

Abstract
Antivirulence agents inhibit the production of disease-causing virulence factors but are neither bacteriostatic nor bactericidal. Antivirulence agents against methicillin-resistant Staphylococcus aureus (MRSA) strain USA300, the most widespread community-associated MRSA strain in the United States, were discovered by virtual screening against the response regulator AgrA, which acts as a transcription factor for the expression of several of the most prominent S. aureus toxins and virulence factors involved in pathogenesis. Virtual screening was followed by similarity searches in the databases of commercial vendors. The small-molecule compounds discovered inhibit the production of the toxins alpha-hemolysin and phenol-soluble modulin α in a dose-dependent manner without inhibiting bacterial growth. These antivirulence agents are small-molecule biaryl compounds in which the aromatic rings either are fused or are separated by a short linker. One of these compounds is the FDA-approved nonsteroidal anti-inflammatory drug diflunisal. This represents a new use for an old drug. Antivirulence agents might be useful in prophylaxis and as adjuvants in antibiotic therapy for MRSA infections.
INTRODUCTION
Staphylococcus aureus is the most widespread bacterial pathogen in the developed world (1). One-third of the population worldwide is thought to be colonized with S. aureus, usually in the nose (2). Colonization is often harmless but sometimes becomes pathogenic. The trigger for this transition is unknown but seems to be associated with the penetration of S. aureus into broken skin, bruises, and wounds, as well as sites of insertion of catheters and other implants, and with host factors such as local or general immunosuppression (3). S. aureus causes a wide range of infections, from skin and soft tissue infections to more-invasive infections, such as pneumonia, endocarditis, meningitis, bacteremia, and sepsis.
The increase in the rate of S. aureus infections has been associated with hospitalization, affecting preferentially immunocompromised individuals. Recently, however, such infections have increasingly occurred in the community, affecting healthy individuals, such as athletes, students, and prisoners. These community-associated infections are generally more virulent than hospital-associated infections (1).
Treatment of S. aureus infections is hampered by the steady increase in resistance to antibiotics. Nowadays, more than two-thirds of S. aureus infections are resistant to methicillin, a second-generation β-lactam antibiotic (2). Vancomycin, linezolid, and daptomycin are the antibiotics of last resort against methicillin-resistant Staphylococcus aureus (MRSA). Alarmingly, strains that are resistant even to vancomycin have emerged recently (4). Thus, the development of new antibacterial agents represents an urgent unmet medical need.
Virulence factor production in S. aureus is regulated by a quorum-sensing mechanism, predominantly under the control of the accessory gene regulator (agr) operon (5). As shown in Fig. 1, the autoinducing peptide (AIP) is the signaling molecule coded for by agrD and processed by agrB. Mature AIP is secreted to the cell surface, where it binds to and activates the histidine kinase AgrC on the same cell or on another cell. Subsequently, AgrC autophosphorylates and transfers its phosphoryl group to Asp 59 on the N-terminal domain of the response regulator protein AgrA. Phosphorylated AgrA undergoes a conformational change to form a dimer, which enables its C-terminal DNA-binding domains to bind to promoter P2 to activate AIP transcription in an autocatalytic fashion. When the AIP concentration reaches a certain threshold, AgrA also binds to the 10-fold-weaker binding promoter P3, driving the expression of a series of toxins and virulence factors in the post-exponential-growth phase (6).
Fig 1.

S. aureus agr operon for toxin production. The cyclic autoinducing peptide (AIP) is the signaling molecule coded for by agrD and processed by AgrB. Mature AIP is secreted to the cell surface, where it binds to and activates the histidine kinase AgrC on the same cell or on a different cell. Subsequently, AgrC autophosphorylates and transfers its phosphoryl group to Asp 59 on the N-terminal domain of the response regulator protein AgrA. Phosphorylated AgrA undergoes a conformational change to form a dimer, which enables its C-terminal DNA-binding domains to bind to promoter P2 to activate AIP transcription in an autocatalytic fashion. When the AIP concentration reaches a certain threshold, AgrA also binds to the 10-fold-weaker promoter P3, which drives the transcription of RNAIII, a master regulator of expression of a series of toxins and virulence factors in the post-exponential-growth phase (6). RNAIII encodes the toxin delta-hemolysin (Hld). The drug discovery target in this work is the inhibition of phosphoryl transfer to AgrA, as indicated by the large shaded X at the top center.
The present work is based on the hypothesis that blocking AgrA phosphorylation by use of small-molecule compounds would inhibit toxin production. Top-scoring compounds identified through virtual screening against the phosphoryl-binding pocket on AgrA were selected for in vitro screening of toxin suppression in MRSA strain USA300 at the protein and RNA levels. More such compounds were discovered by substructure searches in online catalogs of chemical vendors. The best compounds inhibit rabbit blood hemolysis by 98% at a concentration of 10 μg/ml. These compounds may provide the basis for the development of antivirulence agents to combat MRSA.
