

Abstract
Penicillin-binding proteins (PBPs), which catalyze the biosynthesis of the peptidoglycan chain of the bacterial cell wall, are the major molecular target of bacterial antibiotics. Here, we present the crystal structures of the bifunctional peptidoglycan glycosyltransferase (GT)/transpeptidase (TP) PBP4 from Listeria monocytogenes in the apo-form and covalently linked to two β-lactam antibiotics, ampicillin and carbenicillin. The orientation of the TP domain with respect to the GT domain is distinct from that observed in the previously reported structures of bifunctional PBPs, suggesting interdomain flexibility. In this structure, the active site of the GT domain is occluded by the close apposition of the linker domain, which supports the hypothesis that interdomain flexibility is related to the regulation of GT activity. The acylated structures reveal the mode of action of β-lactam antibiotics toward the class A PBP4 from the human pathogen L. monocytogenes. Ampicillin and carbenicillin can access the active site and be acylated without requiring a structural rearrangement. In addition, the active site of the TP domain in the apo-form is occupied by the tartrate molecule via extensive hydrogen bond interactions with the catalytically important residues; thus, derivatives of the tartrate molecule may be useful in the search for new antibiotics to inhibit PBPs.
INTRODUCTION
The bacterial cell wall peptidoglycan is essential for normal cell survival and for proper maintenance of cellular morphology (1). The cell wall consists of glycan chains of alternating N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) cross-linked by short stem peptides attached to the adjacent NAM. The biosynthesis of peptidoglycan is mediated by penicillin-binding proteins (PBPs) through two enzymatic reactions: transglycosylation, which uses glycosyltransferase (GT) to form the glycan backbone by polymerizing disaccharides, and transpeptidation, which uses transpeptidase (TP) to catalyze cross-linking between adjacent glycan chains to create a mesh-like structure (2). As indicated by their name, PBPs are the primary targets of β-lactam antibiotics, including penicillin, which act as mechanism-based inhibitors to mimic the d-alanyl–d-alanine moiety of peptidoglycan precursors (3–5). β-Lactams block the catalytic activity of the TP domain by forming a covalent bond with the catalytic serine residue of the enzyme (6). Based on sequence similarities, the enzymes can be classified into three main classes: A, B, and C (7). Although class A and B PBPs commonly contain C-terminal TP domains, class A PBPs are distinguished from class B PBPs by the structure and catalytic activity of their N-terminal domains. In class A PBPs, the N-terminal domain contains GT, whereas the N-terminal domain of class B PBPs is usually involved in interactions with other proteins related to cell division (8). Class C PBPs are monofunctional enzymes with low-molecular-mass PBPs and have been proposed to play a role in peptidoglycan maturation (9).
Class A PBPs contain both GT and TP domains on the same polypeptide (8) and thus are optimally evolved to synthesize the peptidoglycan bacterial cell wall. In addition, the bifunctional enzymes have garnered attention due to potential applications in drug development. The GT domain has been proposed as an attractive target for new antibacterial agents as a result of the increasing threat of multidrug-resistant bacteria (10). Furthermore, some class A PBPs are involved in the development of β-lactam antibiotic resistance due to mutations in the TP domain (11, 12). Therefore, structural information regarding class A PBPs is important for understanding the two enzymatic activities, as well as for drug development. However, only a few crystal structures of class A PBPs containing both the GT and TP domains have been reported from the Gram-positive Staphylococcus aureus PBP2 (SaPBP2) (13, 14) and the Gram-negative Escherichia coli PBP1b (EcPBP1b) (15), both in complex with moenomycin in the GT domain.
The food-borne pathogen Listeria monocytogenes contains five PBPs, including two class A enzymes (PBP1 and PBP4), two class B enzymes (PBP2 and PBP3), and one class C enzyme (PBP5) (Table 1), which were identified by their ability to bind to radiolabeled β-lactams (16, 17). Among the PBPs of L. monocytogenes, a soluble form of PBP4 (residues 71 to 714) from L. monocytogenes (LmPBP4) contributes most significantly to virulence potential and increased sensitivity to β-lactams (18), indicating that LmPBP4 may be an important target in the treatment of listeriosis. To better understand the function of class A PBPs, we determined the crystal structures of PBP4 in apo-form and in acyl-enzyme complexes with two β-lactams, ampicillin and carbenicillin. Unexpectedly, the active site of apo-form PBP4 is occupied by a tartrate molecule; the binding mode of tartrate is compared with the acylated β-lactams. Our findings will assist in the development of new antibiotics, as well as the understanding of the structural mechanism for the recognition of PBP4 by antibiotics.
