

Abstract
The pharmacokinetics, metabolism, and excretion of dolutegravir, an unboosted, once-daily human immunodeficiency virus type 1 integrase inhibitor, were studied in healthy male subjects following single oral administration of [14C]dolutegravir at a dose of 20 mg (80 μCi). Dolutegravir was well tolerated, and absorption of dolutegravir from the suspension formulation was rapid (median time to peak concentration, 0.5 h), declining in a biphasic fashion. Dolutegravir and the radioactivity had similar terminal plasma half-lives (t1/2) (15.6 versus 15.7 h), indicating metabolism was formation rate limited with no long-lived metabolites. Only minimal association with blood cellular components was noted with systemic radioactivity. Recovery was essentially complete (mean, 95.6%), with 64.0% and 31.6% of the dose recovered in feces and urine, respectively. Unchanged dolutegravir was the predominant circulating radioactive component in plasma and was consistent with minimal presystemic clearance. Dolutegravir was extensively metabolized. An inactive ether glucuronide, formed primarily via UGT1A1, was the principal biotransformation product at 18.9% of the dose excreted in urine and the principal metabolite in plasma. Two minor biotransformation pathways were oxidation by CYP3A4 (7.9% of the dose) and an oxidative defluorination and glutathione substitution (1.8% of the dose). No disproportionate human metabolites were observed.
INTRODUCTION
Since zidovudine was first approved in the United States in 1987 for the treatment of human immunodeficiency virus type 1 (HIV-1) infection, the incorporation of multiple agents into combination antiretroviral therapy (ART) that targets the different phases of the HIV replication cycle has enhanced the management of HIV infection. Inhibitors of HIV-1 integrase have been the last of the three virus-encoded enzymes (reverse transcriptase, integrase, and protease) for which therapeutic agents have been developed, with raltegravir in 2007 being the first licensed HIV-1 integrase inhibitor. The HIV integrase enzyme coordinates the insertion of viral DNA into the host chromosome (1). The 2-metal binding class of integrase inhibitors targets the binding of the divalent metal ions Mg2+ and Mn2+ (primarily Mg2+ under physiological conditions) to prevent the strand transfer step in the integration process, thereby inhibiting viral replication (2–4).
Dolutegravir (S/GSK1349572) (DTG) is a potent tricyclic carbamoyl pyridone integrase inhibitor (5) possessing subnanomolar antiviral 50% effective concentrations (EC50s) in vitro and a distinct resistance profile designed to retain activity against raltegravir- and elvitegravir-resistant strains (A. Sato, M. Kobayashi, T. Seki, C. W. Morimoto, T. Yoshinaga, T. Fujiwara, B. A. Johns, and M, Underwood, presented at the 8th European HIV Drug Resistance Workshop, Sorrento, Italy, 2010). Furthermore, there is a potential for a higher barrier to resistance based on serial-passage experiments, dolutegravir activity against HIV-1 with single and multiple integrase mutation combinations (6), and prolonged binding to mutant integrase proteins in integrase-Mg2+-DNA complexes (7). Dolutegravir has been generally well tolerated, with in vivo efficacy demonstrated in a phase IIa 10-day monotherapy study (8), at 48 weeks of a phase IIb antiretroviral-naïve-adult study (9), and at 24 weeks of a phase IIb treatment-experienced-subject pilot study (10). Dolutegravir exhibits rapid oral absorption and dose-proportional kinetics (2 to 100 mg) from a suspension formulation, and its low apparent clearance and oral terminal half-life (t1/2) of approximately 15 h supports once-daily dosing without the need for a boosting agent (11). Dolutegravir has demonstrated low to moderate pharmacokinetic variability, with a predictable exposure-response relationship (8). In vitro, dolutegravir is primarily metabolized by UDP-glucuronosyltransferase (UGT) 1A1 and cytochrome P450 (CYP) 3A4 and at clinically relevant concentrations is not an inhibitor of CYP or UGT enzymes (12). DTG has demonstrated a limited number of clinically significant drug-drug interactions without dose adjustment for most ART and other commonly coadministered drugs in integrase-naive subjects (I. Song, J. Borland, S. Chen, A. Peppercorn, P. Savina, T. Wajima, S. Min, G. Nichols, and S. Piscitelli, presented at the 13th International Workshop on Clinical Pharmacology of HIV Therapy, Barcelona, Spain, 2012).
This mass balance study identifies the major metabolic profile in humans for the evaluation of cross-species comparisons to assess the systemic exposure to dolutegravir and its metabolites. This comparison provides support for the selection of animal species used in the nonclinical safety assessment. Data from human mass balance studies also provide information on excretory routes that can be used to guide studies in special patient populations, such as individuals with hepatic or renal impairment. In addition, the assessment of exposure to total drug-related material can aid in the design of pharmacodynamic studies, such as a cardiac repolarization study, by helping to identify time points for collecting electrocardiogram data relative to anticipated exposures of the parent drug and key metabolites (13).
The objectives of this investigation were to determine the pharmacokinetics, metabolism, and excretion of dolutegravir following single oral administration of [14C]dolutegravir as a suspension formulation to healthy human subjects.
(This work was previously presented at the 13th Annual International Workshop on Clinical Pharmacology of HIV Therapy, 16 to 18 April 2012, Barcelona, Spain.)
MATERIALS AND METHODS
Chemicals.
[14C]dolutegravir sodium salt (3.63 μCi/mg; radiochemical purity, 99.0%) was provided by the GlaxoSmithKline (GSK) radiochemistry group (Stevenage, Hertfordshire, United Kingdom) as a bulk powder to be reconstituted as a suspension in a vehicle of hypromellose, sodium lauryl sulfate, and sterile water for injection. The reference standard dolutegravir, 15N2H7-dolutegravir stable isotopic label, and dolutegravir glucuronide (GSK2832500; M2) reference standard material were synthesized and supplied by Shionogi and Co., Ltd. (Osaka, Japan). Metabolites are identified with the letter “M” and a number. The chemical structure of dolutegravir with the radiocarbon position is depicted in Fig. 1. Chemicals, solvents of reagent or high-performance liquid chromatography (HPLC) grade, and the scintillation cocktails Ultima Flo M, Ultima Gold XR, Perma Fluor, and Carbosorb were obtained from commercial sources.
Fig 1.
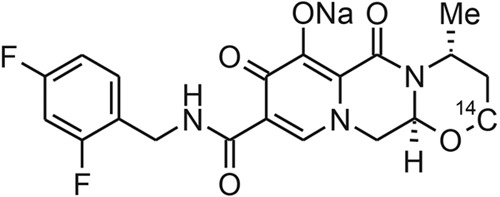
Structure of [14C]dolutegravir (sodium salt).
Subjects.