MATERIALS AND METHODS
Homology modeling of the N-terminal regulatory domain of AgrA.
Since no crystal structure for the N-terminal regulatory domain of the S. aureus response regulator AgrA is available, a model was built by homology to the crystal structure of the regulatory domain of the sigma 54 transcriptional activator NtrC1 from Aquifex aeolicus (Protein Data Bank [PDB] code 1NY5; resolution, 2.4 Å) (7). A model of residues 1 to 125 of AgrA (UniprotKB code C5MZ29; S. aureus strain USA300_TCH959) was built by the Swiss-Model server (8), followed by refinement with the CNS program (9).
Virtual screening.
The National Cancer Institute library of 90,000 small-molecule compounds was screened virtually using the Schrodinger software suite on the High Performance Computing Cluster at Case Western Reserve University. The GLIDE program was used for docking the compounds, one at a time, onto the phosphoryl-binding pocket of AgrA, delineated by a 10 Å cube centered on the phosphoryl acceptor residue Asp 59. The 107 top-scoring compounds were selected for in vitro testing. Additional small-molecule compound candidates for in vitro testing were identified by similarity and substructure searches on the online catalogs of chemical vendors. A total of 250 compounds were acquired and were subjected to in vitro testing.
ELISA for alpha-hemolysin (Hla).
MRSA strain USA300 was cultured overnight at 37°C in 1.5 ml Trypticase soy broth (TSB). The overnight culture was diluted 1 to 100, and aliquots of 2 ml were added to designated incubation tubes, followed by the addition of 40 μl of 0.05 mg/ml, 0.5 mg/ml, or 2.5 mg/ml of a compound in 100% dimethyl sulfoxide (DMSO) to yield a final concentration of 1 μg/ml, 10 μg/ml, or 50 μg/ml of the compound, respectively. Forty microliters of 100% DMSO was added to a control incubation tube. The concentration of DMSO in the growth samples was ≤2%. The tubes were placed in a shaker and were incubated at 37°C for 6 h. Bacterial cultures were drawn into a syringe and were filtered through a 0.22-μm filter. The filtrates were stored at −80°C until use for the enzyme-linked immunosorbent assay (ELISA).
Ninety-six-well microtiter plates (EIA [enzyme immunoassay]/RIA [radioimmunoassay] plates; product no. 9017; Costar) containing 100 μl/well of a polyclonal anti-Hla antibody (ab15948; Abcam) diluted 1:1,000 using phosphate-buffered saline (PBS), pH 7.2, were incubated overnight at 4°C. The supernatant was then removed, and a bovine serum albumin (BSA) block was conducted for 60 min at 4°C using 230 μl of 10-mg/ml BSA in PBS (catalog no. P3688; Sigma). The supernatant was removed, and the plate was washed once with 230 μl of 0.05% Tween 20 in PBS (catalog no. P3563; Sigma). One hundred microliters of the test samples (filtered supernatants of bacterial cultures, diluted 1:8 or 1:16 with PBS) was added to the wells and was incubated for 1 h at room temperature with gentle rocking, followed by three washes with 230 μl of 0.05% Tween 20 in PBS. One hundred microliters of the anti-Hla antibody conjugated to horseradish peroxidase (ab15949; Abcam), diluted 1:1,000 in 10 mg/ml BSA in PBS (catalog no. P3688; Sigma), was then added to the wells and was incubated for 1 h with rocking. Three more washes with 230 μl of 0.05% Tween 20 in PBS were conducted, ensuring that no supernatant was left after the third wash. The plate was subsequently washed twice with 230 μl PBS, pH 7.2, and was allowed to drain on paper towels. One hundred microliters of the substrate solution 3,3′,5,5′-tetramethylbenzidine (TMB) (catalog no. T4444; Sigma) was added to each well, and the plates were incubated at room temperature for 10 min, followed by the addition of 100 μl of stop reagent (catalog no. S5689; Sigma). The optical density at 650 nm (OD650) was measured in a microplate reader, and the percentage of inhibition was calculated relative to the level of growth with the solvent DMSO.
In order to account for differences in bacterial growth in the presence of the compounds, the number of CFU in each sample was measured after 6 h of growth. Cultures were serially diluted, spread onto LB agar plates, and incubated overnight at 37°C. The number of colonies on each plate was counted. Hla inhibition data were normalized to the level of bacterial growth with DMSO devoid of any potential inhibitor.
Rabbit blood hemolytic assay.