Table 1.
Reported PBPs of L. monocytogenes
| PBP | Gene | Mass (kDa) | Class | Putative function |
|---|---|---|---|---|
| PBP1 | lmo1892 | 90.8 | A | Carboxypeptidase Glycosyltransferase Transpeptidase |
| PBP2 | lmo2039 | 81.8 | B | Transpeptidase FtsI |
| PBP3 | lmo1438 | 79.9 | B | Transpeptidase FtsI |
| PBP4 | lmo2229 | 77.9 | A | Carboxypeptidase Glycosyltransferase Transpeptidase |
| PBP5 | lmo2754 | 48.1 | C | Carboxypeptidase Transpeptidase |
MATERIALS AND METHODS
Purification of PBP4.
The truncated version of LmPBP4 (residues 73 to 714) was expressed in the Escherichia coli BL21(DE3) RIL strain and purified as described previously (19). In brief, LmPBP4 was expressed as a 6His-tagged fusion protein and obtained after cleavage by Tev protease. Purification of LmPBP4 required the use of an Ni-nitrilotriacetic acid (NTA) column (Qiagen), a Hitrap Q anion-exchange column (GE Healthcare), and a Superdex 200 column (GE Healthcare). Selenomethionine-substituted protein was prepared by transforming E. coli B834(DE3) RIL methionine auxotroph cells (Novagen) with the pProExHTb vector containing the lmo2229 gene and growing the cells in selenomethionine-containing minimal medium. Purification of selenomethionyl LmPBP4 was performed as described above.
Crystallization and structure determination.
Native crystals were obtained by the sitting-drop vapor diffusion method at 22°C by mixing and equilibrating 2 μl each of the protein solution and a precipitant solution containing 0.2 M ammonium tartrate dibasic, 22% (wt/vol) polyethylene glycol (PEG) 3350, and 100 mM Tris-HCl (pH 8.0), as described previously (19). Acyl complexes were obtained by soaking native crystals in the crystallization solution containing 10 mM β-lactam antibiotics (ampicillin or carbenicillin, all obtained from Sigma-Aldrich Co.). For data collection, the crystals were briefly immersed in the same precipitant containing an additional 15% (wt/vol) glycerol and immediately placed in a 100 K nitrogen gas stream. The single-wavelength anomalous-dispersion data set (SAD) was collected using a native crystal of the selenomethionine-substituted protein on beamline 5C of the Pohang Light Source (PLS) (Republic of Korea). Phenix.autosol was used to locate Se sites and produce a solvent-flattened map (20). Model building and refinement were performed using Coot (21) and CNS (22), respectively. The X-ray diffraction and structure refinement statistics are summarized in Table S1 in the supplemental material.
Protein structure accession numbers.
The atomic coordinates and structure factors for apo-LmPBP4, the LmPBP4-ampicillin complex, and the LmPBP4-carbenicillin complex have been deposited in the Protein Data Bank (PDB) under accession codes 3zg7, 3zg8, and 3zga, respectively.
RESULTS AND DISCUSSION
Overall structure of penicillin-binding protein 4.
The crystal structure of a soluble form of PBP4 (residues 71 to 714) from L. monocytogenes (LmPBP4) was determined to 2.0 Å by the single-wavelength anomalous-dispersion method using selenomethionyl-substituted protein. The asymmetric unit of the crystal contains one PBP4 molecule, and crystal-packing interactions exhibit no sign of oligomer formation of PBP4. Therefore, LmPBP4 would likely function as a monomer physiologically, consistent with previously reported class A PBPs (23, 24). In the refined LmPBP4 structure, electron density is visible for residues 77 to 117 and 186 to 641, which contain three distinct domains: an N-terminal GT domain (residues 92 to 288), a linker domain (residues 77 to 91, 289 to 323, and 518 to 534), and a C-terminal TP domain (residues 324 to 517 and 535 to 641) (Fig. 1A). The overall architecture of LmPBP4 shares the general fold of class A PBPs (Fig. 1B) (14, 23).