Healthy, nonsmoking adult male subjects aged 30 to 55 years and weighing greater than 50 kg with a body mass index of 18.5 to 31 kg/m2 were eligible to participate in this study. Women were excluded from participation. Additional exclusion criteria were similar to those outlined previously (14), including prohibition of the use of multivitamins, iron supplements, antacids, any prescription drugs, herbal and dietary supplements, and grapefruit-containing products within 14 days before the start of the dosing through discharge from the clinical research unit. Subjects were housed in the clinical research unit from the day before dosing until discharge. Discharge criteria were met when less than or equal to 1% of the administered radioactive dose was excreted in 2 consecutive 24-hour collection periods for both urine and feces.
Study design and dosing.
This study was an open-label, phase I, single-dose mass balance study conducted at the Covance Clinical Research Unit, Inc. (Madison, WI). The study was conducted in accordance with Good Clinical Practice and applicable regulatory requirements and conformed to the principles of the Declaration of Helsinki. The recommendations from the 1992 International Commission on Radiological Protection (ICRP) for radiological protection in biomedical research were used to assist the Institutional Review Board in their evaluation of the protocol. Following Institutional Review Board approval and collection of written informed consent, all subjects underwent an initial screening assessment within 30 days of the first dose. Safety was assessed by vital signs, physical assessment, adverse-event (AE) assessments, laboratory tests (chemistry, hematology, and urinalysis), and a 12-lead electrocardiogram. Follow-up visits occurred 7 to 14 days after the last inpatient study assessment.
After an overnight fast of at least 10 h, subjects received a single oral dose of [14C]dolutegravir at 20.9 ± 0.1 mg containing 80 ± 0.4 μCi (0.96 mSv) of radioactivity as a suspension in hypromellose, sodium laural sulfate, and sterile water for injection, immediately followed by water rinses, for a total administration volume of 240 ml. Any radioactivity recovered from the dosing vial was subtracted from the calculated dose. Criteria for selecting the radioactive dose were based on balancing the analytical requirements for meeting the study objectives, minimizing the radioactivity exposure of the volunteers, and a radioactivity exposure not to exceed approximately one-third of the annual average background effective dose to the whole body (including radon) of ∼3 mSv. The effective radioactive dose of 0.96 mSv was selected following review of the effective dose and effective-dose equivalent calculations performed independently of GSK using the Medical Internal Radiation Dose system (15) and the MIRDOSE 3.1 software package obtained from the Radiation Internal Dose Information Center at Oak Ridge, TN. The 20-mg dose was selected to be within the linear range established from a previous study (11) to provide sufficient mass and a characteristic ratio of labeled to unlabeled material to aid in metabolite identification efforts. Except for an hour before and an hour immediately after dosing, subjects were allowed water ad libitum and were provided breakfast following the 4-hour-postdose sample collection.
As part of a separate drug interaction study with efavirenz, informed consent was received from a group of 12 nonsmoking fasted adult male subjects who received 50 mg of unlabeled dolutegravir alone as two 25-mg tablets once daily for 5 days. After 5 days of dosing, residual urine collected over 24 h was used to isolate the target metabolite, M4, for further characterization.
Sample collection.
Following single-dose [14C]dolutegravir administration, blood and plasma samples were collected (EDTA anticoagulant) predose and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, and 72 h postdose, with sample collection continuing until radioactivity in 2 successive samples was less than or equal to twice the background radiation. Blood was stored at 2 to 8°C before analysis, and plasma was stored at −20°C or colder.
Urine was collected predose and at 0 to 6, 6 to 12, and 12 to 24 h and at 24-hour intervals until discharge criteria were met. Urine was stored at 2 to 8°C during collection and pooled over the collection interval, and then portions were subsequently frozen at −20°C or colder. Feces were collected predose and pooled at 24-hour intervals until the subject was discharged. Fecal samples were homogenized with 20% ethanol in deionized water (approximately 3 times the sample weight) by using a probe-type homogenizer, and then portions of the homogenate were stored frozen at −20°C or colder. Scintillation-counting data (counts per minute) were automatically corrected for counting efficiency using the external-standardization technique and an instrument-stored quench curve generated from a series of sealed quenched standards.
Measurement of radioactivity.
Plasma (in duplicate) and urine (in triplicate) sample radioactivity was determined by liquid scintillation counting (LSC). Portions of each sample were mixed with Ultima Gold XR scintillation cocktail and analyzed for radioactivity using a Model 2900TR liquid scintillation counter (Packard Instrument Co., Meriden, CT) for at least 5 min or 100,000 counts.
Whole-blood (in duplicate) and fecal-homogenate (in triplicate) samples were combusted in a Model 307 Sample Oxidizer (Packard Instrument Co.), and the resulting 14CO2 was trapped in a mixture of Perma Fluor and Carbosorb and then analyzed by LSC. Oxidation efficiency was evaluated on each day of sample combustion by analyzing a commercial radiolabeled standard both directly in scintillation cocktail and by oxidation. Acceptance criteria included combustion recoveries of between 95% and 105%.
Quantification of dolutegravir in plasma.
Plasma samples were analyzed for dolutegravir using a validated liquid chromatography and tandem mass spectrometry (LC–MS-MS) method. Plasma samples were extracted by protein precipitation with acetonitrile containing [15N2H7]dolutegravir as the internal standard. The extract was injected onto an Acquity HPLC system (Waters Associates, Milford, MA), and a mobile phase of 39% acetonitrile in aqueous 0.1% formic acid was used to elute components from a 2.1- by 50-mm 3.5-μm XBridge C18 column (Waters Associates). The eluate was detected by using a Sciex API-4000 (AB Sciex, Framingham, MA) equipped with a TurboIonspray ionization source using positive ion mode and multiple reaction monitoring (dolutegravir, ion transition m/z 420 to 277; internal standard, ion transition m/z 428 to 277). Data acquisition and processing were performed with Analyst 1.4.1 software (AB Sciex). The calibration range for dolutegravir was 5 to 5,000 ng/ml. Quality control samples, prepared at three different analyte concentrations and stored with study samples, were analyzed with each batch of samples against separately prepared calibration standards. For the analysis to be acceptable, no more than one-third of the total quality control results and no more than one-half of the results from each concentration level were allowed to deviate from the nominal concentration by more than 15%. The applicable analytical runs met all predefined run acceptance criteria.
Pharmacokinetic analysis of dolutegravir.