The rabbit blood hemolytic assay is a functional assay measuring the release of hemoglobin from erythrocytes due to the hemolytic activity of Hla. Cultures of MRSA strain USA300 were grown at 37°C for 16 to 18 h with shaking to the post-exponential-growth phase (OD600, 2.5; equivalent to 1 × 109 CFU/ml). Compounds were added at various concentrations and were incubated for 6 h at 37°C. Samples (100 μl) of bacterial culture were filtered and were added to 900 μl hemolysin buffer (0.145 M NaCl, 0.02 M CaCl2) and 25 μl of defibrinated rabbit blood (Hemostat Laboratories, Dixon, CA). The solution was incubated for 15 min at 37°C. Unlysed blood cells were pelleted by centrifugation (5,500 × g, room temperature, 1 min). The hemolytic activity of the supernatant was determined by measuring the optical density at 541 nm. Sterile culture medium served as the standard for 0% hemolysis, and a bacterial culture supernatant devoid of any inhibitor (control) was designated as the standard for 100% hemolysis. The percentage of hemolysis inhibition was calculated by comparison with the control culture. Assays were performed in triplicate.
RNA isolation.
Total RNA from MRSA strain USA300 was isolated after 6 h of induction by using the RiboPure-Bacteria RNA isolation kit (Ambion, Life Technologies) according to the manufacturer's instructions with additional DNase I treatment. The RNA yield and purity were assessed by measuring the ratio of UV absorbances at 260 and 280 nm as described by the manufacturer. The integrity of the RNA was assessed on a 1% denaturing agarose gel. High-integrity RNA had two clear bands with no smearing, indicating no fragmentation of 23S and 16S rRNA.
Real-time RT-PCR.
Quantitative reverse transcription-PCR (qRT-PCR) was conducted on RNA isolates, and the levels of hla, psmα, RNAIII, and spa were assessed. The data were analyzed using the ΔΔCT method; samples induced with 2% DMSO were used as the control. The gene corresponding to the DNA-binding protein Hup was used as the reference housekeeping gene.
Real-time RT-PCRs were conducted using a SYBR green mixture (iScript One-Step RT-PCR kit with SYBR green; Bio-Rad). Each reaction tube contained 12.5 μl master mix, 0.5 μl reverse transcriptase, 7 μl pooled forward and reverse primers, and 5 μl (∼1 ng) total RNA. The cycling program was carried out as recommended by the manufacturer with an annealing and extension temperature of 56°C. Primer sequences are listed in Table 1.
Table 1.
Primers used in the qRT-PCR experiments
| Gene | Forward primer | Reverse primer | Reference |
|---|---|---|---|
| hup | AGAAGCTGGTTCAGCAGTAGATG | TACCTCAAAGTTACCGAAACCAA | 29 |
| hla | ATGGATAGAAAAGCATCCAAACA | TTTCCAATTTGTTGAAGTCCAAT | 29 |
| agrA | CCTCGCAACTGATAATCCTTATG | ACGAATTTCACTGCCTAATTTGA | 29 |
| RNAIII | TTCACTGTGTCGATAATCCA | TGATTTCAATGGCACAAGAT | 30 |
| psmα | TATCAAAAGCTTAATCGAACAATTC | CCCCTTCAAATAAGATGTTCATATC | 25 |
| spa | TTAAAGACGATCCTTCAGTGAGC | TGTTGTTGTCTTCCTCTTTTGGT | 29 |
AgrA preparation.
Plasmid pJR28, containing AgrA and an amino-terminal hexahistidine tag, was a gift from Jonathan Reynolds and Sivaramesh Wigneshweraraj of Imperial College London, London, United Kingdom. AgrA was expressed in Escherichia coli by using the pET28b expression vector as described previously (10). Briefly, cells were grown at 25°C, and the protein was expressed without isopropyl-β-d-thiogalactopyranoside (IPTG) induction to prevent accumulation in inclusion bodies. Cells were lysed by osmotic shock in 50 mM HEPES (pH 7.3), 300 mM NaCl, 5% glycerol, and 3 mM β-mercaptoethanol. The lysate was loaded onto a preequilibrated nickel affinity column (HisTrap; GE Healthcare) and was washed with lysis buffer plus 10 mM imidazole. The protein was eluted with same buffer plus 250 mM imidazole, which migrated as a single band of 28 kDa on an SDS-PAGE gel. The identity of the protein was confirmed by mass spectrometry (data not shown).
EMSA.
The electrophoretic mobility shift assay (EMSA) was modified from the method described by Koenig et al. (6). The DNA sequences used were the P3 promoter region (AATTTTTCTTAACTAGTCGTTTTTTATTCTTAACTGTAAATTTTT) and the negative control (CCTGGTTGTCCTCGTCACTATGAAGAGCCTCACACACAAGGTCGTCGA) as given in reference 6. Single strands were synthesized, purified by denaturing polyacrylamide gel electrophoresis, and end labeled with 32P. The complementary oligomers were then annealed, and the duplex was purified by native polyacrylamide gel electrophoresis. To show AgrA binding, limiting concentrations of DNA (1 nM) were allowed to equilibrate with varying concentrations of AgrA at 25°C for 30 min in a 20-μl solution initially containing 50 mM acetyl phosphate and a reaction buffer composed of 10 mM HEPES (pH 7.6), 1 mM EDTA, 2 mM dithiothreitol (DTT), 50 mM KCl, 0.05% Triton X-100, and 5% glycerol. Because it became evident that acetyl phosphate is not required for the formation of a specific AgrA-DNA complex, acetyl phosphate was subsequently left out. EMSAs were performed using 4.5% native polyacrylamide gels in 0.5× Tris-borate-EDTA. Gels were run at 5 W at 4°C. The assay with diflunisal was performed similarly, except that diflunisal was incubated with AgrA, DNA, and the reaction buffer for 5 min prior to the loading of the gel. Finished gels were visualized by phosphorimaging (Storm 840 scanner; Amersham Biosciences) and ImageQuant software.