Fig 1.
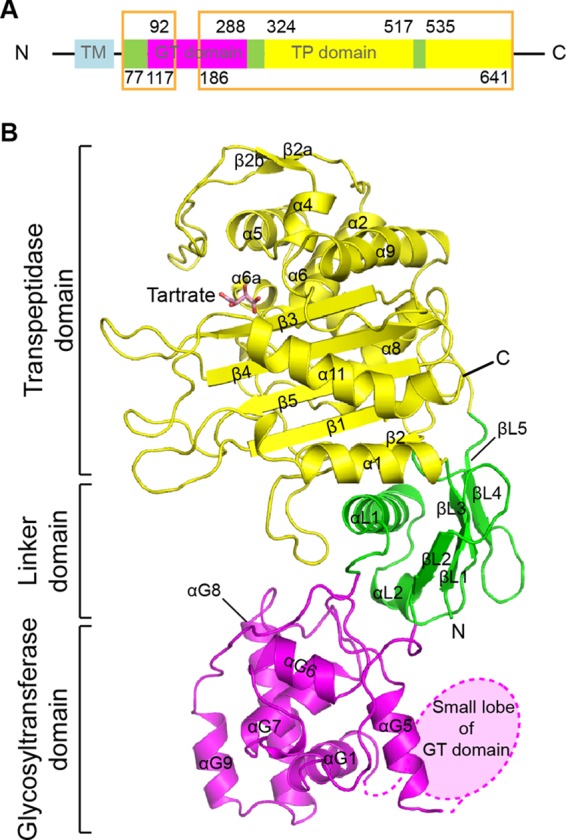
Structure of PBP4 from L. monocytogenes. (A) Schematic representation of the organization of LmPBP4 domains. The transmembrane helix (TM), GT, linker, and TP domains of LmPBP4 are sky blue, magenta, yellow, and green, respectively. The regions presented in the crystal structures are boxed. (B) Ribbon representation of the apo-LmPBP4 structure. The secondary structural elements are colored as in panel A. A tartrate molecule bound to the active site is presented as a stick model. In this orientation, the TP domain is at the top, and the GT domain is at the bottom.
The GT domain is usually composed of nine α-helices organized into large and small lobes that are separated by a deep cleft forming the active site in which the glycan chain binds (23). There was no electron density for the small-lobe region, which consists of residues 118 to 185 and contains the catalytic glutamate residues. In the crystal, the solvent channel around the GT domain does not provide enough space where the small lobe could have moved around as a rigid body. Therefore, we speculated that LmPBP4 was cleaved by contaminated proteases during crystallization and/or protein purification. SDS-PAGE analysis and N-terminal amino acid sequencing of dissolved LmPBP4 crystals clearly revealed the cleavages of LmPBP4 (data not shown). The susceptibility of the small lobe to proteolytic cleavage indicates that there is conformational flexibility of the active-site cleft, likely involving movement of the small lobe toward the other part of the GT domain. However, the N-terminal segment (residues 77 to 117) has remained bound to the main body of the protein as the essential parts of two domains: the GT domain and the linker domain. The association of a short peptide segment after proteolytic cleavages was also observed in the crystal structure (PDB code 2C5W) of bifunctional PBP1a from Streptococcus pneumoniae (25). On the other hand, the large lobe of the GT domain shows well-defined electron density in the structure, with a root mean square deviation (RMSD) of 1.2 Å over 116 C-α carbon atoms from the corresponding domains of SaPBP2 (PDB code 3DWK), indicative of the rigidity of the remaining GT domain.