All pharmacokinetic parameters were calculated by using standard noncompartmental analysis and WinNonlin Pro 5.2 software (Pharsight Corp., Mountain View, CA) based on actual sampling times. After single-dose administration, the following pharmacokinetic parameters were calculated for total radioactivity in whole blood and plasma and for dolutegravir in plasma: area under the concentration-time curve from time zero to the last quantifiable time point (AUC0–t), area under the concentration-time curve from time zero to infinity (AUC0–∞), maximum observed concentration (Cmax), time to Cmax, absorption lag time, and terminal-phase t1/2. The additional parameters apparent oral clearance and apparent volume of distribution were calculated for plasma dolutegravir. Total-radioactivity/dolutegravir ratios were calculated for the terminal-phase t1/2 values and for plasma AUC0–∞ values. The blood-to-plasma ratio of total radioactivity (Cb/Cp) was calculated at each time point, and the percent association with blood cellular components (%Assoc) was calculated as follows: %Assoc = 100 − {[Cp × (1 − Hct)]/Cb} × 100, where Hct is the hematocrit expressed as a fraction, Cb is the concentration of radioactivity in blood, and Cp is the concentration of radioactivity in plasma. Plasma concentration-time profiles for dolutegravir were compared with those for total radioactivity to estimate how much of the total measured radioactivity was due to metabolites. Descriptive statistics were provided for pharmacokinetic parameters.
Sample preparation for radiochemical profiling. (i) Sample pooling.
Three individual pools of plasma were created from the 6-, 24-, and 48-hour-postdose samples by combining equal volumes of plasma from each subject at each time point. Representative samples of urine and fecal homogenates from each subject were pooled proportionally by sample weight to obtain a pool containing 85% or more of the radioactivity excreted via that route.
(ii) Sample pretreatment.
To enhance extraction of radioactivity, each plasma and fecal-sample portion was mixed with a half portion of EDTA disodium salt solution (10 mg/ml in water) before initial extraction.
(iii) Plasma and fecal-homogenate extraction.
A portion of each pooled plasma or fecal-homogenate sample was extracted by adding 1 sample volume of methanol and vortex mixing, followed by the addition of 3 sample volumes of acetonitrile. The extract was centrifuged, and the supernatant was transferred into a fresh tube. The extraction procedure was repeated twice on the residual pellet, and the resulting supernatants were combined with the supernatant from the first extraction. The fecal pellet was further extracted twice with acetonitrile-water-formic acid (50:50:0.1 by volume), and the resulting supernatants were combined with the previous extractions. The total weight of each combined extract was determined, and weighed portions were removed for LSC to determine the efficiency of extraction of radioactivity. The combined supernatants were dried under a stream of nitrogen and reconstituted by adding methanol (200 μl) and water containing EDTA (2 mg/ml; 1,000 μl). The sample extract was centrifuged, and then a portion of each supernatant was removed to determine the recovery of radioactivity upon reconstitution. A portion was analyzed for the metabolic profile by using HPLC with radiochemical detection.
(iv) Urine extraction.
A pooled urine sample from each subject was centrifuged, and a portion was analyzed by using HPLC with radiochemical detection.
Conditions for HPLC radiochemical profiling.
Radiochemical profiles of plasma, urine, and feces were generated by using an Agilent (Palo Alto, CA) 1200 HPLC system. Radiochromatographic peaks were separated following injection onto a Symmetry C18 column (100 by 4.6 mm; 3.5 μm; Waters Associates) at 40°C and eluted under gradient conditions with a mobile phase consisting of 2 solvents: solvent A, 0.1% ammonium acetate in water; and solvent B, 0.1% ammonium acetate in acetonitrile. The gradient conditions at a flow rate of 1.5 ml/min were as follows. Solvent B started at 5% and increased linearly to 25% from 3.3 to 40 min and then to 35% from 40 to 43.3 min and to 95% from 43.3 to 46.7 min. The gradient was held at 95% B for 1.3 min and returned to the initial conditions. The eluate was analyzed with a model 625TR series radiochemical flow detector (PerkinElmer, Waltham, MA) and Ultima Flo M scintillation fluid (3 ml/min). For samples requiring greater sensitivity, radiochemical profiles were generated offline by collecting column effluent fractions into Deepwell LumaPlate 96-well plates (PerkinElmer), drying under a stream of nitrogen, and then analyzing with a TopCount NXT microplate scintillation counter (PerkinElmer). The limit of detection and quantification for offline analysis was set to 2 times background.
Metabolite isolation and identification.
The HPLC method described above for radiochemical profiling was also applied to metabolite analyses. An Agilent 1100 HPLC System was interfaced with an LTQ-Orbitrap hybrid mass spectrometer (ThermoFisher Scientific, San Jose, CA), and LC–MS-MS spectra were acquired by electrospray ionization in the positive ion mode using data-dependent scanning from a mass list, which consisted of all known and likely metabolites. During the LC separation, a postcolumn split was used to direct 20% of the sample to the mass spectrometer. A full-scan mass spectrum (resolution, 30,000) was collected, and the data were interrogated in real time to identify mass peaks (±5 ppm). If present, the metabolite mass peaks were selected as target peaks for subsequent MS-MS scans using either high- or low-resolution settings.
The remaining 80% of LC effluent from the postcolumn split was fractionated by a Collect Pal fraction collector (LEAP Technologies, Carrboro, NC) at 20 s per well. A portion (25 μl) from each fractionated well was transferred into a deep-well LumaPlate-96 solid-scintillant microplate (PerkinElmer) and dried under a stream of nitrogen. The radioactivity in each well was measured by using a Topcount NXT microplate scintillation counter (PerkinElmer). The data from the Topcount NXT were transferred to Excel (Microsoft, Redmond, WA) and used to generate reconstructed radiochemical profiles. The reconstructed radiochemical profiles were compared with the LC–MS-MS data and the definitive radiochemical profiles to ensure proper peak assignment.
Urine from the repeat dose administration was used to isolate adequate amounts of M4 for nuclear magnetic resonance (NMR) characterization. Urine was pooled by volume across all subjects to create a pool of 2.3 liters and then was filtered using a 0.45-μm nylon filter(s) before being freeze-dried in an Advantage Lyophilizer (Virtis, Gardiner, NY). The dried urine sample was reconstituted in 0.1% ammonium acetate in water-0.1% ammonium acetate in acetonitrile, (90:10 [vol/vol]; 230 ml), and portions were centrifuged at ambient temperature before metabolite isolation. To ensure adequate purity for NMR analysis, the isolation of M4 was performed using three separate LC methods. During each LC separation, a postcolumn split was used to direct 10% of the sample to an LTQ mass spectrometer (ThermoFisher Scientific). Data-dependent scanning with a mass list was used to initiate time-based fraction collection for the remaining 90% of the LC effluent. Fractions were combined and reduced to dryness using a rotary concentrator (Genevac Inc., Gardiner, NY). Before each cleanup, LC-MS was performed to provide mass confirmation of M4. Liquid chromatography method 1 employed an Agilent 1100 HPLC System with a Symmetry Prep C18 column (7.8 by 150 mm; 7 μm; Waters Associates) at 40°C and eluted under gradient conditions with a mobile phase consisting of two solvents: solvent A, 0.1% ammonium acetate in water, and solvent B, 0.1% ammonium acetate in acetonitrile. The gradient conditions at a flow rate of 5.0 ml/min were as follows. Solvent B started at 10% and increased linearly to 22% from 0 to 15 min and then to 95% from 15 to 15.1 min. The gradient was held at 95% B for 3 min and returned to the initial conditions. The fractions corresponding to M4 were then subjected to a second LC method (LC method 2), which employed an Agilent 1200 HPLC System with a Luna Phenyl Hexyl column (4.6 by 150 mm; 7 μm; Phenomenex, Torrance, CA) at 40°C, and eluted under gradient conditions with a mobile phase consisting of 2 solvents: water with 0.1% formic acid and solvent B, acetonitrile with 0.1% formic acid. The gradient conditions at a flow rate of 1.0 ml/min were as follows. Solvent B started at 15% and increased linearly to 27% from 0 to 15 min and then to 95% from 15 to 15.1 min. The gradient was held at 95% B for 3 min and returned to the initial conditions. The final purification (LC method 3) was on the same LC system as method 2 but used a Zorbax SB-C8 column (4.6 by 150 mm; 5 μm; Agilent, Walnut Creek, CA) and the following linear gradient. Solvent B started at 5% and increased linearly to 30% from 0 to 25 min and then to 95% from 25 to 25.1 min. The gradient was held at 95% B for 3 min and returned to the initial conditions.