RESULTS
AgrA model.
The target protein for the discovery of small-molecule compounds to inhibit S. aureus virulence factors in this work is the regulatory domain of the response regulator AgrA. Crystal structures of the C-terminal DNA-binding domain of AgrA by itself and in complex with a cognate DNA fragment have been reported (11, 12). However, no crystal structure of the N-terminal regulatory domain of AgrA is available. Therefore, we resorted to homology modeling. A model of the N-terminal regulatory domain of AgrA, residues 1 to 125, was built by homology to the N-terminal domain of the transcriptional regulator NtrC1 from Aquifex aeolicus (PDB code 1ZY2) (7). Although the degree of sequence identity between the two protein domains is only 26%, they share functional similarities, including the aspartic acid phosphoryl receiver (Asp 59 in AgrA) embedded in the conserved sequence motif XDY, where X is a an aliphatic or aromatic residue, D is an aspartic acid, and Y is an aliphatic residue. This motif is conserved in the activation domains of transcriptional regulators of Gram-positive bacteria. Only the structure of the phosphoryl-binding pocket, not the structure of the entire domain, is used for the virtual screening of inhibitors. Therefore, this crystal structure was deemed a valid basis for building a model of the phosphoryl-binding pocket on the N-terminal domain of AgrA. The homology-generated model of the N-terminal domain of AgrA displays a distinct surface pocket centered on Asp 59, as shown in Fig. 2. The dimensions of the pocket are roughly 10 by 10 by 10 Å. Residues lining the binding pocket include Glu 7, Asp 8, Asp 9, Gln 12 (H-bonded to Asp 59), Leu 62, Val 87, and Lys 110 (salt-bridged to Asp 59).
Fig 2.

Ribbon diagram of the homology-built model of the N-terminal domain of AgrA (AgrA_N). Residue Asp 59, shown in stick representation in a pocket on the surface of the protein, is phosphorylated by the histidine kinase AgrC, leading to activation of the C-terminal DNA-binding domain of AgrA, which, in turn, triggers the induction of both the autoinducing peptide and RNAIII, ultimately causing the transcription of a series of toxin genes. The hypothesis is that blockage of the phosphohistidine pocket with a small molecule prevents toxin expression.
High-throughput virtual screening.
The National Cancer Institute library of 90,000 small-molecule compounds was screened virtually for the ability to block the phosphoryl-binding pocket of AgrA. The 107 top-scoring compounds were acquired and were tested for in vitro efficacy at inhibiting the formation of alpha-hemolysin (Hla), the most important S. aureus toxin (13–16).
Inhibition of alpha-hemolysin (Hla) formation by potential inhibitors.
Activation of AgrA leads to increased Hla production. Therefore, as an initial measure of AgrA inhibition, Hla levels were measured by a sandwich ELISA. Seven of the top-scoring 107 compounds from virtual screening were found to inhibit Hla production. Substructure searches were conducted at online catalogs of chemical vendors in order to find additional compounds similar to the initial positive hits. A total of 250 compounds were assayed for inhibition of Hla production. Compounds that inhibited bacterial growth were not examined further. The 11 most active compounds, defined by the highest level of inhibition of Hla, belong to two families, naphthalene derivatives and biaryl compounds, as shown in Fig. 3. At 10 μg/ml, four compounds inhibited Hla formation by more than 70%; all the compounds inhibited Hla in a dose-dependent manner (Table 2).
Fig 3.

Structures of the most active compounds. Compound I, 4-hydroxybenzo[cd]indol-2(1H)-one; compound II, β-oxynaphthoic acid; compound III, 2,3-difluoro-1-naphthoic acid; compound IV, 3-[3-(1,3-benzodioxol-5-yl)prop-2-enoyl]-2-hydroxy-cyclohepta-2,4,6-trien-1-one; compound V, 4-hydroxy-3-methoxy-N-(naphthalen-1-ylmethylideneamino)benzamide; compound VI, 1-(5-ethyl-2,4-dihydroxyphenyl)-2-phenoxyethanone; compound VII, 3′-fluoro-4′-hydroxy-4-biphenylcarboxylic acid; compound VIII, 5-benzoyl-4-hydroxy-2-methoxybenzenesulfonic acid; compound IX, 2′,4′-difluoro-4-hydroxybiphenyl-3-carboxylic acid (diflunisal); compound X, 3-phenyl salicylic acid; compound XI, 4,4′-fluorophenyl benzoic acid.