As observed in other class A PBPs, the linker domain connecting the GT domain to the TP domain is composed of a five-stranded β-sheet and a perpendicular helix (14, 25). The following TP domain, composed of the α-helical subdomain and the α/β-subdomain, shares its overall fold with other PBPs and the serine β-lactamases (Fig. 1B) (26). The α-helical subdomain comprises helices α2, α4 to α6, and α9 and an additional two-stranded antiparallel β-sheet (β2a-β2b). The α/β-subdomain can be described by a central five-stranded antiparallel β-sheet (β3-β4-β5-β1-β2), which is sandwiched between three helices: α1 and α11 on one side and α8 on the opposite side of the sheet. Superposition of the LmPBP4 TP domain with the corresponding domains of SaPBP2 (PDB code 2OLV) (23) and EcPBP1b (PDB code 3FWM) (24) reveals high structural homology, with RMSDs of 1.5 Å and 1.6 Å over 274 C-α carbon atoms, respectively.
Interdomain flexibility of bifunctional PBPs.
The overall conformation of LmPBP4 appears significantly different from the previously reported crystal structures of class A PBPs. This finding may explain why previous attempts using molecular replacement failed when used with existing conformations of the bifunctional PBP folds. When the GT domain of LmPBP4 is superimposed on that of SaPBP2 (PDB codes 2OLV and 3DWK), the LmPBP4 TP domain exhibits rotational movements between 16° and 65° from the SaPBP2 polypeptide structures, and the catalytic Ser394 residue of LmPBP4 is between 14 Å and 55 Å from the corresponding residues of SaPBP2 (Fig. 2A). As observed in the crystal structure of SaPBP2 (PDB code 3DWK, chain A), the αL2 helix (residues 310 to 317) of the LmPBP4 linker domain occludes the active-site cleft of the GT domain (Fig. 2B), where the two sugar rings of the elongating glycan chain bind for the polymerization of the bacterial cell wall (23). Therefore, the GT domain in this LmPBP4 conformation could not access the glycan chain substrate without movement of the TP domain, indicating that the TP domain movement could regulate GT domain activity. As expected from a previous study (14), our crystal structure further supports the hypothesis that interdomain flexibility is a common feature among class A PBPs and is likely involved in the regulation of GT activity.
Fig 2.
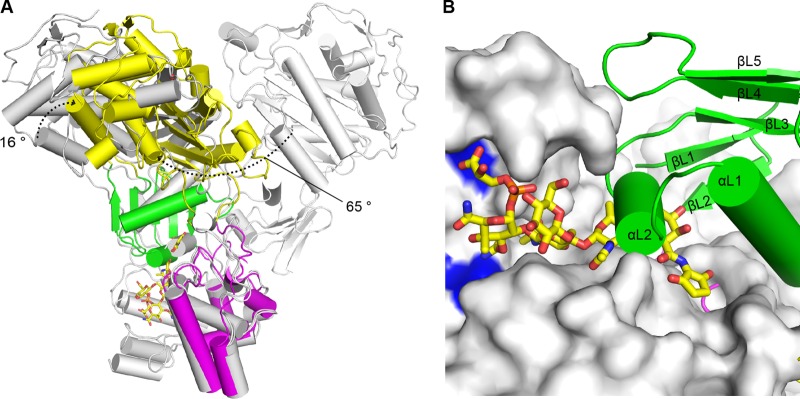
Interdomain flexibility of LmPBP4. (A) TP domain movement. The GT domains of LmPBP4 and those of SaPBP2 (PDB codes 2OLV and 3DWK) were superimposed. The structures of SaPBP2 are in gray for chain B of 2OLV and in cyan for chain A of 3DWK. The dotted arrows indicate the rotational movements of the TP domain. In this superposition, only two conformations of SaPBP2 are shown for clarity. (B) Blocking of the GT active site by the linker domain. The GT domain of the SaPBP2-moenomycin complex (PDB code 2OLV) overlaid on that of LmPBP4 is shown as a surface model, and the moenomycin is shown as a stick model with C atoms colored yellow. The catalytic glutamate residues are in blue. The linker domain of LmPBP4is presented as a ribbon model. The short αL1 helix (residues 310 to 316) of the linker domain occludes the GT active-site cleft, where the growing NAG-NAM polyglycan chain binds.