Purified metabolite M4 was dissolved in 180 μl of d4-methanol (D4 99.8% CIL; Cambridge Isotope Laboratories, Andover, MA), sonicated, and placed in a 3-mm (outside diameter [OD]) NMR tube (Norell S-3-HT-7; Landisville, NJ) before NMR analysis. The NMR experiments were conducted on either an Avance II 700-MHz (5-mm TCI cryoprobe) or 600-MHz (5-mm TFI cryoprobe) NMR spectrometer (Bruker, Billerica, MA). The experiments conducted included 1H with and without 19F decoupling, 19F, 2D-TOCSY (2-dimensional total correlation spectroscopy), 2D-ROESY (rotating-frame Overhauser effect spectroscopy), 1H-13C gHSQC (gradient-selected heteronuclear single-quantum coherence), 1H-13C gHMBC (gradient-selected heteronuclear multiple-bond coherence), and 1H-19F gHSQC as implemented in TopSpin 2.0.4 software (Bruker).
Density functional-theory calculations of 13C chemical shifts for possible regioisomers of M4 were completed using Spartan 10 software (Wavefunction, Irvine, CA). Both geometry optimization and 13C chemical shifts were determined using the B3LYP/6-31G* level of theory and basis sets.
RESULTS
Subjects.
Six healthy, nonsmoking white males aged between 32 and 46 years (mean, 37.5 years) with body mass indices between 23.7 and 28.4 (mean, 26.6) participated in the study.
Safety and tolerability.
All subjects completed the study portion as planned and received the correct treatment in the fasting state. The treatments were well tolerated, and no deaths, nonfatal serious adverse events, pregnancies in female partners of male subjects, or withdrawals due to AEs were reported. Five out of six subjects (83%) reported at least one AE during the study, with diarrhea (2 subjects; 33%) being the most commonly reported drug-related AE. All AEs were mild (grade 1) to moderate (1 subject with grade 2 vomiting) in intensity. No grade 4 or drug-related grade 3 laboratory abnormalities were reported. No clinically significant trends in postdose clinical laboratory values, vital signs, or electrocardiograms were observed.
Excretion and recovery of radioactivity.
After oral administration of [14C]dolutegravir, the recovered radioactivity, which was principally detected in feces, accounted for a mean of 64.0% of the administered dose (range, 59.4% to 72.0%) (Table 1). Urinary excretion accounted for a mean of 31.6% of the administered dose (range, 24.0% to 36.7%). The mean total recovery of radioactivity (Fig. 2) was 95.6% of the dose (range, 93.2% to 97.6%) by 216 h postdose. Most of the dose (94.5%) was recovered in feces and urine by 144 h postdose. Variability in recovery of radioactivity was very low, with coefficient of variation values of 1.5% (total recovery), 7% (feces), and 15% (urine). The radioactivity recovery measurements were based on the actual radioactive dose administered to each volunteer.
Table 1.
Summary of percent radioactivity recovered in urine and feces from healthy male human subjects after a single oral 20-mg (80-μCi) suspension dose of [14C]dolutegravir
| Subject | % of administered dose |
||
|---|---|---|---|
| Feces | Urine | Total | |
| 531001 | 72.0 | 24.0 | 96.1 |
| 531002 | 65.2 | 30.6 | 95.8 |
| 531003 | 61.0 | 34.8 | 95.8 |
| 531004 | 59.4 | 33.8 | 93.2 |
| 531005 | 65.7 | 29.4 | 95.1 |
| 531006a | 60.8 | 36.7 | 97.6 |
| Mean | 64.0 | 31.6 | 95.6 |
| SD | 4.7 | 4.6 | 1.4 |
Subject 531006 withdrew from the study after 144 h postdose. The subject was excluded from descriptive statistics after this time point.
Fig 2.

Overall mean cumulative elimination of radioactivity by six healthy male subjects after a single 20-mg (80-μCi) oral dose of [14C]dolutegravir.
Pharmacokinetics of radioactivity and dolutegravir.
The recovery of radioactive material from the 6-, 24-, and 48-hour pooled plasma samples following solvent extraction ranged from 99.8% to 104%, indicating no noticeable irreversible binding to or sequestration by plasma components.
The mean concentration-time profiles of total radioactivity in blood and plasma and of dolutegravir in plasma are presented in Fig. 3. A summary of select pharmacokinetic parameters is presented in Table 2. Absorption of radioactivity and dolutegravir from the suspension formulation was rapid, with a median time to peak concentration ranging from 0.5 h (plasma dolutegravir and radioactivity) to 1.25 h (blood radioactivity). Plasma concentrations of radioactivity and dolutegravir declined biphasically, falling below the limit of quantification by 168 h postdose. Dolutegravir represented the predominant drug-related component in the plasma, essentially accounting for all the plasma radioactivity through 4 h postdose, with the plasma dolutegravir AUC0–∞ accounting for greater than 97% of the total plasma radioactivity AUC0–∞. The mean blood/plasma radioactivity concentration ratios through 72 h postdose ranged from 0.441 to 0.535, and the association with blood cellular components (hematocrit range, 43.1 to 46.6) was less than 5% at all time points in all subjects, indicating minimal association of dolutegravir or metabolites with blood cells. The estimated terminal t1/2 values were similar among plasma dolutegravir (15.6 h), plasma radioactivity (15.7 h), and blood radioactivity (14.6 h). The geometric mean t1/2 ratios of dolutegravir to blood radioactivity and of dolutegravir to plasma radioactivity were 1.07 and 0.99, respectively.
Fig 3.

Mean concentration-time profiles of plasma dolutegravir and total radioactivity in blood and plasma after a single 20-mg (80-μCi) oral dose of [14C]dolutegravir to six healthy male subjects. (Inset) Data using log scale on the y axis.
Table 2.
