibition was reversible by dilution (Fig. 4 and data not shown), which is in contrast to the irreversible inhibition observed with TbHK1 (M. T. Harris and J. C. Morris, unpublished observation). The reducing reagent dithiothreitol (DTT) was able to prevent but not reverse Eb inhibition of PfHK, likely as a consequence of the preventative DTT binding directly to Eb to block its interaction with the enzyme (14). Last, the presence of the sulfur or selenium atom was important for inhibition of the enzyme, since substitution with either a carbon or oxygen atom at that position (SID 87225568 and 85752767, respectively) ablated inhibition.
Fig 4.
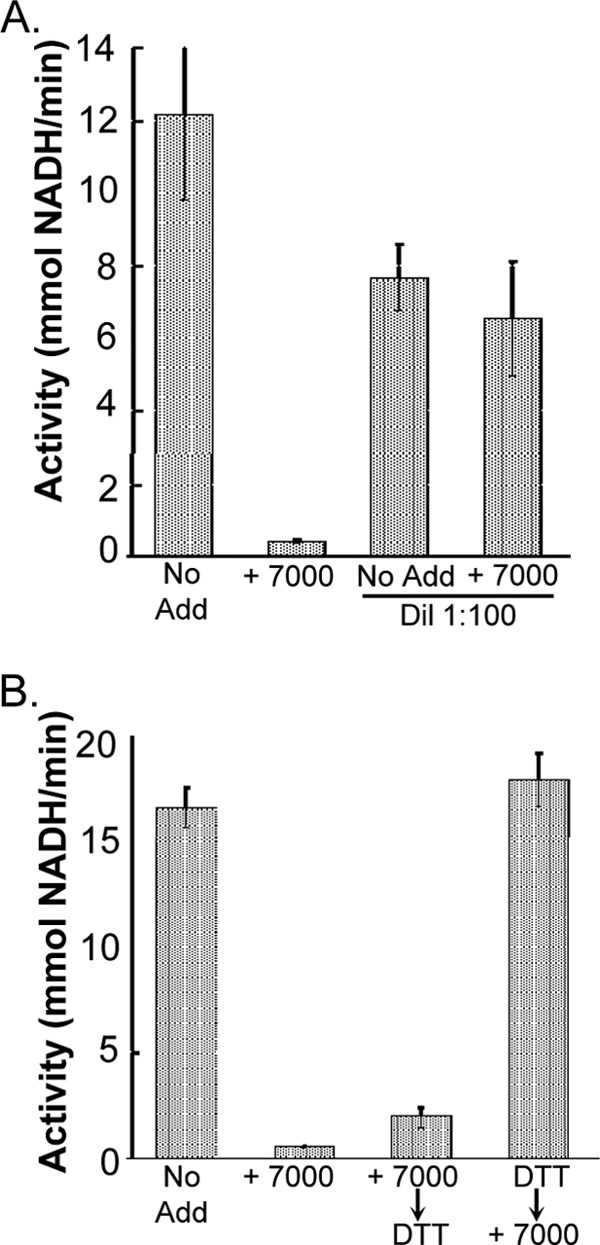
Characterization of PfHK inhibition by SID 17387000. (A) Dilution after inhibition with SID 17387000 recovers enzyme activity. PfHK (200 ng) was incubated with SID 17387000 (0.5 μM) or buffer (10 min, on ice) before addition of other assay components, resulting in an ∼100-fold reduction in the SID 17387000 concentration. (B) Addition of the reducing agent DTT (100 mM) before but not after addition of SID 17387000 (0.5 μM) prevented PfHK inhibition. Standard deviations are indicated.
We also explored inhibition of PfHK with members of a second scaffold identified in the TbHK1 HTS, based around the benzamidobenzoic acid identified in the screen (SID 93575727). Further development of this class of compounds has yielded improved inhibitors of TbHK1, including the probe ML205 (SID 99437306) (Table 1) (11). However, against PfHK, representatives of this class were either poor inhibitors (ML205 was ∼40% inhibitory at 10 μM) or lacked detectable activity against the enzyme.