Tartrate in the active site of the TP domain.
The active site of the TP domain is located in the deep cleft between the α-helical subdomain and the α/β-subdomain, as observed in other PBPs and β-lactamases (Fig. 1B). Three conserved motifs, SXXK, SXN, and KTG, constitute the active site (8). The SXXK motif (Ser394-Thr395-Met396-Lys397) is located at the N-terminal end of α2 and forms the floor of the cleft; Ser394 is the nucleophile that is acylated by the peptide substrate and β-lactam antibiotics, and Lys398 is believed to enhance the nucleophilicity of the catalytic serine by hydrogen bond formation (27). The SXN motif (Ser449-Ile450-Asn451) is found on the loop connecting α4 and α5 and forms one side of the cleft. The third motif (Lys575-Thr576-Gly577) is positioned on strand β3 and forms the opposite side of the active-site cleft.
After several rounds of refinement and before any reconstruction, the Fobs – Fcalc electron density map calculated with phases from the refined model without ligands showed a clear density in the active-site cleft, where the tartrate molecule used in the crystallization solution was nicely fitted (Fig. 3A). The tartrate anion in the catalytic cleft forms hydrogen bonds with the residues Ser394, Ser449, Asn451, Thr576, and Ser578 from the three conserved motifs of PBPs and two water molecules. The oxygen atom of the tartrate carboxylate group occupies the oxyanion hole by forming hydrogen bonds with the main-chain nitrogen atoms of Ser394 and Ser578. In general, the oxyanion hole is occupied by the β-lactam carbonyl oxygen in the acylated PBPs (28, 29). Interestingly, polycarboxylates, such as citrate or tartrate, have been observed in the active sites of other β-lactamases, such as the class A carbapenemase KPC-2 (30), the plasmid-encoded class C β-lactamase CMY-2 (PDB code 1ZC2), a metallo-β-lactamase (PDB code 1MQO) for citrate, and the class D β-lactamase OXA-46 (31) for tartrate. These structural observations support the hypothesis that tartrate or citrate could function as a competitive inhibitor for PBPs or β-lactamases.
Fig 3.

Interaction of LmPBP4 with β-lactam antibiotics or the tartrate molecule. Protein residues interacting with the ligands are shown as a stick model. Water molecules are shown as red spheres, and hydrogen bonds are shown as black dashed lines. The electron density maps (2Fo-Fc) covering the ligands, which are computed with phases from the refined model, are shown at 1 σ contour level. All three structures are shown in stereoview. (A) Tartrate molecule (pink) and interacting residues in the apo-enzyme structure. (B) Ampicillin in the acyl-enzyme complex. (C) Carbenicillin in the acyl-enzyme complex.
Comparison of apo-enzyme and acyl-enzyme complexes.
To understand the structural basis of the action of β-lactam antibiotics, the acylated LmPBP4 was derived by soaking native crystals in mother liquor containing the antibiotic ampicillin or carbenicillin. The crystal structures of the acyl-enzymes were determined: an ampicillin complex structure at 2.1-Å resolution and a carbenicillin complex structure at 2.0-Å resolution. The TP domain of the apo-form structure superimposed on that of the LmPBP4-ampicillin complex and the LmPBP4-carbenicillin complex had RMSDs of 0.20 Å and 0.21 Å over 301 C-α carbon atoms, respectively. This finding shows that the overall conformation of the TP domain was not affected by the acylation. In addition, the active sites of the two acyl complexes shows no substantial conformational changes from the unacylated structure in complex with tartrate. The only difference among the three structures is observed in the residue Gln580, which is hydrogen bonded to the carbonyl group of tartrate in the apo-structure; the benzyl group of ampicillin makes van der Waals contacts with the surrounding residues Val432, Thr589, Gln580, and Thr621 (Fig. 3B), causing the side chain of Gln580 to be shifted to avoid steric hindrance. In the carbenicillin complex, the R1 carboxylate group forms a hydrogen bond with the main-chain amide of Gln580. However, the electron density of the R1 benzyl group of carbenicillin is less clearly defined than that of ampicillin (Fig. 3B and C). These structural differences suggest that the binding affinity of ampicillin to LmPBP4 is higher than that of carbenicillin.