Table 2.
Inhibitors of alpha-hemolysin formation and rabbit blood hemolysis by MRSA USA300
| Compound | % Alpha-hemolysin inhibition as determined by ELISAa at: |
% Inhibition of hemolysis of rabbit blood at: |
||
|---|---|---|---|---|
| 1 μg/ml | 10 μg/ml | 1 μg/ml | 10 μg/ml | |
| I | 28.0 ± 4.4 | 65.0 ± 2.5 | NDb | ND |
| II | 21.3 ± 5.2 | 76.0 ± 1.2 | 42.0 ± 2.8 | 69.9 ± 6.1 |
| III | 20.8 ± 3.5 | 28.3 ± 3.4 | ND | ND |
| IV | 40.2 ± 5.2 | 73.6 ± 2.2 | ND | ND |
| V | 23.0 ± 1.3 | 88.4 ± 1.5 | 9.0 ± 1.3 | 63.6 ± 6.6 |
| VI | 55.2 ± 5.8 | 76.3 ± 11.2 | 9.2 ± 0.7 | 97.9 ± 9.3 |
| VII | 31.3 ± 9.0 | 61.0 ± 14.8 | 38.6 ± 6.4 | 55.6 ± 9.3 |
| VIII | 15.9 ± 4.2 | 41.9 ± 8.2 | 6.0 ± 7.5 | 21.3 ± 6.1 |
| IX | 33.0 ± 3.6 | 62.3 ± 1.8 | 63.0 ± 1.9 | 97.7 ± 4.2 |
| X | 49.4 ± 4.6 | 53.0 ± 5.9 | 34.3 ± 6.9 | 84.7 ± 6.7 |
| XI | 0.0 ± 7.0 | 68.8 ± 11.3 | 49.0 ± 2.2 | 76.6 ± 8.1 |
ELISAs were carried out in 2% DMSO as the solvent. Control measurements with this solvent were arbitrarily assigned values of 0% inhibition and 100% bacterial growth. All other data were normalized to the control solvent data.
ND, not determined.
The ELISA inhibition data were corroborated by a hemolysis assay. Hla creates holes in the membranes of various host cells, such as immune system cells and erythrocytes. Interestingly, Hla does not affect human red blood cells but does damage rabbit and sheep erythrocytes. Rabbit erythrocytes were used for the hemolysis assay. The amount of hemoglobin released from lysed red blood cells was taken as a measure of Hla production. Of the tested compounds, all resulted in marked decreases in the level of hemolysis at both 1 μg/ml and 10 μg/ml, and all appeared to inhibit hemolysis in a dose-dependent manner (Table 2). The highest degree of inhibition was observed with compounds VI and IX, which decreased hemolysis by more than 97% at a concentration of 10 μg/ml.
These compounds are neither bactericidal nor bacteriostatic. None of these compounds inhibited growth at 1 or 10 μg/ml.
Inhibition of transcription of agr-regulated toxin genes by antivirulence compounds.
To shed light on the mechanism of Hla inhibition, RT-qPCR experiments were carried out in the presence of selected antivirulence compounds. When MRSA was incubated in the presence of 50 μg/ml of the compounds, a marked decrease in hla expression was observed, in particular with compounds IX, X, and XI (Fig. 4A). Furthermore, a decrease of at least 2-fold in hla expression was observed for all the compounds at 10 μg/ml (except for compound V). Compound IX elicited the most dramatic inhibition of the Hla transcript, by a factor of about 1,000 at a concentration of 50 μg/ml. This inhibition of hla is consistent with the model of AgrA inhibition.
Fig 4.

Levels of transcription of hla, psmα, and RNAIII in MRSA strain USA300 exposed to 10 μg/ml (filled bars) or 50 μg/ml (open bars) of compounds. No gene expression was determined for compound VI at 50 μg/ml because growth inhibition prevented RNA isolation. Values are averages for three separate experiments; error bars indicate standard deviations. ***, P < 0.0005; **, P < 0.005; *, P < 0.05. (A) hla expression is displayed as log2 of relative gene expression. (B) psmα expression is displayed as log2 of relative gene expression. (C) RNAIII expression displayed as the fold change from the value with DMSO, which was set at 1. Values above 1 indicate an increase in expression, while values below 1 signify inhibition of expression. (D) spa expression is displayed as log2 of relative gene expression.
hla transcription is indirectly upregulated by phosphorylated AgrA, whereas the regulator RNAIII and the leukolytic protein phenol-soluble modulin α (PSMα) are directly upregulated by the binding of phosphorylated AgrA to their respective promoters (5, 17). Therefore, RNAIII and psmα expression is thought to be directly correlated to AgrA activation and, subsequently, inhibition. When MRSA was incubated with select antivirulence compounds at 50 μg/ml, psmα expression decreased significantly (Fig. 4B). All of the compounds investigated inhibited the transcription of Hla and PSMα in a dose-dependent manner, as shown in Fig. 4A and B (except for compound XI).