PfHK inhibitors are toxic to P. falciparum.
Glycolysis is important to P. falciparum during the erythrocytic infection, suggesting that inhibitors of the enzyme could be promising lead compounds for therapeutic development. To explore this possibility, we grew cultured Plasmodium parasites in the presence of compound and monitored cell density after 72 h. Both the isobenzothiazolinones and benzamidobenzoic acid-based compounds were tested in this assay, with 50% effective concentrations (EC50s) determined for those that inhibited cell growth >50% at 10 μM (Fig. 5 and Table 2). Compounds from the isobenzothiazolinone group included two of the most potent antimalarial compounds, SID 92126115 and SID 85747718. While all of the compounds that inhibited PfHK were toxic to parasites, one molecule (SID 85742034) was toxic to parasites while not inhibiting PfHK. With the exception of SID 87225569, the antiparasitic compounds had EC50s that were 4- to 680-fold higher than the PfHK IC50s. This discrepancy could result from the inability of the molecules to reach their target as a consequence of limited RBC and/or parasite cell permeability or trafficking. The identified PfHK inhibitors have minimal impact on human cell lines, with EC50s of >12.5 μM (12), suggesting at least 20-fold-greater toxicity toward parasites for the most potent compound (Table 2).
Fig 5.
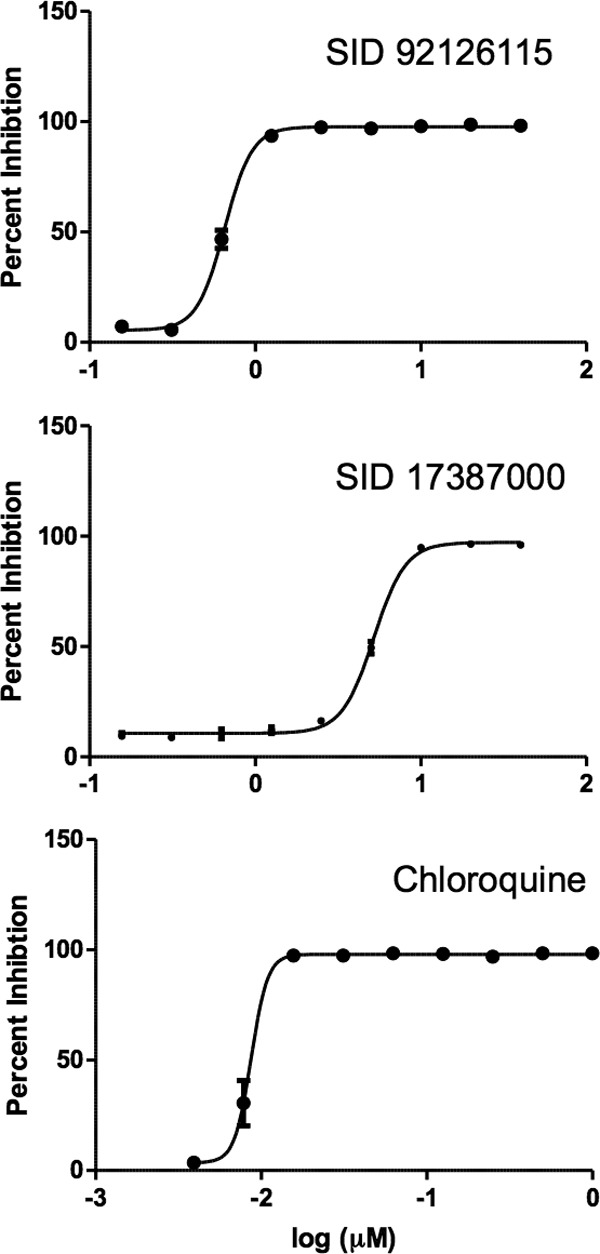
Isobenzothiazolinones inhibited the growth of P. falciparum 3D7 parasites in a dose-dependent manner. Twofold dilution series of the compounds SID 92126115 and SID 17387000 were incubated with parasites, and percent inhibition was calculated by comparison to results for parasites grown in the DMSO controls from each plate. Chloroquine was included as a positive control. Averages for experiments performed in triplicate were calculated and EC50s were determined using Prism (GraphPad Software), with standard deviations indicated.