Although the position of the antibiotics covalently bound to Ser394 is similar to that observed in many acyl-PBP complexes, the carboxylate group of the β-lactam antibiotic moiety forms a distinct hydrogen bond with Ser623 on the loop β5-α11 (Fig. 4). In general, the carboxylate group hydrogen bonds with a serine or threonine residue immediately following the KTG motif of the β3 strand, and the interaction probably contributes to recovering the antiparallel nature of β3 and β4 in the acylated form (29, 32). Interestingly, the conformation of β3 in the apo-form of LmPBP4 is stably positioned in antiparallel fashion to β4, and the loop connecting the two strands is located in the open conformation of the active-site groove. These differences explain why the conformation of the β3 strand of LmPBP4 is not changed by acylation. Therefore, the antibiotics can easily access the active site to form the Michaelis complex and transit to the acyl-LmPBP4 complex; this transition can be accomplished without structural rearrangement of the active site, which incurs an energy cost. By lowering the transition state energy barrier, the acylation rate will be accelerated. These implications might explain the observation that the acylation rate of LmPBP4 by ampicillin was 6-fold higher than that of EcPBP1b (17, 24), because the absence of the residue corresponding to Ser623 in EcPBP1b might require the structural rearrangement of the β3 strand for ampicillin binding (Fig. 4).
Fig 4.
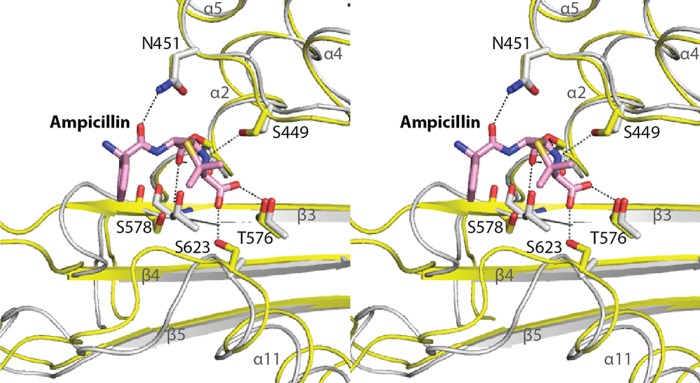
Comparison of β-lactam recognition between LmPBP4 and EcPBP1b (PDB code 3FWM). The two structures are superimposed and shown in stereoview. Atoms of LmPBP4 are colored as described in the legend to Fig. 1. EcPBP1b is colored gray, and the secondary structure is presented as a ribbon model. The bound ampicillin is shown as a stick model.
Comparison of the two acyl-complexes and the tartrate-bound form reveals that many of the key residues involved in β-lactam antibiotic recognition also form hydrogen bonds with the tartrate molecule in the same conformation (Fig. 3). The residues involved in the noncovalent interactions with tartrate in the LmPBP4 enzyme are highly conserved in other PBPs; thus, tartrate may interact similarly with other PBPs. For example, the interacting residues of LmPBP4 are well superimposed on the corresponding residues of class A PBP2 (PDB code 3DWK) from S. aureus and also on the residues of class B PBP3 (PDB code 3OCL) from Pseudomonas aeruginosa, with RMSDs of 0.45 Å and 0.46 Å, respectively, for the overlapped main-chain atoms (see Fig. S1 in the supplemental material). Based on the conserved interactions and the shape complementarity of tartrate for the narrow active-site groove of the TP domain, the tartrate molecule could be a starting point for the design of noncovalent inhibitors of PBPs. Noncovalent, non-β-lactam compounds would function as effective inhibitors when bound tightly to the active site without acylation (33). Beck et al. also identified derivatives of citrate or isocitrate as novel inhibitors of the class D β-lactamase OXA-10 (34). Therefore, the tartrate binding mode may provide valuable information for the development of novel antibiotics.
Conclusions.