RNAIII expression was only moderately inhibited when MRSA was incubated with compounds V, VII, and IX at 50 μg/ml (Fig. 4C). Only compound IX decreased RNAIII expression at 10 μg/ml. In contrast to the downregulation of the toxins, expression of the antiphagocytic surface protein A was increased in the presence of the compounds (Fig. 4D).
Inhibition of the binding of AgrA to promoter P3 by an antivirulence compound.
In an effort to gain insight into the mechanism of action of the antivirulence compounds, DNA-binding experiments were carried out with purified AgrA and an oligonucleotide corresponding to promoter P3, which drives the expression of virulence factors in S. aureus. As shown in Fig. 5, lane 2, a strong band appears upon the addition of AgrA to the DNA fragment, corresponding to the AgrA-P3 complex. The band corresponding to this complex is not present when diflunisal (compound IX) is added (Fig. 5, lane 3), indicating inhibition of specific binding of AgrA to promoter P3 by the antivirulence compound. Similar DNA-binding inhibition was observed with other compounds (data not shown) and will be described in a follow-up publication. Thus, the compounds inhibit the transcription of virulence factors by preventing the response regulator transcription factor from binding to the promoter driving the expression of virulence factors.
Fig 5.
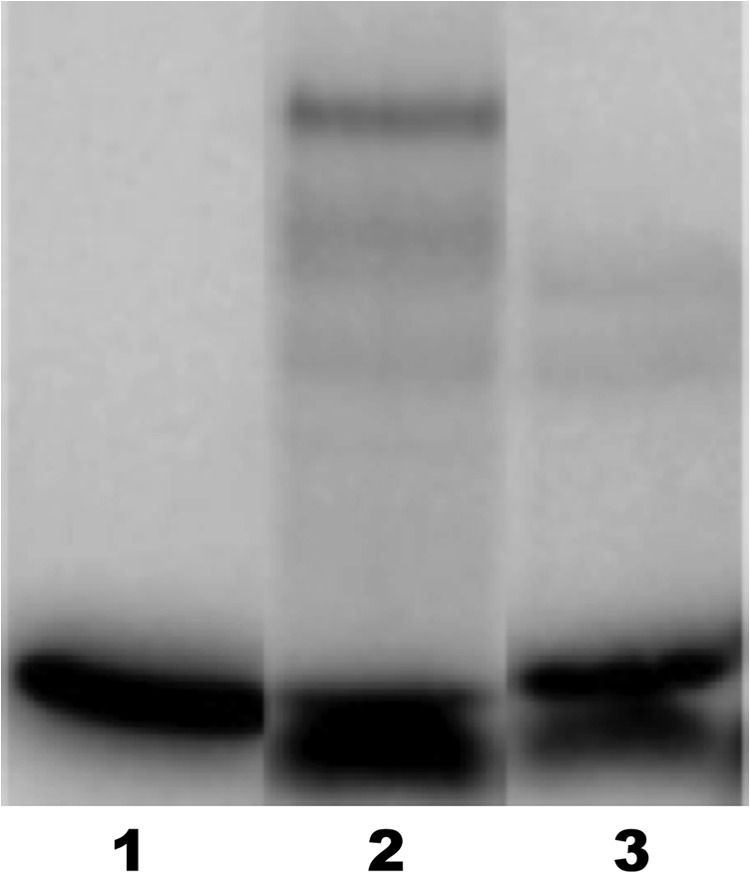
Electrophoretic mobility shift assay of a 33P-labeled oligonucleotide corresponding to the sequence promoter P3. Lane 1, DNA alone; lane 2, DNA plus 2 μM AgrA; lane 3, DNA plus 2 μM AgrA plus 200 μM diflunisal (compound IX). The strong band in lane 2 at a higher molecular weight corresponds to the AgrA-P3 complex. This band is not present when diflunisal is added. The weak bands in lanes 2 and 3 may correspond to nonspecific AgrA-DNA complexes.
DISCUSSION
Antibiotics are currently the treatment of choice for MRSA infections, but increasing resistance to these agents, coupled with the decline in the commercial development of new antibiotics, has created an urgent need and a window of opportunity to introduce new treatment options. The World Health Organization has identified antimicrobial resistance as one of the three greatest threats to human health. Antivirulence agents offer an alternative to antibiotics. Will resistance to antivirulence agents develop? Virulence factors empower the pathogen with chemical weapons, which impair the ability of the host immune system to fight an infection. Since the survival of bacteria is not threatened by antivirulence treatment, it is conceivable that resistance to these agents will not significantly impede the efficacy of such drugs over time. At present there are no data to support this assumption. One might argue that antivirulence therapy, by suppressing pathogen-host interactions, does create selective pressure, since it renders the pathogen susceptible, once again, to innate immunity. The key is that this selective pressure is present only in areas where innate immunity is active. There is no selective pressure on the commensal flora of nonvirulent bacteria or when the drug is released into the environment (18). Thus, antivirulence therapy could help to preserve the efficacy of conventional antibiotics.