Summary of selected pharmacokinetic parameters for plasma dolutegravir, plasma radioactivity, and blood radioactivity in healthy male human subjects after a single oral 20-mg (80-μCi) dose of [14C]dolutegravir as a suspensiona
| Analyte | n | Cmax (μg/ml) | Tmaxb (h) | AUC0–t (μg · h/ml) | AUC0–∞ (μg · h/ml) | CL/F (liter/h) | Vz/F (liter) | t1/2 (h) |
|---|---|---|---|---|---|---|---|---|
| Plasma dolutegravir | 6 | 2.57 (24) | 0.50 (0.50–2.00) | 35.7 (12) | 35.9 (12) | 0.56 (12) | 12.5 (9) | 15.6 (16) |
| Plasma radioactivity | 6 | 2.46 (24) | 0.50 (0.50–1.50) | 35.9 (11) | 36.1 (11) | NR | NR | 15.7 (14) |
| Blood radioactivity | 6 | 1.13 (25) | 1.25 (0.50–2.00) | 17.7 (13) | 18.4 (13) | NR | NR | 14.6 (12) |
Values are geometric mean (percent coefficient of variation) unless otherwise stated; NR, not reported. CL/F, apparent oral clearance; Tmax, time to Cmax; Vz/F, apparent volume of distribution.
Median (range).
Metabolite profiling.
Representative radiochromatograms from pooled 24-hour plasma samples, the 0- to 72-hour urine collection pool, and the 0- to 96-hour fecal homogenate pool are presented in Fig. 4. A summary of the mean relative abundances of the principal quantifiable radiochromatographic peaks is presented in Table 3. Recovery of radioactivity from each excreta sample following the extraction procedures ranged from 95% to 101%.
Fig 4.

Representative radiochromatograms of plasma, urine, and feces after a single oral 20-mg (80-μCi) dose of [14C]dolutegravir to an individual healthy male subject. (A) Plasma at 24 h. (B) Urine at 0 to 72 h. (C) Feces at 0 to 96 h.
Table 3.
Radiochromatographic analyses of urine and feces following a single oral administration of [14C]dolutegravir to healthy male human subjects at a target dose of 20 mg (80 μCi)
| Peak ID | Retention time (min) | % Matrix radioactivity (mean ± SD; n = 6) [% dose (mean ± SD)]a |
|
|---|---|---|---|
| Urine | Feces | ||
| M1 | 12.2–12.8 | 11.8 ± 2.0 [3.6 ± 0.9] | 2.2 ± 0.7 [1.3 ± 0.4] |
| M2 | 24.4–24.8 | 62.5 ± 3.9 [18.9 ± 3.0] | ND |
| M3b | 37.2 + 40.2 | 10.1 ± 2.8 [3.0 ± 0.9] | ND |
| M4 | 24.0–24.4 | NQ | 3.1 ± 1.3 [1.8 ± 1.3] |
| Dolutegravir | 44.4 | 2.2 ± 0.2 [0.7 ± 0.1] | 89.1 ± 3.3 [53.1 ± 4.4] |
| Total radioactive material assigned | 86.6 ± 2.3 [26.2 ± 3.9] | 94.4 ± 3.5 [56.3 ± 3.9] | |
| % Dose in matrix pool analyzed | 30.2 ± 4.3 | 59.7 ± 5.5 | |
| Total % dose excreted in matrix (all time points) | 31.6 ± 4.6 | 64.0 ± 4.7 | |
ND, not detected (below lower limit of detection of 3 times background); NQ, not quantifiable (radioactivity was present at this retention time, but the concentration was too low for complete structural identification).
The reported values for M3 are the sum of both diastereomers.
(i) Plasma.
Dolutegravir accounted for 95.2%, 96.8%, and 99.8% of the radioactivity in the 6-, 24-, and 48-hour pooled plasma radiochromatograms, respectively. The glucuronide, M2, was a minor component, corresponding to 2.4% of the 6-hour and 1.5% of the 24-hour plasma pool radiochromatograms, and was not quantifiable in the 48-hour plasma pool radiochromatogram. Combined, dolutegravir and M2 accounted for approximately 98.6% of the total radioactivity in each of the three plasma pools.
(ii) Urine.
The principal quantifiable radiochromatographic peaks identified (Table 3) were unchanged dolutegravir, its glucuronide conjugate (M2), and a product of oxidation (M3) with its hydrolysis product (M1). The predominant component in urine was M2, which represented a mean of 62.5% of the radioactivity (18.9% of the dose). The renal excretion of unchanged dolutegravir was low (0.7% of the dose). Small radiochromatographic peaks present, which collectively accounted for <5% of the administered dose, were not fully characterized.
(iii) Feces.
Three quantifiable radiochromatographic peaks were detected, with dolutegravir accounting for 89.1% of the radioactivity (53.1% of the dose). The two minor radioactivity peaks corresponded to M1 and M4. Together, these three drug-related components accounted for 94.4% of the total radioactivity in the fecal homogenates (Table 3).
Metabolite identification.
The metabolites observed in plasma or excreta were structurally characterized by high-resolution mass spectrometry. In addition, M3 and M4 required NMR methods for full characterization (Table 4). The primary MS-MS fragmentation of dolutegravir differentiates the difluorobenzyl group from the tricyclic moiety and therefore is useful in characterizing the biotransformations. The most prominent metabolite in plasma and urine, M2, was characterized as an ether glucuronide conjugate of dolutegravir and matched the chromatographic and mass spectral data of the synthetic standard. Mass spectral data for the two diastereomers of M3 indicated the addition of an oxygen atom on the difluorobenzyl portion of the molecule. Further, characterization by 1H NMR (unpublished data) indicated that the prochiral benzylic methylene group was oxidized to form a hemiaminal. The N-dealkylated metabolite M1, which is related to M3, was readily characterized by MS analysis.
Table 4.