We determined the crystal structure of bifunctional class A PBP4 from L. monocytogenes, the first published PBP structure from the pathogen. The structure comprises three distinct domains: a GT domain, a linker domain, and a TP domain. However, the overall conformation is distinct from those of the previously reported class A PBPs, indicating that interdomain flexibility is an intrinsic property of class A PBPs. In this conformation, the linker domain occupies the active-site cleft of the GT domain; this finding supports the hypothesis that interdomain flexibility is related to the regulation of GT domain activity. Another finding is that the tartrate molecule was observed in the active site of the TP domain in the apo-form structure. All of the residues involved in tartrate binding belong to the three conserved motifs, which play a role in the recognition of β-lactam. In addition, the residues involved in tartrate binding are not reoriented upon acylation of PBP4 by β-lactams. These structural observations indicate that the interaction mode of the tartrate molecule in the active site resembles the core of the β-lactam antibiotics, and the LmPBP4-tartrate structure might provide a foundation for the design of a new type of antibiotic.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Marine Extreme Genome Research Center Program of the Ministry of Land, Transport and Maritime Affairs and by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (grant 2011-0007201).
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 13 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00144-13.
REFERENCES
- 1. Popham DL, Young KD. 2003. Role of penicillin-binding proteins in bacterial cell morphogenesis. Curr. Opin. Microbiol. 6:594–599 [DOI] [PubMed] [Google Scholar]
- 2. van Heijenoort J. 2001. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology 11:25R–36R [DOI] [PubMed] [Google Scholar]
- 3. Tipper DJ, Strominger JL. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. U. S. A. 54:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee W, McDonough MA, Kotra L, Li ZH, Silvaggi NR, Takeda Y, Kelly JA, Mobashery S. 2001. A 1.2-A snapshot of the final step of bacterial cell wall biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 98:1427–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee M, Hesek D, Suvorov M, Lee W, Vakulenko S, Mobashery S. 2003. A mechanism-based inhibitor targeting the DD-transpeptidase activity of bacterial penicillin-binding proteins. J. Am. Chem. Soc. 125:16322–16326 [DOI] [PubMed] [Google Scholar]
- 6. Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. 2006. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 30:673–691 [DOI] [PubMed] [Google Scholar]
- 7. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:234–258 [DOI] [PubMed] [Google Scholar]
- 8. Goffin C, Ghuysen JM. 1998. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62:1079–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghosh AS, Chowdhury C, Nelson DE. 2008. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 16:309–317 [DOI] [PubMed] [Google Scholar]
- 10. Halliday J, McKeveney D, Muldoon C, Rajaratnam P, Meutermans W. 2006. Targeting the forgotten transglycosylases. Biochem. Pharmacol. 71:957–967 [DOI] [PubMed] [Google Scholar]
- 11. Smith AM, Klugman KP. 2003. Site-specific mutagenesis analysis of PBP 1A from a penicillin-cephalosporin-resistant pneumococcal isolate. Antimicrob. Agents Chemother. 47:387–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hakenbeck R, Konig A, Kern I, van der Linden M, Keck W, Billot-Klein D, Legrand R, Schoot B, Gutmann L. 1998. Acquisition of five high-Mr penicillin-binding protein variants during transfer of high-level beta-lactam resistance from Streptococcus mitis to Streptococcus pneumoniae. J. Bacteriol. 180:1831–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wright GD. 2007. Biochemistry. A new target for antibiotic development. Science 315:1373–1374 [DOI] [PubMed] [Google Scholar]
- 14. Lovering AL, De Castro L, Strynadka NC. 2008. Identification of dynamic structural motifs involved in peptidoglycan glycosyltransfer. J. Mol. Biol. 383:167–177 [DOI] [PubMed] [Google Scholar]