There is currently no antivirulence agent in clinical use against bacterial infections. However, data are available on animal models of S. aureus infections that shed light on the usefulness of antivirulence agents for combating infections. Hla is an essential virulence factor of S. aureus, and its level correlates with virulence in a mouse model of S. aureus pneumonia (19). Monoclonal antibodies against Hla provide protection against lethal S. aureus pneumonia in an experimental murine model and prevent human lung cell injury in vitro (20). However, once the animals are infected, treatment is effective when the antibodies are administered up to 8 h postinfection. Likewise, a β-cyclodextrin derivative that blocks the oligomeric assembly of Hla and inhibits its lytic function reduces the severity of MRSA infections in a murine model of S. aureus pneumonia (21). As in the case of monoclonal anti-Hla antibodies, treatment with the β-cyclodextrin derivative at 10 mg/kg is of limited value postinfection, since the efficacy drops when the compound is administered beyond 2 h postinfection. However, active or passive immunization against Hla in a murine model reduces the severity of skin and soft tissue infections caused by MRSA strain USA300, the leading cause of community-associated MRSA infections in the United States (15).
In contrast to anti-Hla agents, which neutralize the action of a single toxin, the compounds discovered in this work inhibit the expression of Hla, Psmα, and, to some extent, RNAIII. Along with Hla, Psmα is a key virulence factor in S. aureus infections (14). Hla and Psmα are toxins with complementary lytic activities against the host. Whereas Psmα is more efficacious in lysing neutrophils (22), Hla is more lytic to erythrocytes in rabbits, mice, and sheep but not in humans (23). The major contribution of Hla to pathogenesis in humans is its interaction with the metalloprotease ADAM10, which leads to disruption of epithelial barrier function (13).
The agr operon is the most important operon for virulence production in S. aureus. Knockout of the agr operon in MRSA strain USA300 drastically reduces the size of abscesses in a rabbit model of skin infections (14).
Inhibition of the agr system can, in principle, be achieved by blocking any step along the pathway shown in Fig. 1. The receptor histidine kinase AgrC was the target of a synthetic approach using cyclic peptide mimics of the autoinducing peptide to inhibit receptor activation (17). Inhibitory antibodies have been shown to cause agr quenching by sequestering the autoinducing peptide (24). The target of agr inhibition in this work was the transcription factor AgrA, which controls the expression of all the virulence factors under the control of the agr system. Hla is upregulated by AgrA. In the presence of the antivirulence compounds described in this work, there is a marked decrease in Hla production, as shown by ELISA and rabbit blood hemolysis. Furthermore, a significant decrease in the transcription of Hla was observed by RT-qPCR. In addition, some of the compounds were shown to significantly inhibit the expression of PSMα, a toxin directly upregulated by phosphorylated AgrA in an RNAIII-independent manner (25). Interestingly, the decrease in RNAIII expression was not as large as that for Hla and PSMα. This may be explained by differential binding of AgrA to promoters in other operons, such as sar, that trigger higher expression levels (26, 27). This finding is corroborated by a report that induction of Hla transcription is not coupled to higher concentrations of RNAIII (28). Whereas the expression of the toxins Hla and PSMα is inhibited, the expression of the immune evasion protein A (Spa) is upregulated by the antivirulence compounds. This finding is in accordance with a previous report of downregulation of protein A when agr is upregulated (25). Whereas an increase in immune evasion is an undesirable effect, it is counterbalanced by a marked decrease in disease-causing toxins. Thus, the overall consequence of antivirulence therapy may be a reduction in the severity of the infection.
Compound IX, diflunisal, is an FDA-approved nonsteroidal anti-inflammatory drug. This represents a new use for an old drug. Diflunisal inhibits the binding of AgrA to an oligonucleotide corresponding to the DNA sequence of promoter P3, as shown in Fig. 5. In principle, the formation of the specific protein-DNA complex could be blocked by the binding of diflunisal either to DNA or to AgrA. The latter is considered more likely. It is conceivable that diflunisal causes a conformational change on AgrA that impairs its DNA-binding capacity. Perhaps diflunisal prevents the proper dimerization of AgrA required for specific promoter binding (10).
The drug discovery target for this project was the phosphoryl-binding pocket on the N-terminal domain of AgrA. However, the binding of AgrA to promoter P3 does not require phosphorylation (6), a finding corroborated in this work by the formation of a specific AgrA-DNA complex in the absence of acetyl phosphate or any other phosphate donor (Fig. 5). Thus, diflunisal may or may not bind to the phosphoryl-binding pocket on AgrA. Recently a ligand-binding pocket has been identified on the C-terminal DNA-binding domain of AgrA (12). Thus, it is possible that diflunisal binds to the C-terminal domain, or perhaps to both domains, of AgrA. Localization of the diflunisal-binding site on the surface of AgrA will have to await a cocrystal structure of AgrA and diflunisal.