Structural characterization data for dolutegravir and its metabolites from high-resolution mass spectrometric and NMR analyses
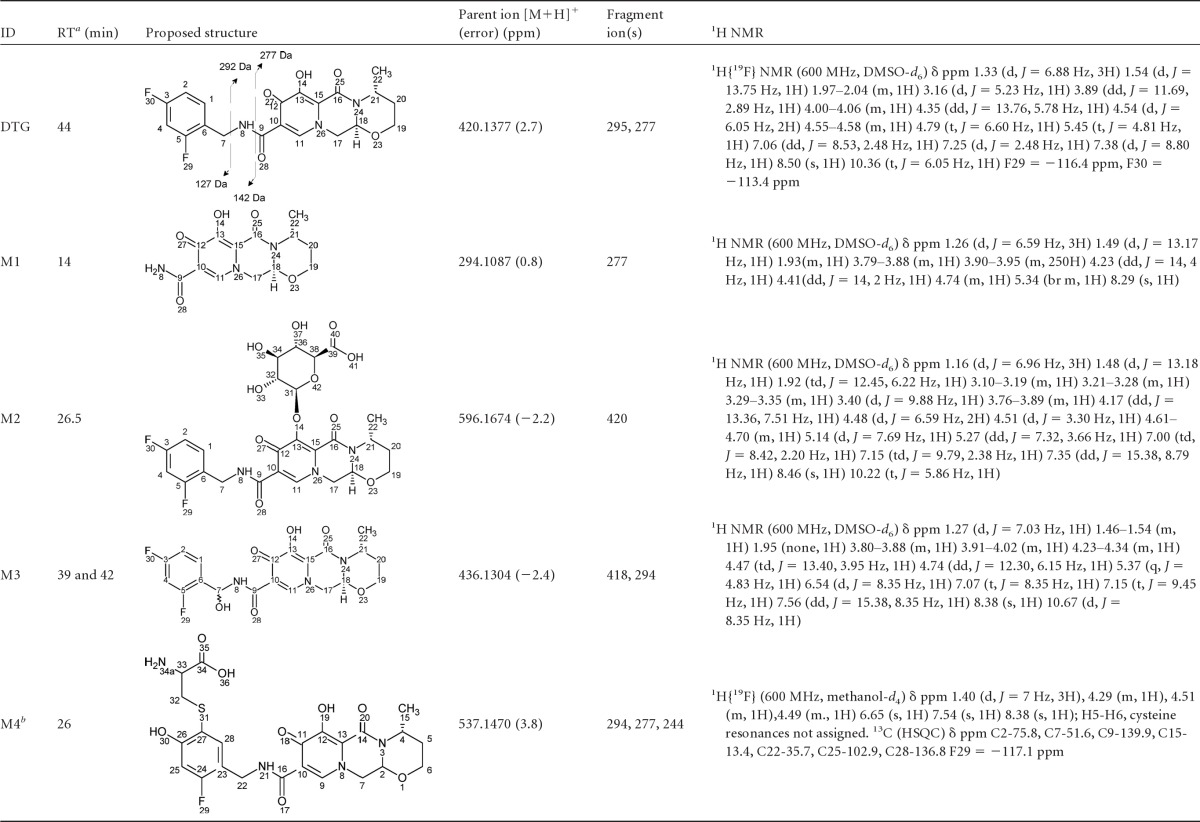
RT, retention time.
A putative structural isomer is shown.
The most challenging metabolite structure elucidation was that of M4. The decision to initiate a detailed characterization of M4, even though in urine it was below the limits of quantification and represented on average 1.8% of the dose in feces, was based on the preliminary observation that it was derived from a glutathione pathway and linked to an initial bioactivation step. Based on accurate mass data, M4 formation consisted of formal loss of a fluorine atom and the additions of an oxygen atom and a cysteine residue. A total of 12 possible regioisomers would be consistent with these data if the biotransformations occurred exclusively on the aromatic ring. An additional two isomers must be considered if oxidation of the benzylic carbon is included. Our strategy relied on the isolation of sufficient quantities for NMR interrogation using dolutegravir chemical shift and coupling constants as a reference. Even though the concentration of M4 in human urine from a repeat dose study was lower than that observed in feces from the [14C]dolutegravir single-dose study, the repeat dose urine samples were deemed an easier biological matrix to isolate and purify metabolites. We estimate that <10 μg of M4 was isolated for NMR analysis. The 1H NMR spectrum acquired with 19F decoupling displayed two singlet aromatic resonances at 6.65 and 7.54 ppm. The absence of observable 1H-1H coupling is consistent with these two protons having a 1,4 relationship on the aromatic ring. Both the TOCSY and ROESY spectra indicate that the proton at 7.54 ppm has a correlation with the benzyl protons at 4.51 ppm, establishing the benzyl group adjacent or ortho to this proton. These data exclude all but four possible structural isomers (Fig. 5). Carbon chemical shift data for protonated carbons were acquired from a gHSQC experiment. Unfortunately, too little material was present to collect quaternary carbon chemical shift data using the gHMBC pulse sequence. Carbon chemical shifts were calculated using ab initio methods for the four remaining isomers (Fig. 5). The observed carbon chemical shift data are most consistent with both isomers I and III based on the comparison with the calculated values.
Fig 5.
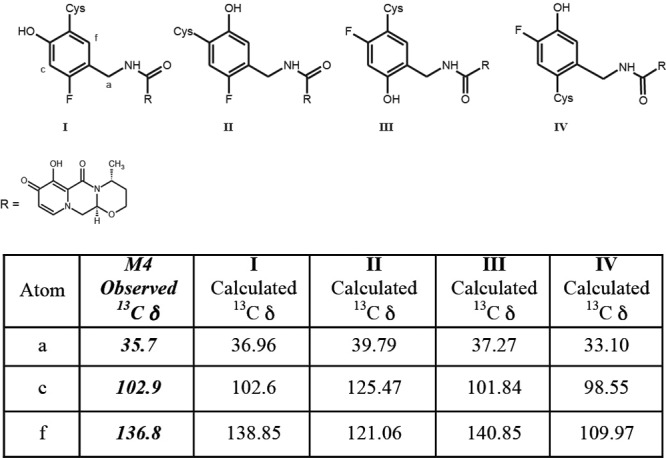
Nuclear magnetic resonance characterization of M4 regioisomers.
The single fluorine atom of M4 has a chemical shift of −117.1 ppm, which is similar to the ortho-fluorine chemical shift of dolutegravir that was observed at −116.4 ppm (the para-fluorine chemical shift of DTG was −113.4 ppm). However, 19F chemical shifts vary with concentration, solvent, and endogenous components; therefore, the observed 19F chemical shift for M4 could not be used to definitively identify a specific regioisomer. In dolutegravir, a large H-F coupling constant was observed between the ortho-fluorine and the benzylic protons. This was not observed in the case of M4, but this negative evidence was not compelling enough to make a definitive assignment between isomers. Consequently, the experimental ambiguity associated with both the 19F chemical shift and the coupling constant to the benzylic protons did not permit us to determine if M4 contains an ortho- or para-fluorine and, thus, differentiate between isomers I and III (Fig. 5).
DISCUSSION
The pharmacokinetics, metabolism, and excretion of dolutegravir after a single oral-suspension dose (20 mg or 80 μCi) of [14C]dolutegravir were investigated in this study. Dolutegravir was well tolerated in these healthy male subjects, with a safety profile consistent with other phase I dolutegravir studies and which have been previously reported in patients who have received a dose of 50 mg that resulted in a significant drop in plasma HIV-1 RNA (8–10). Mass balance was achieved, with most of the radioactivity recovered in urine and feces within 6 days after oral administration, consistent with the observed oral t1/2 and indicative of the expected metabolic stability of the radiolabel. By its mode of action, dolutegravir binds multivalent cations, and the mass balance that was attained, in addition to the complete extraction from sample matrices, indicates no notable sequestration or covalent binding of dolutegravir or metabolites. During an initial rising-dose pharmacokinetic study, the AUC of dolutegravir increased proportionally with single oral-suspension doses of 2 to 100 mg and showed time-independent pharmacokinetics during administration of repeat oral-suspension doses of 10 to 50 mg (11). The results from this initial pharmacokinetic study, combined with the pharmacokinetics from our current study, indicate that a 20-mg dose can be extrapolated directly to the anticipated clinical dose of 50 mg.