DISCUSSION
The intraerythrocytic malaria parasite requires extracellular glucose for survival, largely due to the significant consumption of the hexose as a consequence of ATP generation by glycolysis. Inhibitors of this pathway have demonstrated potent antimalarial activity. These include small molecules that interfere with hexose transport (15), as well as glucose analogs like 2-deoxyglucose (6, 16).
PfHK, the first enzyme in glycolysis, has heretofore been considered an interesting potential target but has not been exploited due to an inability to produce active recombinant protein for in vitro characterization (17). Using an E. coli codon-optimized gene, we have for the first time generated active PfHK. Characterization of PfHK revealed it to share a number of features with other HKs, including the finding that the reaction products G6P and ADP were inhibitors of the enzyme, suggesting the Plasmodium protein may be regulated via the feedback mechanism described for vertebrate HKs (18). This observation was somewhat surprising given that an HK from the related apicomplexan parasite Toxoplasma gondii (which is 44% identical to PfHK) is not sensitive to substrate inhibition (19). PfHK is also kinetically similar to other HKs, having glucose, ATP, and MgCl2 Km values similar to those of HKs from other systems, including T. brucei and Homo sapiens. The PfHK Km value for glucose was similar to that described by Roth from activity associated with infected RBC lysates (620 μM and 431 μM, respectively) (20). These values are ∼5-fold higher than the Km reported from HK activity associated with uninfected RBC lysates, suggesting that the host enzyme should outcompete the parasite enzyme for glucose under limiting conditions. However, glucose transport in the intraerythrocytic parasite is highly efficient, with the concentration of the substrate inside the parasite rapidly reaching levels higher than that found in the external medium (15).
The second PfHK substrate, ATP, is required for activity but can also be an inhibitor of the enzyme. PfHK has a Km value for ATP well below the intraerythrocytic ATP concentration (∼3 mM) and is inhibited at ATP levels approaching those found in the host cytoplasm. While we anticipate that PfHK utilizes a pool of ATP distinct from that found in the host cytoplasm, there could be conditions under which the parasite has generated sufficient ATP to satisfy its needs. To continue to synthesize ATP could be damaging to the host cell, either directly as a consequence of usurping host glucose or by depletion of host purines required for ATP synthesis, since the parasite cannot synthesize purines and must salvage them from the host. The finding that ATP is a regulator of HK activity is not novel among HKs (21), though its role in the malaria parasite remains unclear.
Taking advantage of a preexisting library of small molecules designed around two scaffolds known to inhibit a trypanosome HK, we searched for inhibitors of PfHK. The two enzymes share limited sequence identity (28%) (Fig. 1), but nevertheless we found that members of one of the two classes of analogues, the isobenzothiazolinones, were more potent against PfHK than against TbHK1 (Table 2). Additionally, the isobenzothiazolinone analog Eb was extremely potent against PfHK (IC50 = 0.01 ± 0.00). Eb can promiscuously modify cysteine residues, and this nonspecific interaction is known to be the mechanism of its inhibition of some enzymes, such as human indoleamine 2,3-dioxygenase (22). However, site-directed mutagenesis of TbHK1 cysteines, including the two conserved in PfHK (residues 273 and 398 in PfHK), did not alter TbHK1 sensitivity to Eb inhibition, indicating that either cysteine residues are not involved in Eb inhibition or multiple cysteines must be bound in order for inhibition to occur. Unlike the trypanosome enzyme, PfHK inhibition by Eb and the isobenzothiazolinone SID 17387000 was reversible by dilution, suggesting that the inhibitors interact with the two enzymes differently.