- 15. Terrak M, Sauvage E, Derouaux A, Dehareng D, Bouhss A, Breukink E, Jeanjean S, Nguyen-Disteche M. 2008. Importance of the conserved residues in the peptidoglycan glycosyltransferase module of the class A penicillin-binding protein 1b of Escherichia coli. J. Biol. Chem. 283:28464–28470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vicente MF, Perez-Daz JC, Baquero F, Angel de Pedro M, Berenguer J. 1990. Penicillin-binding protein 3 of Listeria monocytogenes as the primary lethal target for beta-lactams. Antimicrob. Agents Chemother. 34:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zawadzka-Skomial J, Markiewicz Z, Nguyen-Disteche M, Devreese B, Frere JM, Terrak M. 2006. Characterization of the bifunctional glycosyltransferase/acyltransferase penicillin-binding protein 4 of Listeria monocytogenes. J. Bacteriol. 188:1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guinane CM, Cotter PD, Ross RP, Hill C. 2006. Contribution of penicillin-binding protein homologs to antibiotic resistance, cell morphology, and virulence of Listeria monocytogenes EGDe. Antimicrob. Agents Chemother. 50:2824–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeong JH, Bae JE, Kim YG. 2011. Purification, crystallization and preliminary X-ray crystallographic analysis of PBP4 from Listeria monocytogenes. Acta Crystallogr. F 67:1247–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Terwilliger TC, Adams PD, Read RJ, Mccoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. 2009. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D 65:582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921 [DOI] [PubMed] [Google Scholar]
- 23. Lovering AL, de Castro LH, Lim D, Strynadka NC. 2007. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 315:1402–1405 [DOI] [PubMed] [Google Scholar]
- 24. Sung MT, Lai YT, Huang CY, Chou LY, Shih HW, Cheng WC, Wong CH, Ma C. 2009. Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 106:8824–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Contreras-Martel C, Job V, Di Guilmi AM, Vernet T, Dideberg O, Dessen A. 2006. Crystal structure of penicillin-binding protein 1a (PBP1a) reveals a mutational hotspot implicated in beta-lactam resistance in Streptococcus pneumoniae. J. Mol. Biol. 355:684–696 [DOI] [PubMed] [Google Scholar]
- 26. Paetzel M, Danel F, de Castro L, Mosimann SC, Page MG, Strynadka NC. 2000. Crystal structure of the class D beta-lactamase OXA-10. Nat. Struct. Biol. 7:918–925 [DOI] [PubMed] [Google Scholar]
- 27. Gordon E, Mouz N, Duee E, Dideberg O. 2000. The crystal structure of the penicillin-binding protein 2x from Streptococcus pneumoniae and its acyl-enzyme form: implication in drug resistance. J. Mol. Biol. 299:477–485 [DOI] [PubMed] [Google Scholar]
- 28. Kawai F, Clarke TB, Roper DI, Han GJ, Hwang KY, Unzai S, Obayashi E, Park SY, Tame JR. 2010. Crystal structures of penicillin-binding proteins 4 and 5 from Haemophilus influenzae. J. Mol. Biol. 396:634–645 [DOI] [PubMed] [Google Scholar]
- 29. Sainsbury S, Bird L, Rao V, Shepherd SM, Stuart DI, Hunter WN, Owens RJ, Ren J. 2011. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: comparison of native and antibiotic-bound forms. J. Mol. Biol. 405:173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petrella S, Ziental-Gelus N, Mayer C, Renard M, Jarlier V, Sougakoff W. 2008. Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A beta-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob. Agents Chemother. 52:3725–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. 2010. Crystal structure of the narrow-spectrum OXA-46 class D beta-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob. Agents Chemother. 54:2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Macheboeuf P, Di Guilmi AM, Job V, Vernet T, Dideberg O, Dessen A. 2005. Active site restructuring regulates ligand recognition in class A penicillin-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 102:577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turk S, Verlaine O, Gerards T, Zivec M, Humljan J, Sosic I, Amoroso A, Zervosen A, Luxen A, Joris B, Gobec S. 2011. New noncovalent inhibitors of penicillin-binding proteins from penicillin-resistant bacteria. PLoS One 6:e19418. 10.1371/journal.pone.0019418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beck J, Vercheval L, Bebrone C, Herteg-Fernea A, Lassaux P, Marchand-Brynaert J. 2009. Discovery of novel lipophilic inhibitors of OXA-10 enzyme (class D beta-lactamase) by screening amino analogs and homologs of citrate and isocitrate. Bioorg. Med. Chem. Lett. 19:3593–3597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.