Recovery from excreta and radiochromatograms showed low intersubject variability, with only small quantitative differences across each of the six subjects complementing the low pharmacokinetic variability of dolutegravir. Fecal excretion was the primary route of elimination and reflects both elimination of unabsorbed material and biliary secretion of dolutegravir or its metabolic products.
Absorption of dolutegravir from the suspension formulation was rapid, consistent with earlier observations (11). Based on the fractions of the dose renally eliminated (31.6%) and excreted in the feces as oxidative metabolic products (3.1%), at least 34% of the administered dose was absorbed. The expected biliary secretion of dolutegravir glucuronide, the principal metabolite, suggests that the absorption of dolutegravir is higher. Although bile was not collected as part of this study, biliary secretion of dolutegravir and dolutegravir conjugates (i.e., M2) in these subjects is inferred from the analysis of bile samples collected from rats and monkeys (unpublished data) and from molecular weight hypotheses of biliary secretion (16). The mean whole-gut (colonic) transit time (the time from ingestion to passage) in humans has been reported to be approximately 30 to 40 h, with an upper limit of normal of 70 h and a lower value of 14 h (17). Therefore, if the mean percentages of the dose at 48 and 72 h postdose recovered in the feces of 16% and 31%, respectively, represent unabsorbed material, then the 33% to 48% remaining to be excreted after this time would be indicative of dolutegravir conjugates that undergo biliary secretion and are excreted as parent dolutegravir. Thus, absorption of dolutegravir is expected to be higher than that reflected by fecal elimination, and the total systemic burden of absorbed material would be approximately 70% to 80%. Food was noted to modestly increase systemic exposure to dolutegravir, but the effect is not clinically significant, and dolutegravir can be taken without regard to meals (11, 18). Therefore, the results from this study are applicable to the nonfasted or fasted state, as food is not expected to alter the metabolic profile of dolutegravir.
The similarity of the dolutegravir concentration and the radioactivity concentration plasma profiles indicates that unchanged dolutegravir was the predominant drug-related component in the plasma, accounting for more than 97% of the total plasma radiocarbon AUC0–∞. This was confirmed by LC–MS-MS analysis, where unchanged dolutegravir represented the entire radioactivity content at Cmax. Metabolic products appeared at later time points, consistent with low to negligible presystemic clearance. The parallel terminal-phase t1/2 values of radioactivity and of dolutegravir indicated that the products that were present in the systemic circulation were formation rate limited and did not persist. This observation supported the electrocardiographic sampling scheme for a cardiac repolarization study, which primarily focused on the anticipated maximum concentration and duration in plasma of the parent compound (14).
The pharmacokinetic parameters from the suspension dose indicated low variability and were consistent with the parameter estimates previously reported for doses of 2 to 100 mg when administered as a suspension (11). The mean concentration, 0.502 μg/ml, of dolutegravir at 24 h after a single 20-mg dose was 8-fold higher than the protein-adjusted 90% inhibitory concentration (0.064 μg/ml) and, together with the observed oral dolutegravir t1/2 (15.6 h), further supports a once-daily regimen. The low exposure to total circulating metabolites representing less than 5% of the dolutegravir-related components would be expected not to contribute significantly to either the pharmacological activity of or intolerance for dolutegravir.
Dolutegravir was extensively metabolized with observed biotransformations following three primary pathways: glucuronidation of dolutegravir principally by UGT1A1, carbon oxidation via CYP3A4 (12), and what appears to be a sequential oxidative defluorination and glutathione conjugation (Fig. 6). The ether glucuronide metabolite, M2, was the only metabolite characterized in plasma and represented the predominant metabolism pathway based on the mean percentage of the dose recovered (18.9% in urine). The formation of the ether glucuronide disrupts the ability of the molecule to bind to metal ions, and thus, the metabolite is inactive.
Fig 6.

Metabolic scheme for dolutegravir after a single oral 20-mg (80-μCi) dose of [14C]dolutegravir to six healthy male subjects.
The oxidation of the benzylic methylene generated two diastereomeric hemiaminal intermediates (M3) detected in urine. The reduced basicity of the amide nitrogen prevents complete chemical decomposition to the corresponding amide, M1, and the corresponding difluorobenzaldehyde. Consequently, the significance of this biotransformation is best reflected in the sum of the M3 diastereomers and M1 as a mean percentage of the dose (6.6%). It was noted during the course of sample manipulation and repeated chromatographic runs that the radio peak areas corresponding to the two diastereomers changed, suggesting that some decomposition to M1 could occur during sample processing.
Refinement beyond isomer I or III to a definitive structure could not be achieved for M4 due to the limited amount of sample and ambiguous 19F NMR data. From a mechanism perspective, isomer I can be rationalized as the result of an initial enzyme-mediated addition of an oxygen atom to generate either a fluoro-hydroxyl cyclohexyl diene or the corresponding epoxide intermediate, followed by glutathione addition and HF elimination (Fig. 7). In contrast, formation of isomer III would require two separate biotransformations: oxidative defluorination to generate a stable 2-hydroxy-4-fluorobenzyl group and glutathione opening of an epoxide intermediate where water rather than HF is eliminated. Both biotransformations in the formation of isomer III appear to involve epoxide intermediates. The mechanism leading to isomer III is more difficult to rationalize based on the biotransformation literature (19–21). Thus, although there are not enough spectroscopic data from this study to definitively characterize M4 as isomer I, mechanistic considerations favor this structure. Although the proposed mechanism of formation for M4 proceeds through an arene oxide, the low fractional clearance through this pathway, lack of evidence for covalent binding, and the low to moderate dose administered suggest the risk for drug-induced toxicities via this potential bioreactive species is low. This analysis indicating the low body burden of systemic metabolites is consistent with the strategic analysis recently reviewed (22).
Fig 7.

Proposed mechanism for the formation of M4 isomer I.