Previous research has demonstrated that Eb is toxic to P. falciparum, with EC50s similar to those found in our study (14 μM and 6.8 μM, respectively) (23). Eb may have a polypharmacological impact on the parasite, given the likely reactivity of the selenium atom with cellular cysteines. However, it is nontoxic for humans and is currently in phase III clinical trials for the treatment of stroke survivors (Stroke Trials Registry Home Page as of 27 February 2013), suggesting that the consequences of off-target effects in the human host may be tolerable.
The isobenzothiazolinone SID 92126115 was previously identified in a screen performed by GlaxoSmithKline of more than 2 million compounds for molecules with anti-Plasmodium activity, inhibiting parasite growth 97% at 2 μM (http://www.ebi.ac.uk/chemblntd) (13). SID 92126115 was a potent PfHK inhibitor, with an IC50 of 0.16 ± 0.04 μM. Isobenzothiazolinone inhibitors of PfHK were also effective at inhibiting the growth of cultured parasites, with EC50s below 10 μM. The EC50s were higher that IC50s found for PfHK, likely due to issues with cell permeability and off-target interactions. With the exception of SID 85742034, compounds that did not inhibit PfHK did not kill parasites.
In conclusion, several observations support the value of PfHK as a therapeutic target. First, the single-copy gene product shares limited (26%) identity with the human-equivalent enzyme. Second, the enzyme is responsible for catalyzing the first reaction in two critical pathways, glycolysis and the PPP, and is likely indispensable for parasite viability. Last, inhibitors of the enzyme can inhibit parasite proliferation, as demonstrated here. Together, these findings suggest the value of further consideration of the isobenzothiazolinone scaffold, as well as identification of additional new chemotypes, as a potential lead in therapeutic development.
ACKNOWLEDGMENTS
We thank Meredith T. Morris for her comments on the manuscript.
This work was supported in part by the U.S. National Institutes of Health, grants 2R15AI075326 to J.C.M. and U54HG005031 to the University of Kansas Specialized Chemistry Center.
Footnotes
Published ahead of print 28 May 2013
REFERENCES
- 1. Vaidya AB, Mather MW. 2009. Mitochondrial evolution and functions in malaria parasites. Annu. Rev. Micro. 63:249–267 [DOI] [PubMed] [Google Scholar]
- 2. Olszewski KL, Mather MW, Morrisey JM, Garcia BA, Vaidya AB, Rabinowitz JD, Llinas M. 2010. Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature 466:774–778 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Roth EF, Jr, Raventos-Suarez C, Perkins M, Nagel RL. 1982. Glutathione stability and oxidative stress in P. falciparum infection in vitro: responses of normal and G6PD deficient cells. Biochem. Biophys. Res. Commun. 109:355–362 [DOI] [PubMed] [Google Scholar]
- 4. Pfaller MA, Krogstad DJ, Parquette AR, Nguyen-Dinh P. 1982. Plasmodium falciparum: stage-specific lactate production in synchronized cultures. Exp. Parasitol. 54:391–396 [DOI] [PubMed] [Google Scholar]
- 5. Vander Jagt DL, Hunsaker LA, Campos NM, Baack BR. 1990. D-lactate production in erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 42:277–284 [DOI] [PubMed] [Google Scholar]
- 6. van Schalkwyk DA, Priebe W, Saliba KJ. 2008. The inhibitory effect of 2-halo derivatives of D-glucose on glycolysis and on the proliferation of the human malaria parasite Plasmodium falciparum. J. Pharmacol. Exp. Ther. 327:511–517 [DOI] [PubMed] [Google Scholar]