The antivirulence compounds discovered in this work may lead to the development of adjuvants to conventional antibiotic therapy or perhaps to novel antimicrobials in monotherapy for topical applications.
ACKNOWLEDGMENTS
This work was supported by grant-in-aid 09GRNT2380131 from the American Heart Association Great Rivers Affiliate and by a grant from the Steris Foundation.
Robert Bonomo is thanked for invaluable advice and thoughtful discussions. Jonathan Reynolds and Sivaramesh Wigneshweraraj of Imperial College London, London, United Kingdom, are thanked for the AgrA expression plasmid.
Footnotes
Published ahead of print 20 May 2013
REFERENCES
- 1. Otto M. 2010. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 64:143–162 [DOI] [PubMed] [Google Scholar]
- 2. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 3. Rasmussen RV, Fowler VG, Jr, Skov R, Bruun NE. 2011. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 6:43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pantosti A, Sanchini A, Monaco M. 2007. Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol. 2:323–334 [DOI] [PubMed] [Google Scholar]
- 5. Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S, Novick RP. 1995. The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol. Gen. Genet. 248:446–458 [DOI] [PubMed] [Google Scholar]
- 6. Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK. 2004. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 186:7549–7555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee SY, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. 2003. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 17:2552–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201 [DOI] [PubMed] [Google Scholar]
- 9. Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921 [DOI] [PubMed] [Google Scholar]
- 10. Reynolds J, Wigneshweraraj S. 2011. Molecular insights into the control of transcription initiation at the Staphylococcus aureus agr operon. J. Mol. Biol. 412:862–881 [DOI] [PubMed] [Google Scholar]
- 11. Sidote DJ, Barbieri CM, Wu T, Stock AM. 2008. Structure of the Staphylococcus aureus AgrA LytTR domain bound to DNA reveals a beta fold with an unusual mode of binding. Structure 16:727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leonard PG, Bezar IF, Sidote DJ, Stock AM. 2012. Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding. Biochemistry 51:10035–10043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. 2011. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17:1310–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Wardenburg JB, Schneewind O, Otto M, DeLeo FR. 2011. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 204:937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilke GA, Wardenburg JB. 2010. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 107:13473–13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. George EA, Novick RP, Muir TW. 2008. Cyclic peptide inhibitors of staphylococcal virulence prepared by Fmoc-based thiolactone peptide synthesis. J. Am. Chem. Soc. 130:4914–4924 [DOI] [PubMed] [Google Scholar]
- 18. Rasko DA, Sperandio V. 2010. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 9:117–128 [DOI] [PubMed] [Google Scholar]
- 19. Bubeck Wardenburg J, Schneewind O. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ragle BE, Bubeck Wardenburg J. 2009. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 77:2712–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ragle BE, Karginov VA, Bubeck Wardenburg J. 2010. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 54:298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loeffler B, Hussain M, Grundmeier M, Brueck M, Holzinger D, Varga G, Roth J, Kahl BC, Proctor RA, Peters G. 2010. Staphylococcus aureus Panton-Valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 6:e1000715. 10.1371/journal.ppat.1000715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bernheimer AW. 1965. Staphylococcal alpha toxin. Ann. N. Y. Acad. Sci. 128:112–123 [DOI] [PubMed] [Google Scholar]
- 24. Park J, Jagasia R, Kaufmann GF, Mathison JC, Ruiz DI, Moss JA, Meijler MM, Ulevitch RJ, Janda KD. 2007. Infection control by antibody disruption of bacterial quorum sensing signaling. Chem. Biol. 14:1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. 2008. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol. Cell 32:150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheung AL, Ying P. 1994. Regulation of alpha- and beta-hemolysins by the sar locus of Staphylococcus aureus. J. Bacteriol. 176:580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saïd-Salim B, Dunman PM, McAleese FM, Macapagal D, Murphy E, McNamara PJ, Arvidson S, Foster TJ, Projan SJ, Kreiswirth BN. 2003. Global regulation of Staphylococcus aureus genes by Rot. J. Bacteriol. 185:610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohlsen K, Koller KP, Hacker J. 1997. Analysis of expression of the alpha-toxin gene (hla) of Staphylococcus aureus by using a chromosomally encoded hla::lacZ gene fusion. Infect. Immun. 65:3606–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Even S, Charlier C, Nouaille S, Ben Zakour NL, Cretenet M, Cousin FJ, Gautier M, Cocaign-Bousquet M, Loubiere P, Le Loir Y. 2009. Staphylococcus aureus virulence expression is impaired by Lactococcus lactis in mixed cultures. Appl. Environ. Microbiol. 75:4459–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaudaux P, Francois P, Bisognano C, Kelley WL, Lew DP, Schrenzel J, Proctor RA, McNamara PJ, Peters G, Von Eiff C. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect. Immun. 70:5428–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]