In conclusion, following oral administration, the recovery of dolutegravir and dolutegravir-related components from urine and feces was nearly complete, with feces being the principal route of excretion (Fig. 8). Dolutegravir was the predominant circulating compound in plasma and was consistent with minimal presystemic clearance. Radioactivity was minimally associated with the blood cellular components. At least 34% of the dose was absorbed, with an additional 33% to 48% fraction undergoing enterohepatic recirculation. Dolutegravir was extensively metabolized, and the disposition kinetics of the metabolites was formation rate limited, indicating no long-lived metabolites. An inactive ether glucuronide, formed primarily via UGT1A1, was the principal biotransformation pathway (at least 18.9% of the dose, with an additional amount secreted in bile and deconjugated), and minor biotransformation pathways included oxidation by CYP3A4 (7.9% of the dose) and a correlated oxidative defluorination and glutathione substitution (1.8% of the dose). These biotransformations were also observed in nonclinical studies, thereby supporting selection of the nonclinical species for safety coverage in humans. Thus, this study provides an understanding of the clearance pathways via the routes of metabolism and excretion of dolutegravir, underpinned by the nonclinical safety assessment and the low risk of idiosyncratic reactions by bioreactive metabolites. The metabolic profile and fractional clearances obtained from this study, in conjunction with the in vitro investigations (14), provide the mechanistic basis for understanding potential effects when dolutegravir is coadministered with other therapeutic products.
Fig 8.

Summary of the mean excretion and mass balance after a single oral administration of [14C]dolutegravir to six healthy male subjects at a target dose of 20 mg (80 μCi).
ACKNOWLEDGMENTS
We all meet the criteria for authorship set forth by the International Committee for Medical Journal Editors and are all employees of GlaxoSmithKline.
We are grateful to the following study contributors for their support of this investigation: Stephen D. Flach, principal investigator, and the staff at the Covance Clinical Research Unit; Nate Ensrud and Vikash Patel for radioanalysis at Covance Laboratories; and the subject participants without whom this study would not be possible. We also acknowledge Glenn Tabolt for quantitative MS assistance, Ernest Schubert and Andy Roberts for helpful NMR discussions, and Eri Kanaoka of Shionogi & Co., Ltd., for review of the experimental protocols and reports.
This study was supported by ViiV Healthcare. Editorial assistance was provided under our direction by Chris Lawrence, with funding from ViiV Healthcare.
J. Borland, S. C. Piscitelli, I. Song, S. Chen, Y. Lou, S. S. Min, and P. M. Savina participated in study design; D. Wagner, L. Moss, I. Goljer, and A. Culp conducted experiments; J. Borland, S. C. Piscitelli, I. Song, S. Chen, S. S. Min, Y. Lou, S. Castellino, and P. M. Savina performed data analysis; and J. Borland, S. C. Piscitelli, I. Song, S. Chen, Y. Lou, S. S. Min, P. M. Savina, S. Castellino, D. Wagner, L. Moss, I. Goljer, and A. Culp wrote or contributed to the writing of the manuscript.
Footnotes
Published ahead of print 13 May 2013
REFERENCES
- 1. Engelman A, Craigie R. 1992. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66:6361–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. 2006. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol. Pharmacol. 69:1454–1460 [DOI] [PubMed] [Google Scholar]
- 3. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. 2010. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 107:20057–20062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hare S, Smith SJ, Métifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P. 2011. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 80:565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 55:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL, III, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR. 2011. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob. Agents Chemother. 55:4552–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Min S, Sloan L, DeJesus E, Hawkins T, McCurdy L, Song I, Stroder R, Chen S, Underwood M, Fujiwara T, Piscitelli S, Lalezari J. 2011. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS 25:1737–1745 [DOI] [PubMed] [Google Scholar]
- 9. van Lunzen J, Maggiolo F, Arribas JR, Rakhmanova A, Yeni P, Young B, Rockstroh JK, Almond S, Song I, Brothers C, Min S. 2012. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect. Dis. 12:111–118 [DOI] [PubMed] [Google Scholar]
- 10. Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, Poizot-Martin I, Richmond G, Soriano V, Ait-Khaled M, Fugiwara T, Huang J, Min S, Vavro C, Yeo J. 2013. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING study. J. Infect. Dis. 10.1093/infdis/jis750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Min S, Song I, Borland J, Chen S, Lou Y, Fujiwara T, Piscitelli SC. 2010. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 54:254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E, Webster LO, Harmon KA, Clarke JD, Polli JW. 2013. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 41:353–361 [DOI] [PubMed] [Google Scholar]
- 13. Food and Drug Administration Center for Drug Evaluation and Research Center for Biologics Evaluation and Research 2005. Guidance for industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. U. S. Department of Health and Human Services, Rockville, MD [Google Scholar]
- 14. Chen S, Min SS, Peppercorn A, Borland J, Lou Y, Song I, Fujiwara T, Piscitelli SC. 2012. Effect of a single supratherapeutic dose of dolutegravir on cardiac repolarization. Pharmacotherapy 32:333–339 [DOI] [PubMed] [Google Scholar]
- 15. Loevinger RL, Budinger TF, Watson EE. 1999. MIRD primer for absorbed dose calculations, revised ed Society of Nuclear Medicine, New York, NY [Google Scholar]
- 16. Yang X, Gandhi YA, Duignan DB, Morris ME. 2009. Prediction of biliary excretion in rats and humans using molecular weight and quantitative structure-pharmacokinetic relationships. AAPS J. 11:511–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao SSC, Camilleri M, Hasler WL, Maurer AH, Parkman HP, Saad R, Scott MS, Simren M, Soffer E, Szarka L. 2011. Evaluation of gastrointestinal transit in clinical practice: position paper of the American and European Neurogastroenterology and Motility Societies. Neurogastroenterol. Motil. 23:8–23 [DOI] [PubMed] [Google Scholar]
- 18. Song I, Borland J, Chen S, Patel P, Wajima T, Peppercorn A, Piscitelli S. 2012. Effect of food on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob. Agents Chemother. 56:1627–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dear GJ, Ismail IM, Mutch PJ, Plumb RS, Davies LH, Sweatman BC. 2000. Urinary metabolites of a novel quinoxaline non-nucleoside reverse transcriptase inhibitor in rabbit, mouse and human: identification of fluorine NIH shift metabolites using NMR and tandem MS. Xenobiotica 30:407–426 [DOI] [PubMed] [Google Scholar]
- 20. Yergey JA, Trimble LA, Silva J, Chauret N, Li C, Therien M, Grimm E, Nicoll-Griffith DA. 2001. In vitro metabolism of the COX-2 inhibitor DFU, including a novel glutathione adduct rearomatization. Drug Metab. Dispos. 29:638–644 [PubMed] [Google Scholar]
- 21. Samuel K, Yin W, Stearns RA, Tang YS, Chaudhary AG, Jewell JP, Lanza T, Jr, Lin LS, Hagmann WK, Evans DC, Kumar S. 2003. Addressing the metabolic activation potential of new leads in drug discovery: a case study using ion trap mass spectrometry and tritium labeling techniques. J. Mass Spectrom. 38:211–221 [DOI] [PubMed] [Google Scholar]
- 22. Smith DA, Obach RS. 2009. Metabolites in safety testing (MIST): considerations of mechanisms of toxicity with dose, abundance, and duration of treatment. Chem. Res. Toxicol. 22:267–279 [DOI] [PubMed] [Google Scholar]