- 7. Slavic K, Straschil U, Reininger L, Doerig C, Morin C, Tewari R, Krishna S. 2010. Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol. Microbiol. 75:1402–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muller S. 2004. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol. Microbiol. 53:1291–1305 [DOI] [PubMed] [Google Scholar]
- 9. Morris MT, DeBruin C, Yang Z, Chambers JW, Smith KS, Morris JC. 2006. Activity of a second Trypanosoma brucei hexokinase is controlled by an 18-amino-acid C-terminal tail. Eukaryot. Cell 5:2014–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misset O, Opperdoes FR. 1984. Simultaneous purification of hexokinase, class-I fructose-bisphosphate aldolase, triosephosphate isomerase and phosphoglycerate kinase from Trypanosoma brucei. Eur. J. Biochem. 144:475–483 [DOI] [PubMed] [Google Scholar]
- 11. Sharlow E, Golden JE, Dodson H, Morris M, Hesser M, Lyda T, Leimgruber S, Schroeder CE, Flaherty DP, Weiner WS, Simpson D, Lazo JS, Aube J, Morris JC. 2010. Identification of inhibitors of Trypanosoma brucei hexokinases. Probe reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information, Bethesda, MD: [PubMed] [Google Scholar]
- 12. Sharlow ER, Lyda TA, Dodson HC, Mustata G, Morris MT, Leimgruber SS, Lee KH, Kashiwada Y, Close D, Lazo JS, Morris JC. 2010. A target-based high throughput screen yields Trypanosoma brucei hexokinase small molecule inhibitors with antiparasitic activity. PLoS Neg. Trop. Dis. 4:e659. 10.1371/journal.pntd.0000659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310 [DOI] [PubMed] [Google Scholar]
- 14. Borges VC, Rocha JB, Nogueira CW. 2005. Effect of diphenyl diselenide, diphenyl ditelluride and ebselen on cerebral Na(+), K(+)-ATPase activity in rats. Toxicology 215:191–197 [DOI] [PubMed] [Google Scholar]
- 15. Saliba KJ, Krishna S, Kirk K. 2004. Inhibition of hexose transport and abrogation of pH homeostasis in the intraerythrocytic malaria parasite by an O-3-hexose derivative. FEBS Lett. 570:93–96 [DOI] [PubMed] [Google Scholar]
- 16. Udeinya IJ, Van Dyke K. 1981. 2-Deoxyglucose: inhibition of parasitemia and of glucosamine incorporation into glycosylated macromolecules, in malarial parasites (Plasmodium falciparum). Pharmacology 23:171–175 [DOI] [PubMed] [Google Scholar]
- 17. Olafsson P, Matile H, Certa U. 1992. Molecular analysis of Plasmodium falciparum hexokinase. Mol. Biochem. Parasitol. 56:89–101 [DOI] [PubMed] [Google Scholar]
- 18. Easterby JS, Qadri SS. 1982. Hexokinase type II from rat skeletal muscle. Methods Enzymol. 90:11–15 [DOI] [PubMed] [Google Scholar]
- 19. Saito T, Maeda T, Nakazawa M, Takeuchi T, Nozaki T, Asai T. 2002. Characterisation of hexokinase in Toxoplasma gondii tachyzoites. Int. J. Parasitol. 32:961–967 [DOI] [PubMed] [Google Scholar]
- 20. Roth EF., Jr 1987. Malarial parasite hexokinase and hexokinase-dependent glutathione reduction in the Plasmodium falciparum-infected human erythrocyte. J. Biol. Chem. 262:15678–15682 [PubMed] [Google Scholar]
- 21. Dodson HC, Morris MT, Morris JC. 2011. Glycerol 3-phosphate alters Trypanosoma brucei hexokinase activity in response to environmental change. J. Biol. Chem. 286:33150–33157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terentis AC, Freewan M, Sempertegui Plaza TS, Raftery MJ, Stocker R, Thomas SR. 2010. The selenazal drug ebselen potently inhibits indoleamine 2,3-dioxygenase by targeting enzyme cysteine residues. Biochemistry 49:591–600 [DOI] [PubMed] [Google Scholar]
- 23. Huther AM, Zhang Y, Sauer A, Parnham MJ. 1989. Antimalarial properties of ebselen. Parasitol. Res. 75:353–360 [DOI] [PubMed] [Google Scholar]